

Abstract
Transplant glomerulopathy (TxG) can show secondary focal and segmental glomerulosclerosis (FSGS). FSGS in native kidneys is caused by podocytopenia. This study examines podocytopenia and the role of decreased paracrine Met activation on podocytes by decreased glomerular hepatocyte growth factor (HGF) levels in the development of podocytopenia in TxG. Podocytes were counted in 10 zero-hour biopsies and 10 specimens each with and without TxG. HGF/Met was examined with immunostains and quantitative RT-PCR in a set of three consecutive biopsies from 10 patients with TxG, including the diagnostic biopsy (DiagnBx) and the two previous biopsies (1stPrevBx and 2ndPrevBx). Antiapoptotic effects of HGF on podocytes were examined in vitro. Mean podocyte numbers per glomerulus were lower and glomerular volume higher in TxG. Fewer of the two preceding biopsies of the patients than of the controls contained phospho-Met(Tyr1349)–positive podocytes (2 of 8 versus 7 of 7, P = 0.0070; 4 of 9 versus 9 of 9, P = 0.0294). Glomerular HGF mRNA levels were lower in the 1stPrevBx of the patients (0.049 ± 0.083 versus 0.284 ± 0.331; P = 0.0155). In vitro, HGF stimulation of podocytes resulted in antiapoptotic phosphorylation of AKT and extracellular signal-regulated kinase (ERK) and induction of X-linked inhibitor of apoptosis protein (XIAP). Decreased antiapoptotic Met signaling in podocytes, probably due to decreased HGF secretion by glomerular epithelial cells, could contribute to podocyte loss and FSGS in TxG.
Despite improvements in immunosuppressive therapy, which have successfully reduced the incidence of acute allograft rejection in transplant kidneys, chronic rejection remains a clinical challenge.1 Although interstitial fibrosis and tubular atrophy (IFTA) is multifactorial in origin, transplant vasculopathy and transplant glomerulopathy (TxG) are considered specific lesions of chronic allograft rejection.2–7 The prevalence of TxG increases over time after transplantation from 5% at 1 year to 20% at 5 years.8 Early, subdiagnostic findings include an “activated” aspect of glomerular endothelial cells (GECs), with accumulation of mitochondria, Golgi apparati, and ribosomes, and a transition from fenestrated to continuous endothelium.9 Diagnostic findings are double contours of the glomerular basement membrane, visible by light microscopy, involving 10% or more of at least one glomerulus with mesangial proliferation.2 Similar changes are frequently present in the peritubular capillaries.10–12 This suggests a pathomechanism with primary endothelial damage throughout the kidney. This chronic endothelial damage is thought to be due to alloantibody fixation.8,12–14
Whereas the primary insult in TxG seems to be directed against GECs, other features of TxG, such as proteinuria8 and focal and segmental glomerulosclerosis15 are typical indicators of podocyte damage. Proteinuria suggests podocyte damage, because podocytes are considered to be the most important component of the glomerular filter.16 FSGS suggests podocyte damage, because podocytopenia is considered necessary for its development.17,18 Podocytopenia has been observed in native kidneys in various glomerulopathies such as primary FSGS,19 IgA glomerulonephritis,20 hypertensive nephrosclerosis,21 diabetic glomerulosclerosis,22,23 Heymann nephritis,24 and recently, also in a rat model of chronic allograft nephropathy.25 These observations suggest that during the development of TxG, primary damage to the GECs causes secondary podocyte damage that ultimately leads to podocytopenia and FSGS, since podocytes are considered postmitotic cells that are unable to replicate.26,27 The mechanism of how primary damage to the GECs could cause secondary podocyte damage is currently unknown. A possible mechanism could be disruption of cytokine signaling from GECs to podocytes. Based on literature results, hepatocyte growth factor (HGF) could be a candidate. HGF was originally discovered as a potent mitogen for hepatocytes.28,29 The active form of HGF is a heterodimer30 derived from a pre-proprecursor by proteolytic cleavage.31,32 HGF is also known as a renotrophic factor, and the kidney is the organ with the highest HGF expression.33,34 Mesenchymal cells such as GECs, mesangial cells, fibroblasts, and macrophages secrete renal HGF,34,35 which has a promitogenic,33 antiapoptotic,36 cytoprotective,37 and differentiation-preserving effect38 on a variety of renal cell types. HGF is the only known agonist of its receptor Met that becomes phosphorylated at tyrosine residues.39 Met is expressed in podocytes.40,41 Met activation leads to phosphorylation of downstream mediators AKT42 and extracellular signal-regulated kinase (ERK),43 which are both known to have antiapoptotic effects in general44 and also in podocytes.45,46 The present study tests the hypothesis that podocytopenia occurs in TxG and that disruption of the antiapoptotic HGF signal from GECs to podocytes is a possible cause of this podocytopenia. The association of TxG with podocytopenia is examined by glomerular morphometry in human zero-hour biopsies, regular transplant biopsies without TxG, and transplant nephrectomy specimens with TxG. The possible causative role of diminished HGF signaling is then examined retrospectively in human transplant biopsies with TxG and the two preceding biopsies, and in matched controls without TxG and in vitro in immortalized murine podocytes.
Materials and Methods
Measurement of Podocyte Density in Zero-Hour Biopsies, Regular Transplant Biopsies, and Transplant Nephrectomy Specimens with TxG
First, we tried to demonstrate podocytopenia in TxG. To this end, a set of each 10 zero-hour biopsies, 10 regular transplant biopsies without TxG, and 10 transplant nephrectomy specimens with severe TxG (cg3 according to Banff criteria2 with secondary FSGS), each containing at least 30 glomeruli, were selected from the archives of the Institute of Pathology, Hannover Medical School. These specimens are characterized in Table 1 and derived from 30 different patients. Podocyte density could not be measured in the biopsy collective below in which the role of HGF signaling was examined, because most of these biopsies contained only between 10 and 20 glomeruli. The scarce tissue remaining in these blocks was mostly used for assessment of the HGF/Met signaling axis and did not suffice to estimate podocyte density.
Table 1.
Characterization of the Specimens That Served to Determine Podocyte Density in Renal Transplants With and Without TxG
|
n = 10 |
|||
|---|---|---|---|
| Zero-hour Bx | Regular Bx without TxG | TxG | |
| Recipient sex (female:male) | 4:6 | 5:5 | 5:5 |
| Donor type (living:cadaveric) | 3:7 | 6:4 | 0:9 (1 unknown) |
| Donor sex (female:male) | 5:5 | 7:3 | 3:3 (4 unknown) |
| Time after transplantation | 0 ± 0.0 months | 3 ± 3.4 months | 165 ± 177.5 months |
| ai | 0.1 ± 0.32 | 0.4 ± 0.52 | 0.9 ± 0.99 |
| at | 0.0 ± 0.00 | 0.3 ± 0.67 | 0.5 ± 0.97 |
| av⁎ | 0.0 ± 0.00 | 0.0 ± 0.00 | 1.5 ± 0.85 |
| ag⁎ | 0.0 ± 0.00 | 0.0 ± 0.00 | 1.2 ± 1.03 |
| ptc | 0.0 ± 0.00 | 0.0 ± 0.00 | 0.8 ± 1.23 |
| ct⁎ | 0.1 ± 0.32 | 0.7 ± 0.48 | 3.0 ± 0.00 |
| ci⁎ | 0.1 ± 0.32 | 0.4 ± 0.52 | 3.0 ± 0.00 |
| cv | 0.0 ± 0.00 | 0.0 ± 0.00 | 1.7 ± 1.25 |
| cg⁎ | 0.0 ± 0.00 | 0.0 ± 0.00 | 2.8 ± 0.42 |
| mm⁎ | 0.0 ± 0.00 | 0.0 ± 0.00 | 2.5 ± 0.70 |
| ah | 0.6 ± 0.70 | 0.8 ± 0.79 | 1.8 ± 1.23 |
| Banff C4d peritubular⁎ | 0.0 ± 0.00 | 0.0 ± 0.00 | 1.3 ± 1.49 |
| C4dglomerular | 0.0 ± 0.00 | 0.0 ± 0.00 | 0.9 ± 1.10 |
Specimens included 10 each of the zero-hour biopsies (zero-hour Bx), transplant biopsies without TxG taken later after transplantation (regular Bx without TxG), and transplant nephrectomy specimens (TxG) from 30 different patients.
Bx, biopsy; ai, acute extent of interstitial infiltrate at severity of acute tubulitis; av, severity of acute endothelialitis; ag, severity of acute glomerulitis; ptc, severity of peritubular capillaritis; ct, extent of tubular atrophy; ci, extent of interstitial fibrosis; cv, severity of chronic transplant vasculopathy; cg, severity of transplant glomerulopathy; mm, extent of mesangial expansion; ah, severity of arteriolar hyalinosis.
P < 0.05.
WT1 IHC and Morphometry
Mean glomerular volume, mean podocyte number per glomerulus, and mean podocyte volume density were calculated according to a method recently described, which counts Wilms' Tumor-1 (WT1)-positive podocytes by evaluation of thick and thin sections.47 Thick sections were cut at 8 μm, thin sections at 4 μm. Both thick and thin sections were stained in a Bond III automatic immunostainer according to the manufacturer‘s instructions using a mouse monoclonal primary antibody (WT1-562-L-CE; Leica Microsystems, Wetzlar, Germany). Representative micrographs are displayed in Figure 1. JPEG images of 30 glomeruli were acquired from each specimen. The number of WT1-positive podocytes and also the glomerular area were determined with the public domain software ImageJ (http://rsbweb.nih.gov/ij/index.html, last accessed March 19, 2011). Podocyte nuclei in the glomerular tufts were identified by positive WT1 staining. Mean glomerular volume, mean podocyte number per glomerulus, and podocyte volume density were calculated as described previously.47 Calculations of the glomerular area and the different podocyte counts in the thick and the thin sections gave the podocyte volume density. Multiplication of the mean glomerular volume with the podocyte volume density yielded the mean podocyte number per glomerulus.47
Figure 1.
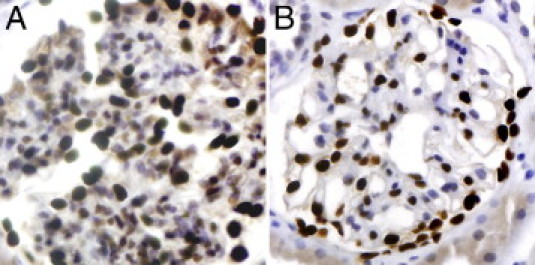
A: example of a glomerulus, podocyte nuclei with brown immunostaining for WT1 on a thick section (8 μm). B: Glomerulus from the same specimen with the same immunostain on a thin section (4 μm). Both, immunoperoxidase stain; original magnification, ×400.
Examination of HGF/Met Signaling in Patient and Control Biopsies
Next, we examined the possible role of decreased HGF/Met signaling on podocytes in the development of podocytopenia in TxG. Patient biopsies (PBx) and control biopsies (CBx) were chosen from the archives of the renal transplant protocol biopsy program of the Hannover Medical School. Inclusion criterion for the PBx group was biopsy-proven transplant glomerulopathy (at least cg1 and mm1 according to the Banff components2) within 4 years after renal transplantation. Exclusion criteria were any other de novo or recurrent glomerular diseases. Ten TxG patients fulfilling these criteria were selected. As set out in Figure 2, from each of these 10 TxG patients, the first biopsy that was diagnostic of TxG (DiagnBx) and the two preceding biopsies, the first and second previous biopsies (1stPrevBx and 2ndPrevBx), were included in the study. For each of these biopsies, a matched control biopsy (DiagnBx, the 1stPrevBx, and the 2ndPrevBx), which was taken at the same interval after transplantation, was chosen from a cohort of patients in which the development of TxG within 4 years after transplantation was excluded by biopsy. Thus, 30 biopsies from 10 TxG patients (10 each DiagnBx, 1stPrevBx, and 2ndPrevBx) were compared with 30 biopsies from 22 control patients, that were matched for time after transplantation.
Figure 2.
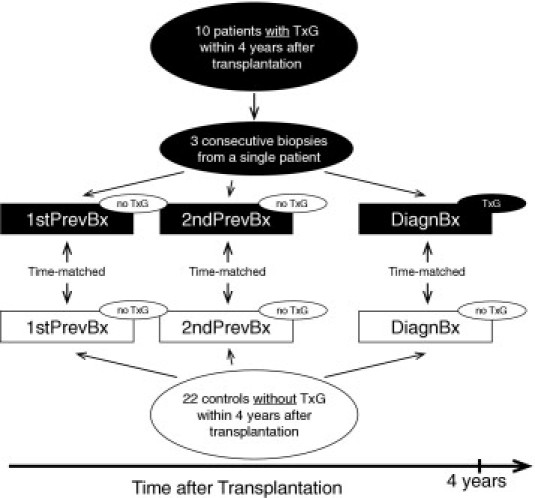
Selection of the patient biopsy (PBx) and the control biopsy (CBx): 10 patients with transplant glomerulopathy (TxG) within the first 4 years after transplantation were selected with their diagnostic biopsies (DiagnBx) and their two previous, nondiagnostic biopsies, the first and the second previous biopsy (1stPrevBx and 2ndPrevBx). For each of these 10 biopsy sets, time-matched control biopsies (CBx) were selected from a pool of 22 controls in which TxG within the first 4 years after transplantation was excluded by biopsy. Thus, 3 times 10 biopsies from patients were compared to 3 times 10 biopsies from controls.
Clinical Data
Clinical data from the biopsies were retrieved from the registry of the renal transplant protocol biopsy program of the Hannover Medical School. Parameters included in the analysis were donor sex, donor age, donor serum creatinine level at explantation, cold ischemia time, number of human leukocyte antigen (HLA) mismatches, presence of panel reactive antibodies, recipient sex, recipient age, initial graft function, lowest serum creatine level within the first 6 weeks after transplantation, and immunosuppressive regimen.
Routine Histological Evaluation According to Banff Criteria
All biopsies were subjected to our routine histological work-up including multiple H&E-, PAS-, and Jones-stained level sections and immunostains for C4d of formalin-fixed and paraffin-embedded tissue. In every biopsy that was taken more than 1 year after transplantation, glomerulonephritis was excluded by IHC for IgA, IgG, IgM, C1q, and C3c and also by the ultrastructural examination described below. Results were reported according to the current definition of the Banff components.2,48–50 The currently unclassified C4d staining of GECs was graded as C4dglomerular0 to C4dglomerular3 with 0%, <10%, <50%, and >50% in analogy to Banff grading of peritubular capillary staining.
Immunohistochemical Evaluation of HGF/Met-Signaling
Formalin-fixed and paraffin-embedded kidney sections were deparaffinized in xylol and graded ethanols. Endogenous peroxidase and nonspecific avidin/biotin binding were blocked with 3% H2O2 and Avidin/Biotin Blocking Kit (Vector Laboratories, Burlingame, CA). Sections were incubated overnight at 4°C with goat anti-human HGF IgG (AF-294-NA; R&D Systems, Minneapolis, MN) at 1:10 dilution. Bound primary antibody was labeled with a biotinylated secondary rabbit anti-goat-antibody at 1:2000 (81-1640; Invitrogen, Carlsbad, CA) followed by incubation with streptavidin–horseradish peroxidase conjugate [ZytoChemPlus (HRP) Broad Spectrum Bulk Kit; Zytomed Systems, Berlin, Germany] for 30 minutes. Diaminobenzidine (DAB High Contrast Kit; Zytomed Systems) was used as the substrate. Immunohistochemical staining for phospho-Met(Tyr1349) was performed similarly as previously described.51 Phosphorylation of tyrosine at position 1349 is essential to bind adapter proteins such as Gab1, which activate further pathways responsible for cell survival and proliferation.52,53 After antigen retrieval in citraconic acid anhydride at 98°C for 40 minutes, kidney sections were incubated overnight at 4°C with phospho-Met(Tyr1349) polyclonal rabbit antibody (#3121; Cell Signaling Technology, Danvers, MA) at 1:50 dilution. A horseradish peroxidase detection system with DAB as the substrate [ZytoChemPlus (HRP) Polymer Bulk Kit and DAB High Contrast Kit; both Zytomed Systems] was used to visualize the bound primary antibody. Slides from the manufacturer of the primary antibody (Cell Signaling Technology) were used as positive and negative control samples. HGF and phospho-Met(Tyr1349) immunostainings were scored as negative or positive. All sections were examined in a blinded fashion by two renal pathologists.
Quantitative Real-Time PCR
Formalin-fixed and paraffin-embedded renal biopsies were cut into 4-μm sections. About 50 glomerular transections of each biopsy were microdissected on an mmi CellCut Plus System (Olympus, Hamburg, Germany) and collected for RNA isolation. Tissue was digested immediately in 50 μL of Proteinase K solution at 20 mg/mL (Merck, Darmstadt, Germany), 50 μL of RNA digestion solution (4.2 mol/L guanidinium thiocyanate, 30 mmol/L Tris-HCl of pH 7.6, and 2% sodium N-lauroylsarcosine), and 0.5 μL of 2-mercaptoethanol at 55°C overnight. Then 10 μL of 3 mol/L sodium acetate (pH 5.2), 63 μL of Roti-Aqua-Phenol (Roth, Karlsruhe, Germany), and 27 μL of chloroform were added to precipitate RNA. After phase separation through centrifugation, the aqueous phase (95 μL) was carefully removed and added to 96 μL of 2-propanol and 1 μL of glycogen (20 mg/mL; Roche, Mannheim, Germany) as precipitation carrier. The precipitated RNA was then stored at −20°C.
cDNA synthesis was performed for 2 hours at 37°C as prescribed in the manual of the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA). cDNA was preamplified using TaqMan PreAmp Master Mix (2×) (Applied Biosystems) according to the manufacturer‘s manual. The primer pool used for preamplification consisted of six TaqMan Assays (Applied Biosystems) including HGF, WT1, nephrin, and the three reference genes glyceraldehyde 3-phosphate dehydrogenase (GAPDH), β-glucuronidase (GUSB), and polymerase (RNA) II (DNA directed) polypeptide A (POLR2A). Twenty-five microliters of TaqMan PreAmp Master Mix as well as 12.5 μL of the assay pool were added to 12.5 μL of cDNA and subjected to the following thermocycler program: 95°C for 10 minutes, 14 cycles of 95°C for 15 seconds, and 60°C for 4 minutes. Preamplified cDNA was stored at −20°C.
Five microliters of the preamplified cDNA (1:10 dilution) were added to 15 μL of master mix consisting of 10 μL of Gene Expression Master Mix (Applied Biosystems), 4 μL of purified water, and 1 μL of the respective TaqMan Gene Expression Assay (Applied Biosystems). Real-time PCR primers were used to amplify HGF (Hs 00900070_m1), WT1 (Hs 01103751_m1), nephrin (Hs 00190446_m1), POLR2A (Hs 00172187_m1), GAPDH (Hs 99999905_m1), and GUSB (Hs 99999908_m1). Real-time PCR was performed with a 7500 Fast Real-Time PCR System (Applied Biosystems). Relative expression levels of target genes HGF, WT1 and nephrin were calculated as 2mean(Ct reference genes)−Ct target gene.
Ultrastructural Examination
Ultrastructural damage to GECs and podocytes was evaluated only in the PBx and the CBx. Minute samples of the renal cortex were embedded in Araldit CY212 (PLANO, Wetzlar, Germany) and analyzed with a transmission electron microscope (Zeiss EM10; Carl Zeiss, Oberkochen, Germany). Glomerular parameters in the ultrastructural examination were double contours of the lamina densa, widening of lamina rara interna, cytoplasmic swelling, and/or loss of fenestrae of GECs, and podocyte foot process effacement. Foot process effacement was estimated as a percentage in 10% increments; each of the other parameters was scored as 0 (absent), 1 (only one segment), 2 (≤50% of glomeruli), or 3 (>50% of glomeruli). Peritubular capillaries were also evaluated regarding swelling of endothelial cells (absent or present) and multilamination of the peritubular basement membrane (one lamella: 0, two to three lamellae: 1, three to five lamellae: 2, >five lamellae: 3).
TUNEL
Glomerular apoptosis rates were examined with terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) stains of formalin-fixed and paraffin-embedded tissue sections with the ApopTag Peroxidase In Situ Detection Kit (S7101; Millipore, Billerica, MA) according to the manufacturer‘s instructions. Secondary follicles in formalin-fixed and paraffin-embedded human tonsil tissue served as positive controls.
In Vitro Experiments using Podocyte Culture
The HGF-driven activation of antiapoptotic signaling cascades by HGF in podocytes was examined in vitro. To this end, conditionally immortalized mouse podocytes (a gift from Peter Mundel, Mt. Sinai School of Medicine, New York, NY) were cultivated as described by Mundel and Kriz.54 To induce proliferation, cells were cultivated on type I collagen (BD Biosciences, Bedford, MA) at 33°C in the presence of 10 U/mL mouse recombinant γ-IFN (Cell Sciences, Canton, MA). A temperature change to 37°C and the absence of γ-IFN, maintained for 6 days, prompted the differentiation of the cells. Podocytes were stimulated with recombinant human HGF (Cell Sciences) at a concentration of 50 ng/μL on day 7 followed by harvesting as described below.
Western Blots
Cells were harvested for time-course analysis of proteins at the following time points: 0 minutes, 30 minutes, 60 minutes, 2 hours, 4 hours, 8 hours, and 24 hours. Podocytes were lysed on ice in radioimmunoprecipitation assay buffer [50 mmol/L Tris (pH 7.5); 150 mmol/L NaCl; 0.5% sodium deoxycholate; 1% Nonidet P-40, and 0.1% SDS) containing protease inhibitor (Complete Mini; Roche), 100 mmol/L sodium orthovanadate, 1 mol/L NaF, and 200 μg/L okadaic acid. Lysates were centrifuged at 4°C and 11,000 rpm for 15 minutes to remove insoluble material. Protein concentration in the lysates was determined using the BCA Protein Assay Kit (Thermo Fisher Scientific, Rockford, IL). The samples were separated by 12% SDS-PAGE and transferred to polyvinylidene difluoride membranes (Immobilon-P; Millipore). The membranes were incubated overnight with anti–phospho-Met(Tyr1349) polyclonal rabbit antibody at 1:1000 dilution (#3121, Cell Signaling Technology), anti–phospho-Akt(Ser473) monoclonal rabbit antibody at 1:2000 dilution (#4060, Cell Signaling Technology), and anti–phospho-p44/42 MAPK (ERK1/2) (Thr202/Tyr204) monoclonal rabbit antibody at 1:1000 dilution (#4370, Cell Signaling Technology). We recently found that X-linked inhibitor of apoptosis (XIAP), which is known to inhibit caspase 3, 7,55 and 9,56 is present in podocytes (unpublished data). Therefore, this antiapoptotic protein was examined with anti-XIAP monoclonal mouse antibody at 1:1000 dilution (#610762, BD Biosciences, Franklin Lakes, NJ). HRP-labeled anti-mouse or anti-rabbit secondary antibodies (1:10,000), followed by enhanced chemiluminescence reagents (SuperSignalWest Pico STable Peroxide Solution and SuperSignalWest Pico Luminal/Enhancer Solution; Pierce Inc., Thermo Fisher Scientific) were used to visualize the antibody–antigen complexes.
Statistical Analysis
Statistics were calculated with JMP or StatView (SAS Institute, Cary, NC). For comparison of continuous variables, two-sided Kruskal-Wallis or Mann-Whitney U-tests were used; for nominal variables, two-sided χ2 or, when applicable, Fisher's exact tests were used. For dependent nominal variables, the two-sided McNemar test, implemented in StatXact 7.0 (Cytel, Cambridge, MA) was used. Since this is an exploratory study, the P values were assessed descriptively.
Ethical Approval
All examinations were conducted according to the latest revision of the Declaration of Helsinki57 and were approved by the Ethics Committee of Hannover Medical School.
Results
Mean Glomerular Volume, Mean Podocyte Number per Glomerulus, and Podocyte Volume Density
The morphometric data regarding mean glomerular volume, podocyte volume density, and mean podocyte number per glomerulus are given in Figures 3, 4, and 5. The mean glomerular volume was smaller in regular biopsies without TxG (2.23 ± 0.368 × 106 μm3) than in zero-hour biopsies (2.88 ± 0.814 × 106 μm3; P = 0.0355)) and in nephrectomy specimens with TxG (3.60 ± 1.222 × 106 μm3; P = 0.0001). No difference regarding glomerular volume was found between zero-hour biopsies and nephrectomy specimens with TxG (Figure 3). Mean podocyte numbers per glomerulus were significantly lower in nephrectomy specimens with TxG (265 ± 181) than in zero-hour biopsies (684 ± 121; P = 0.0001) and in regular biopsies without TxG (573 ± 223; P = 0.0052). No significant difference was found between zero-hour biopsies and regular biopsies without TxG (Figure 4).
Figure 3.
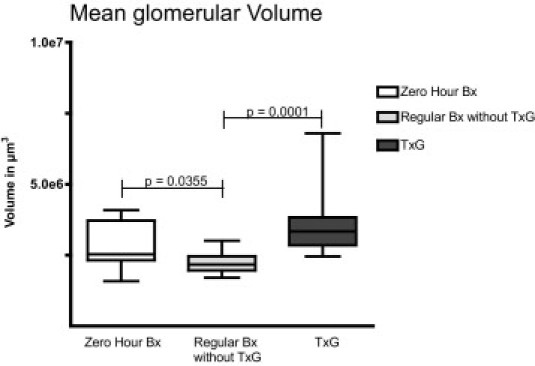
Mean glomerular volume in relation to specimen type: significantly lower mean volume was found in regular transplant biopsies without TxG. Also significantly larger mean glomerular volume was found in nephrectomy specimens with TxG than in specimens without. Depicted are the median, quartiles, minimum, and maximum. Bx, biopsy.
Figure 4.
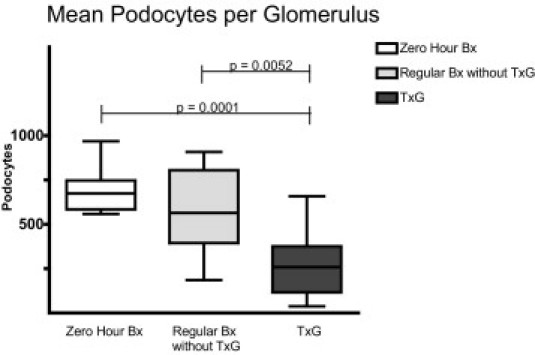
Mean podocyte number per glomerulus: significantly lower mean podocyte number per glomerulus was found in nephrectomy specimens with transplant glomerulopathy (TxG) than in zero-hour biopsies and regular transplant biopsies without TxG. Depicted are the median, quartiles, minimum, and maximum.
Figure 5.
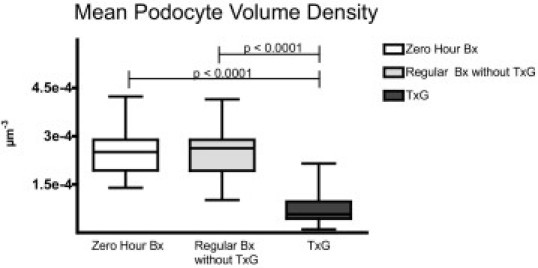
Podocyte volume density in relation to specimen type: significantly lower podocyte volume density was found in specimens with transplant glomerulopathy (TxG) than in zero-hour biopsies and regular transplant biopsies without TxG (regular Bx without TxG). Depicted are the median, quartiles, minimum, and maximum.
Consequently, podocyte volume density was significantly lower in nephrectomy specimens with TxG (0.8 ± 0.56 × 10−4 podocytes/μm3) than in zero-hour biopsies (2.5 ± 0.76 × 10−4 podocytes/μm3; P < 0.0001) and in regular biopsies without TxG (2.5 ± 0.83 × 10−4 podocytes/μm3; P < 0.0001). No significant difference regarding podocyte volume density was found between zero-hour biopsies and regular biopsies without TxG (Figure 5).
Clinical Data
Clinical data of the 10 TxG patients and the 22 matched control patients are given in Table 2. Donor age and the number of HLA mismatches were significantly higher in the patients than in the controls. Although no significant difference regarding the donor serum creatinine could be found between patients and controls, the lowest serum creatinine level within the first 6 weeks after transplantation was higher in the patients than in the controls. No significant difference could be found in the other examined parameters between the patients and controls.
Table 2.
Clinical Parameters of the TxG Patients and the Control Patients
| TxG patients (n = 10) | Control patients (n = 22) | |
|---|---|---|
| Donor sex (female/male) | 5:5 | 9:13 |
| Donor age | 65 ± 10.7 years⁎ | 53 ± 13.5 years⁎ |
| Donor serum creatinine at explantation | 94.5 ± 24.26 μmol/L | 97.7 ± 41.01 μmol/L |
| Cold ischemia time | 11.7 ± 7.73 hours | 15.2 ± 6.40 hours |
| Number of HLA mismatches | 3.5 ± 1.43⁎ | 2.0 ± 1.68⁎ |
| Panel reactive antibodies present at time of transplantation | 0 (0%) | 1 (5%) |
| Recipient sex (female/male) | 4:6 | 15:7 |
| Recipient age | 54 ± 11.4 years | 50 ± 13.0 years |
| Initial graft function | 6 (60%) | 13 (59%) |
| Lowest serum creatinine within the first 6 weeks after transplantation | 190.2 ± 63.41 μmol/L⁎ | 128.2 ± 59.01 μmol/L⁎ |
| Initial immunosuppression with cyclosporin A | 8 (80%) | 17 (77%) |
| Initial immunosuppression with tacrolimus | 2 (20%) | 4 (18%) |
| Initial immunosuppression with rapamycin | 1 (10%) | 1 (5%) |
| Initial immunosuppression with mycophenolate mofetil | 7 (70%) | 12 (55%) |
| Initial immunosuppression with corticosteroids | 9 (90%) | 21 (95%) |
P < 0.05 in Mann-Whitney U-test.
Banff Components and Glomerular C4d
A representative example of split glomerular basement membranes, reflected in the Banff cg component, and of mesangial matrix expansion reflected in the mm component, is given in Figure 6B. A synopsis of the Banff scores is shown in Table 3. Differences in the Banff components between PBx and CBx did not reach statistical significance, except for cg and mm, which were defining criteria for the cohorts. C4d staining of glomerular or peritubular capillaries was not different between the groups (Table 3). When considering the presence of at least one of the three indicators of humoral rejection (transplant glomerulitis, peritubular capillaritis, or positive C4d staining in at least 10% of the peritubular capillaries) as indicators of acute humoral rejection, no significant difference could be found between PBx and CBx in the 1stPrevBx (4 of 10 versus 2 of 10), the 2ndPrevBx (6 of 10 versus 3 of 10), or the DBx (6 of 10 versus 2 of 10).
Figure 6.

A: Normal glomerular morphology in one of the regular biopsies without TxG shows regular contours of the glomerular basement membrane and no mesangioproliferation. Jones stain; original magnification, ×600. B: Typical histological findings of transplant glomerulopathy (TxG) with double contours of glomerular basement membrane (arrow) and mesangioproliferation (double arrow) in one of the transplant nephrectomy specimens with TxG. Jones stain; original magnification, ×600. C: Glomerulus with HGF-positive (brown) glomerular endothelial cells (arrows) from one of the control cohort‘s (CBx) second previous biopsies (2ndPrevBx). D: Glomerulus with HGF-negative GECs and HGF-positive mesangial cells (brown) from one of the second previous biopsies (2ndPrevBx) of the patient cohort (PBx). Immunoperoxidase with hematoxylin counterstain; original magnification, ×600. E: Phospho-Met (Tyr1349)–positive (brown) podocytes (arrow) in a second previous biopsy (2ndPrevBx) from the control cohort (CBx). Immunoperoxidase with hematoxylin counterstain; original magnification, ×600. F: Phospho-Met (Tyr1349)–negative podocyte (arrow) in a first previous biopsy (1stPrevBx) from the patient cohort. Two biopsies later, transplant glomerulopathy (TxG) was histologically confirmed in this patient. Immunoperoxidase with hematoxylin counterstain; original magnification, ×600.
Table 3.
Banff Components in the Patient Biopsies That Were Diagnostic for TxG and the Two Preceding Biopsies, 1stPrevBx and 2ndPrevBx
|
n = 10 |
||||||
|---|---|---|---|---|---|---|
| 1stPrevBx |
2ndPrevBx |
DiagnBx |
||||
| PBx | CBx | PBx | CBx | PBx | CBx | |
| ai | 1.0 ± 0.82 | 0.8 ± 0.79 | 0.5 ± 0.71 | 0.4 ± 0.52 | 0.7 ± 0.48 | 1.0 ± 0.94 |
| at | 0.5 ± 0.97 | 0.4 ± 0.70 | 0.3 ± 0.48 | 0.3 ± 0.48 | 0.1 ± 0.32 | 0.4 ± 0.70 |
| av | 0.1 ± 0.32 | 0.1 ± 0.32 | 0.3 ± 0.67 | 0.1 ± 0.32 | 0.0 ± 0.0 | 0.0 ± 0.0 |
| ag | 0.5 ± 1.08 | 0.0 ± 0.0 | 0.3 ± 0.48 | 0.3 ± 0.67 | 0.1 ± 0.32 | 0.0 ± 0.0 |
| ptc | 0.3 ± 0.67 | 0.0 ± 0.0 | 0.1 ± 0.32 | 0.0 ± 0.0 | 0.6 ± 0.84 | 0.0 ± 0.0 |
| ct | 0.7 ± 0.67 | 0.8 ± 0.79 | 0.4 ± 0.52 | 0.7 ± 0.48 | 1.3 ± 0.82 | 0.9 ± 0.32 |
| ci | 0.6 ± 0.70 | 0.9 ± 0.99 | 0.3 ± 0.48 | 0.5 ± 0.53 | 1.1 ± 0.99 | 0.6 ± 0.52 |
| cv | 0.2 ± 0.63 | 0.2 ± 0.63 | 0.1 ± 0.32 | 0.0 ± 0.0 | 0.2 ± 0.42 | 0.0 ± 0.0 |
| cg* | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 1.9 ± 0.99 | 0.0 ± 0.0 |
| mm* | 0.0 ± 0.0 | 0.3 ± 0.67 | 0.3 ± 0.48 | 0.3 ± 0.48 | 1.3 ± 0.67 | 0.7 ± 0.95 |
| ah | 0.1 ± 0.32 | 0.6 ± 0.97 | 0.7 ± 0.82 | 0.4 ± 0.70 | 0.7 ± 0.95 | 0.4 ± 0.70 |
| C4d peritubular | 0.4 ± 0.97 | 0.4 ± 0.84 | 0.6 ± 1.07 | 0.4 ± 0.84 | 0.6 ± 0.97 | 0.4 ± 0.84 |
| C4dglomerular | 0.8 ± 1.03 | 0.0 ± 0.0 | 0.9 ± 1.29 | 0.3 ± 0.95 | 0.7 ± 1.16 | 0.2 ± 0.63 |
Data shown include C4d staining of peritubular capillary endothelial cells and GECs. Glomerular endothelial C4d staining (C4dglomerular) is not among the presently described Banff components; it is defined in analogy to the C4d scoring in the peritubular capillaries. Components are shown in comparison to control biopsies (CBx) that were matched for time after transplantation. None of the components except for inclusion criteria cg and mm were different between the groups.
CBx, control biopsy; PBx, patient biopsy; ai, acute extent of interstitial infiltrate at severity of acute tubulitis; av, severity of acute endothelialitis; ag, severity of acute glomerulitis; ptc, severity of peritubular capillaritis; ct, extent of tubular atrophy; ci, extent of interstitial fibrosis; cv, severity of chronic transplant vasculopathy; cg, severity of transplant glomerulopathy; mm, extent of mesangial expansion; ah, severity of arteriolar hyalinosis.
Met and HGF IHC
Representative examples of phospho-Met(Tyr1349) and HGF immunostains are given in Figure 6, C-F. Results of the phospho-Met(Tyr1349) immunostains of podocytes are given in Figure 7. Significantly less PBx than CBx had phospho-Met(Tyr1349)–positive podocytes in the 1stPrevBx (2 of 8 versus 7 of 7; P = 0.0070) and also in the 2ndPrevBx (4 of 9 versus 9 of 9; P = 0.0294). In the DiagnBx, no significant difference could be found between CBx and PBx (4 of 7 versus 7 of 9).
Figure 7.
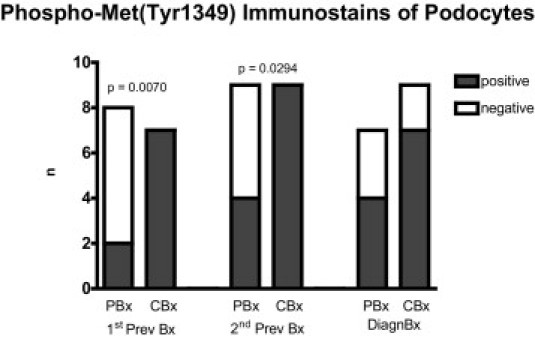
Frequency of biopsies with phospho-Me t(Tyr1349)–positive podocytes is lower compared to time-matched controls in the first (1stPrevBx) and the second previous biopsies (2ndPrevBx) but not in the third consecutive biopsy in which transplant glomerulopathy (TxG) was diagnosed (DiagnBx). Results from immunoperoxidase stains.
There was no difference found in the phospho-Met(Tyr 1349) immunostains of mesangial cells between PBx and CBx at all time points examined (1stPrevBx, 0 of 8 versus 0 of 7; 2ndPrevBx, 4 of 9 versus 4 of 9; DiagnBx, 2 of 7 versus 2 of 9).
Similar to the mesangial cells, no significant differences were found in phospho-Met(Tyr1349) immunostaining of GECs between PBx and CBx at all time points examined (1stPrevBx, 3 of 8 versus 2 of 7; 2ndPrevBx, 3 of 9 versus 1 of 9; DiagnBx, 3 of 7 versus 2 of 9).
Also, no significant differences could be found in the ratio of biopsies with positive HGF immunostaining of GECs or mesangial cells between PBx and CBx in the 1stPrevBx (GECs, 5 of 9 versus 3 of 7; mesangial cells, 1 of 9 versus 0 of 7), the 2ndPrevBx (GECs, 1 of 9 versus 4 of 9; or mesangial cells, 5 of 9 versus 1 of 9), or the DiagnBx (GECs, 5 of 9 versus 7 of 10; or mesangial cells, 6 of 9 versus 3 of 10). However, in the PBx, a significant increase of biopsies with positive mesangial cells was apparent from the 1stPrevBx (1 of 9) to the 2ndPrevBx (5 of 9) and the DiagnBx (6 of 9).
Quantitative Real-Time PCR
Results of the quantitative real-time PCR analysis of HGF mRNA are given in Figure 8. Glomerular HGF mRNA was significantly lower in PBx than in CBx only in the 1stPrevBx (mean, 0.05 ± 0.083 versus 0.28 ± 0.331; P = 0.0155), whereas in the 2ndPrevBx (0.06 ± 0.095 versus 0.04 ± 0.053) and the DiagnBx (0.16 ± 0.190 versus 0.03 ± 0.063), differences between PBx and CBx did not reach statistical significance.
Figure 8.
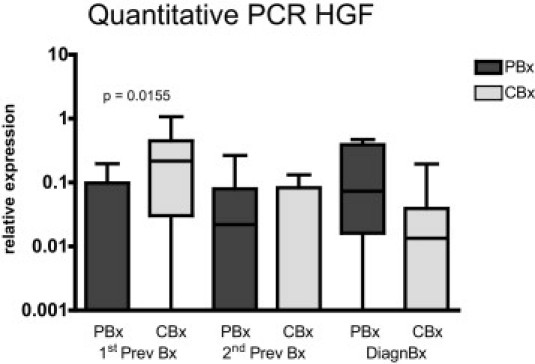
Relative levels of HGF-mRNA are significantly lower compared to controls in the first (1stPrevBx) of three consecutive biopsies from patients in which the third was diagnostic of transplant glomerulopathy (TxG). Results from quantitative real-time PCR experiments, relative to levels of the mean of three reference genes. Depicted are the median, quartiles, minimum, and maximum.
No significant difference in the levels of WT1 could be found between PBx and CBx (1stPrevBx, 4.09 ± 2.396 versus 3.87 ± 4.078; 2ndPrevBx, 8.66 ± 10.538 versus 9.70 ± 5.801; DiagnBx, 3.83 ± 2.724 versus 6.41 ± 3.348).
Also no significant difference could be found between the groups regarding expression levels of nephrin between PBx and CBx (1stPrevBx, 24.45 ± 9.678 versus 25.17 ± 27.959; 2ndPrevBx, 36.01 ± 45.848 versus 50.52 ± 34.654; DiagnBx, 30.70 ± 42.809 versus 18.14 ± 25.220).
TUNEL Stains
In none of the specimens were TUNEL-positive cells found in the glomeruli (data not shown).
Ultrastructural Examination
No significant differences were found in the ultrastructural parameters double contours of the lamina densa, widening of the lamina rara interna, cytoplasmic swelling and/or loss of fenestrae of GECs, or extent of foot process effacement between PBx and CBx. These parameters were as follows: Double contours of the lamina densa: 1stPrevBx score 0, 7 of 8 versus 6 of 9; score 1, 0 of 8 versus 3 of 9; score 2, 1 of 8 versus 0 of 9; score 3, 0 of 8 versus 0 of 9; 2ndPrevBx score 0, 5 of 7 versus 6 of 10; score 1, 0 of 7 versus 4 of 10; score 2, 1 of 7 versus 0 of 10; score 3, 1 of 7 versus 0 of 10; DiagnBx score 0. 4 of 7 versus 7 of 8; score 1, 1 of 7 versus 0 of 8; score 2, 0 of 7 versus 0 of 8; score 3, 2 of 7 versus 1 of 8. Widening of lamina rara interna: 1stPrevBx score 0, 4 of 8 versus 4 of 9; score 1, 3 of 8 versus 4 of 9; score 2, 1 of 8 versus 1 of 9; score 3, 0 of 8 versus 0 of 9; 2ndPrevBx score 0, 2 of 7 versus 4 of 10; score 1, 2 of 7 versus 4 of 10; score 2, 2 of 7 versus 1 of 10; score 3, 1 of 7 versus 1 of 10; DiagnBx score 0, 1 of 7 versus 4 of 8; score 1, 4 of 7 versus 3 of 8; score 2, 2 of 7 versus 0 of 8; score 3, 0 of 7 versus 1 of 8. Cytoplasmic swelling and/or loss of fenestrae of GECs: 1stPrevBx score 0, 1 of 8 versus 1 of 9; score 1, 2 of 8 versus 7 of 9; score 2, 2 of 8 versus 1 of 9; score 3, 3 of 8 versus 0 of 9; 2ndPrevBx score 0, 0 of 7 versus 1 of 10; score 1, 2 of 7 versus 5 of 10; score 2, 4 of 7 versus 4 of 10; score 3, 1 of 7 versus 0 of 10; DiagnBx score 0, 0 of 7 versus 2 of 8; score 1, 4 of 7 versus 5 of 8; score 2, 1 of 7 versus 1 of 8; score 3, 2 of 7 versus 0 of 8. Podocyte foot process effacement: 1stPrevBx, 13% ± 13.1% versus 5% ± 3.5%; 2ndPrevBx, 25% ± 34.2% versus 16% ± 17.6%; DiagnBx mean, 10% ± 4.1% versus 9% ± 10.9%.
In Vitro Experiments
Western blots showed sustained phosphorylation of Met on stimulation with human recombinant HGF at sites Tyr1234/5, lasting for 8 hours, and Tyr1349, lasting for about 8 hours. Activating phosphorylation of downstream antiapoptotic mediators AKT and ERK could be detected for about 4 to 8 hours. The antiapoptotic XIAP was induced after 30 minutes, lasting for 2 hours (Figure 9).
Figure 9.
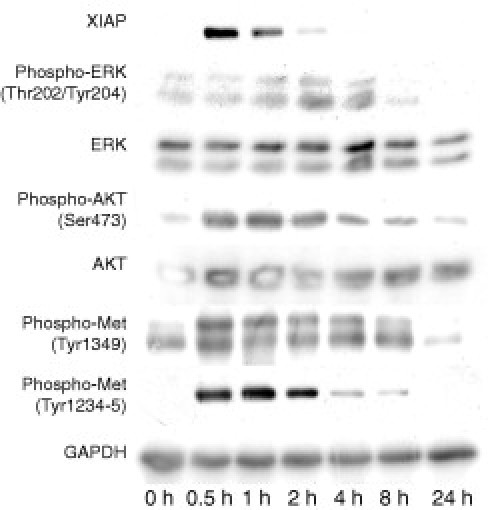
Western blot of cultivated mouse podocytes on stimulation with HGF: increased phosphorylation of Met at sites Tyr1234-5 and Tyr1349 beginning 0.5 hours after stimulation and lasting for about 8 hours. Further downstream in the signaling cascade, activating phosphorylation of antiapoptotic ERK at sites Thr202/Tyr204 and AKT at Ser473 occurs. Induction of XIAP, lasting for 2 hours, also can be observed.
Discussion
The present study is the first, to our knowledge, to demonstrate podocytopenia in human TxG. A recent study in rats has shown the same in a model of chronic allograft nephropathy.25 Both studies expand the paradigm that podocytopenia is a prerequisite of glomerular scarring,17,58,59 derived from studies of glomerulopathies in native kidneys to chronic rejection in transplant kidneys. Podocytopenia can be absolute, with decreased podocyte numbers per glomerulus, or relative, with stable numbers of podocytes in hypertrophic glomeruli. Mixed forms with decreased podocyte numbers in hypertrophic glomeruli probably also exist. Our findings support the hypothesis that podocytes are lost during the development of TxG and that absolute podocytopenia contributes to the development of secondary FSGS in TxG. Interestingly, glomerular volume was lower in the regular biopsies without TxG compared to both zero-hour biopsies and specimens with TxG. This is surprising, since glomerular hypertrophy could be expected to develop in a single kidney transplant. Studies in the 1990s have shown that, in nondiabetic recipients, glomerular volume does not increase but rather decreases after transplantation.60 Since podocyte volume density was not different between zero-hour biopsies and regular biopsies without TxG, this apparent glomerular “shrinkage” cannot simply be explained by prior “inflation” of the glomeruli with preservation solution as the authors speculated.60 Also, prevention of glomerular hypertrophy due to cyclosporin A, as has been shown in uninephrectomized rats,61 is an unlikely explanation for the lack of initial hypertrophy, since glomerular hypertrophy occurs later during the development of TxG under the same immunosuppression. This is reflected in the volume increase between the biopsies without TxG and the nephrectomy specimens with TxG. This increase in glomerular volume can be explained by compensatory hypertrophy as laid out in the Brenner hypothesis62,63 and already discussed before as a cause of renal transplant failure.64,65 In summary, it can be concluded that podocytopenia in TxG is neither purely absolute nor purely relative; it is, rather, a mixed form, with decreased podocyte numbers in hypertrophic glomeruli. The podocyte loss that leads to the “absolute” component of podocytopenia in TxG can potentially be due to necrosis, apoptosis, and/or detachment. Glomerular necrosis is not usually seen in transplant kidneys. Therefore, podocyte loss in TxG likely occurs only by apoptosis, by detachment with subsequent loss in the urine or a combination of both mechanisms. To the best of our knowledge, apoptosis of podocytes has only been described in vitro and in animal models but never in human specimens. Therefore, it was not surprising that TUNEL stains gave negative results in all examined glomeruli. Consequently, the results from the TUNEL stains do not exclude podocyte apoptosis as a mechanism of podocyte loss in TxG, since podocyte apoptosis is probably a slowly evolving process, resembling the slowly evolving changes in the glomerular basement membrane.
Decreased Met activation in podocytes was observed in the two biopsies preceding the diagnostic biopsy. Decreased Met signaling in podocytes could contribute to both mechanisms of podocyte loss, apoptosis and detachment. The studies of Dai et al.46 in podocyte-specific Met-knockout mice have shown that HGF diminishes the rate of podocyte apoptosis after administration of adriamycin, a specific podocyte toxin. Their in vitro experiments in human podocytes have shown the activation of antiapoptotic AKT and ERK on HGF stimulation.46 Our in vitro experiments could confirm the activation of the antiapoptotic AKT and ERK by HGF. In addition to this, we could also demonstrate that increased expression of XIAP seems to be responsible for the antiapoptotic effects of HGF on podocytes. Therefore, it is conceivable that a decrease in HGF signaling from GECs to podocytes likely causes a continuously but minimally increased rate of podocyte apoptosis that is not apparent in TUNEL stains of standard renal biopsies. The second mechanism of podocyte loss, detachment and shedding in the urine, has already been described in native kidneys.24,66 Decreased Met signaling in podocytes could also be relevant for podocyte shedding because podocytes are anchored by α3β1 integrins to the glomerular basement membrane.67 HGF/Met signaling has been shown to increase the avidity of various integrins, including β1 through β5.68 Therefore, decreased Met signaling in podocytes could loosen the integrin anchoring to the glomerular basement membrane, causing podocyte detachment. Since urine samples were not available for examination in the present study, podocyte shedding needs further evaluation in prospective studies with carefully selected examination methods, since not all of them deliver valid results.69 Of course, the present retrospective study does not allow us to prove a causal relationship between the podocyte loss and the observed perturbation in the glomerular HGF/Met signaling. Nevertheless, based on the results above, the following pathophysiologic sequence is proposed: antiapoptotic Met activation is decreased in podocytes during the early development of TxG, but no longer at the time of diagnosis. The immunohistochemical stains suggest that, in this phase, glomerular HGF is mostly produced by GECs, and the glomerular HGF mRNA levels indicate a decreased expression during the early phase of the development of TxG. Therefore, the present results support the hypothesis that reduced HGF production by damaged GECs disrupts Met signaling in podocytes. This suggests a novel pathophysiologic pathway, how primary damage to GECs could lead to secondary podocyte damage by disruption of the cytokine cross talk. This cross talk between GECs and podocytes has recently found much interest. However, research has focused only on the signaling from podocytes to GECs, which is mainly mediated by VEGF-A and its various isoforms.70–73 According to our data, a reciprocal signal mediator, from GECs to podocytes, could indeed be HGF. According to the already mentioned study in podocyte-specific Met-knockout mice, the Met signaling does not seem to be required for maturation, differentiation, or proper function of podocytes in physiological states. However, on podocyte stress, an intact HGF/Met axis seems to confer protection against apoptosis.46 Podocytes in transplants are also subject to stressors, among them cyclosporin A and profibrotic cytokines. In this situation, podocyte survival is probably dependent on intact HGF signaling from GECs. Indeed, HGF has been shown to counteract the proapoptotic effects of cyclosporin A.40 In addition, Met signaling also counteracts the effects of TGF-β,74 a profibrotic cytokine pivotal in fibrosis of native75,76 and transplant77,78 kidneys, that induces apoptosis in podocytes.45 Disruption of HGF/Met signaling from GECs to podocytes could also be of importance in other glomerular diseases with secondary FSGS, in which GECs are a target, namely all forms of thrombotic microangiopathies and membranoproliferative glomerulonephritis type I. Furthermore, possible changes in the reciprocal signaling patterns from podocytes to GECs that are induced by decreased Met signaling in podocytes could be of interest. These changes could establish a vicious circle of GECs damaging podocytes damaging GECs, which will be addressed in future experiments.
Among the demographic parameters evaluated in the present study, only donor age and lowest serum creatinine level within the first 6 weeks after transplantation were higher in the TxG patients. These two parameters could reflect a limited regenerative potential of the kidney transplant, potentially predisposing to TxG. None of the examined histological or ultrastructural parameters, including C4d staining, were significantly different between the patient and the control cohorts (Table 2); this could be the result of the rather small sample size. Clearly, the lack of evidence for a humoral contribution to TxG development is a limitation of the present study. We were not able to provide any serological data regarding donor-reactive antibodies, which are considered a key factor in the development of TxG.8,79,80 Also, no difference could be found in the prevalence of panel reactive antibodies at the time of transplantation between the patients and the controls. Still, the higher number of HLA mismatches in the TxG cohort supports the role of donor-reactive antibodies for TxG in our cohorts.
Apart from donor-reactive antibody-mediated injury, other types of injury against GECs, for example calcineurin inhibitor toxicity or hypertension, could also contribute to the lesion that is called TxG. This is best illustrated in the transplant nephrectomy specimens, which also showed severe chronic calcineurin inhibitor arteriolopathy and benign nephrosclerosis. Therefore, examination of final common pathways as reflected in the HGF/Met signaling from GECs to podocytes seem to be a more integrative approach for risk assessment of TxG development. Ancillary techniques such as the immunohistochemical examination of Met phosphorylation in podocytes or quantification of glomerular HGF mRNA via real-time PCR could turn into promising tools to identify patients at risk for earlier TxG development. This approach could provide information beyond the humoral focus of the Banff components. However, these tools need further evaluation in prospective studies before they could become part of the pathologist's routine armamentarium.
The activation of Met on mesangial cells as seen in both patient and control biopsies, which could lead to mesangioproliferation, a common but nonspecific finding in renal transplants, shows the possible adverse effects of glomerular Met activation. HGF secretion is probably rather tightly regulated in the glomerulus. This limits future therapeutic options aimed at stimulation of HGF secretion or activation of Met to prevent podocyte loss in renal transplants. Besides the potential unwanted effect of mesangioproliferation, increased Met signaling has been shown to be involved in crescent formation,81 proteinuria,82 and down-regulation of nephrin.83 Met is also known as a proto-oncogene,84 linked to hereditary papillary renal cell carcinoma85 and other neoplasms.39 All therapeutic approaches aiming at a systemic activation of Met could increase the risk of neoplasia. Nevertheless, promising results have been obtained with HGF therapy in renal fibrosis models of native kidneys,38,86,87 in calcineurin inhibitor nephropathy,88,89 and transplanted kidneys.90,91 The risks, benefits, and modalities of HGF therapy in kidney transplants should be further examined.
Footnotes
Supported by a grant from the German Federal Ministry of Education and Research (reference number: 01EO0802); the contents of this article are the sole responsibility of the authors.
References
- 1.Meier-Kriesche H.U., Schold J.D., Srinivas T.R., Kaplan B. Lack of improvement in renal allograft survival despite a marked decrease in acute rejection rates over the most recent era. Am J Transplant. 2004;4:378–383. doi: 10.1111/j.1600-6143.2004.00332.x. [DOI] [PubMed] [Google Scholar]
- 2.Racusen L.C., Solez K., Colvin R.B., Bonsib S.M., Castro M.C., Cavallo T., Croker B.P., Demetris A.J., Drachenberg C.B., Fogo A.B., Furness P., Gaber L.W., Gibson I.W., Glotz D., Goldberg J.C., Grande J., Halloran P.F., Hansen H.E., Hartley B., Hayry P.J., Hill C.M., Hoffman E.O., Hunsicker L.G., Lindblad A.S., Yamaguchi Y. The Banff 97 working classification of renal allograft pathology. Kidney Int. 1999;55:713–723. doi: 10.1046/j.1523-1755.1999.00299.x. [DOI] [PubMed] [Google Scholar]
- 3.Cosio F.G., Gloor J.M., Sethi S., Stegall M.D. Transplant glomerulopathy. Am J Transplant. 2008;8:492–496. doi: 10.1111/j.1600-6143.2007.02104.x. [DOI] [PubMed] [Google Scholar]
- 4.Colvin R.B. Antibody-mediated renal allograft rejection: diagnosis and pathogenesis. J Am Soc Nephrol. 2007;18:1046–1056. doi: 10.1681/ASN.2007010073. [DOI] [PubMed] [Google Scholar]
- 5.Nankivell B.J., Chapman J.R. Chronic allograft nephropathy: current concepts and future directions. Transplantation. 2006;81:643–654. doi: 10.1097/01.tp.0000190423.82154.01. [DOI] [PubMed] [Google Scholar]
- 6.Solez K., Colvin R.B., Racusen L.C., Sis B., Halloran P.F., Birk P.E., Campbell P.M., Cascalho M., Collins A.B., Demetris A.J., Drachenberg C.B., Gibson I.W., Grimm P.C., Haas M., Lerut E., Liapis H., Mannon R.B., Marcus P.B., Mengel M., Mihatsch M.J., Nankivell B.J., Nickeleit V., Papadimitriou J.C., Platt J.L., Randhawa P., Roberts I., Salinas-Madriga L., Salomon D.R., Seron D., Sheaff M., Weening J.J. Banff '05 Meeting Report: differential diagnosis of chronic allograft injury and elimination of chronic allograft nephropathy (‘CAN') Am J Transplant. 2007;7:518–526. doi: 10.1111/j.1600-6143.2006.01688.x. [DOI] [PubMed] [Google Scholar]
- 7.Li C., Yang C.W. The pathogenesis and treatment of chronic allograft nephropathy. Nat Rev Nephrol. 2009;5:513–519. doi: 10.1038/nrneph.2009.113. [DOI] [PubMed] [Google Scholar]
- 8.Gloor J.M., Sethi S., Stegall M.D., Park W.D., Moore S.B., Degoey S., Griffin M.D., Larson T.S., Cosio F.G. Transplant glomerulopathy: subclinical incidence and association with alloantibody. Am J Transplant. 2007;7:2124–2132. doi: 10.1111/j.1600-6143.2007.01895.x. [DOI] [PubMed] [Google Scholar]
- 9.Wavamunno M.D., O'Connell P.J., Vitalone M., Fung C.L., Allen R.D., Chapman J.R., Nankivell B.J. Transplant glomerulopathy: ultrastructural abnormalities occur early in longitudinal analysis of protocol biopsies. Am J Transplant. 2007;7:2757–2768. doi: 10.1111/j.1600-6143.2007.01995.x. [DOI] [PubMed] [Google Scholar]
- 10.Monga G., Mazzucco G., Novara R., Reale L. Intertubular capillary changes in kidney allografts: an ultrastructural study in patients with transplant glomerulopathy. Ultrastruct Pathol. 1990;14:201–209. doi: 10.3109/01913129009076124. [DOI] [PubMed] [Google Scholar]
- 11.Ivanyi B., Fahmy H., Brown H., Szenohradszky P., Halloran P.F., Solez K. Peritubular capillaries in chronic renal allograft rejection: a quantitative ultrastructural study. Hum Pathol. 2000;31:1129–1138. doi: 10.1053/hupa.2000.16677. [DOI] [PubMed] [Google Scholar]
- 12.Sis B., Campbell P.M., Mueller T., Hunter C., Cockfield S.M., Cruz J., Meng C., Wishart D., Solez K., Halloran P.F. Transplant glomerulopathy, late antibody-mediated rejection and the ABCD tetrad in kidney allograft biopsies for cause. Am J Transplant. 2007;7:1743–1752. doi: 10.1111/j.1600-6143.2007.01836.x. [DOI] [PubMed] [Google Scholar]
- 13.Regele H., Bohmig G.A., Habicht A., Gollowitzer D., Schillinger M., Rockenschaub S., Watschinger B., Kerjaschki D., Exner M. Capillary deposition of complement split product C4d in renal allografts is associated with basement membrane injury in peritubular and glomerular capillaries: a contribution of humoral immunity to chronic allograft rejection. J Am Soc Nephrol. 2002;13:2371–2380. doi: 10.1097/01.asn.0000025780.03790.0f. [DOI] [PubMed] [Google Scholar]
- 14.Einecke G., Sis B., Reeve J., Mengel M., Campbell P.M., Hidalgo L.G., Kaplan B., Halloran P.F. Antibody-mediated microcirculation injury is the major cause of late kidney transplant failure. Am J Transplant. 2009;9:2520–2531. doi: 10.1111/j.1600-6143.2009.02799.x. [DOI] [PubMed] [Google Scholar]
- 15.Perkowska-Ptasinska A., Ciszek M., Chmura A., Galazka Z., Paczek L., Durlik M. Transplant glomerulopathy: clinical and pathological correlations. Transplant Proc. 2009;41:141–149. doi: 10.1016/j.transproceed.2008.10.052. [DOI] [PubMed] [Google Scholar]
- 16.Patrakka J., Tryggvason K. New insights into the role of podocytes in proteinuria. Nat Rev Nephrol. 2009;5:463–468. doi: 10.1038/nrneph.2009.108. [DOI] [PubMed] [Google Scholar]
- 17.Fries J.W., Sandstrom D.J., Meyer T.W., Rennke H.G. Glomerular hypertrophy and epithelial cell injury modulate progressive glomerulosclerosis in the rat. Lab Invest. 1989;60:205–218. [PubMed] [Google Scholar]
- 18.Kriz W., Kretzler M., Nagata M., Provoost A.P., Shirato I., Uiker S., Sakai T., Lemley K.V. A frequent pathway to glomerulosclerosis: deterioration of tuft architecture-podocyte damage-segmental sclerosis. Kidney Blood Press Res. 1996;19:245–253. doi: 10.1159/000174083. [DOI] [PubMed] [Google Scholar]
- 19.Chen C.A., Hwang J.C., Guh J.Y., Chang J.M., Lai Y.H., Chen H.C. Reduced podocyte expression of alpha3beta1 integrins and podocyte depletion in patients with primary focal segmental glomerulosclerosis and chronic PAN-treated rats. J Lab Clin Med. 2006;147:74–82. doi: 10.1016/j.lab.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 20.Lemley K.V., Lafayette R.A., Safai M., Derby G., Blouch K., Squarer A., Myers B.D. Podocytopenia and disease severity in IgA nephropathy. Kidney Int. 2002;61:1475–1485. doi: 10.1046/j.1523-1755.2002.00269.x. [DOI] [PubMed] [Google Scholar]
- 21.Wang G., Lai F.M., Kwan B.C., Lai K.B., Chow K.M., Li P.K., Szeto C.C. Podocyte loss in human hypertensive nephrosclerosis. Am J Hypertens. 2009;22:300–306. doi: 10.1038/ajh.2008.360. [DOI] [PubMed] [Google Scholar]
- 22.Pagtalunan M.E., Miller P.L., Jumping-Eagle S., Nelson R.G., Myers B.D., Rennke H.G., Coplon N.S., Sun L., Meyer T.W. Podocyte loss and progressive glomerular injury in type II diabetes. J Clin Invest. 1997;99:342–348. doi: 10.1172/JCI119163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Steffes M.W., Schmidt D., McCrery R., Basgen J.M. Glomerular cell number in normal subjects and in type 1 diabetic patients. Kidney Int. 2001;59:2104–2113. doi: 10.1046/j.1523-1755.2001.00725.x. [DOI] [PubMed] [Google Scholar]
- 24.Petermann A.T., Krofft R., Blonski M., Hiromura K., Vaughn M., Pichler R., Griffin S., Wada T., Pippin J., Durvasula R., Shankland S.J. Podocytes that detach in experimental membranous nephropathy are viable. Kidney Int. 2003;64:1222–1231. doi: 10.1046/j.1523-1755.2003.00217.x. [DOI] [PubMed] [Google Scholar]
- 25.Pippin J., Kumar V., Stein A., Jablonski P., Shankland S.J., Davis C.L. The contribution of podocytes to chronic allograft nephropathy. Nephron Exp Nephrol. 2009;111:e1–e10. doi: 10.1159/000178762. [DOI] [PubMed] [Google Scholar]
- 26.Nagata M., Nakayama K., Terada Y., Hoshi S., Watanabe T. Cell cycle regulation and differentiation in the human podocyte lineage. Am J Pathol. 1998;153:1511–1520. doi: 10.1016/s0002-9440(10)65739-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adamczak M., Gross M.L., Krtil J., Koch A., Tyralla K., Amann K., Ritz E. Reversal of glomerulosclerosis after high-dose enalapril treatment in subtotally nephrectomized rats. J Am Soc Nephrol. 2003;14:2833–2842. doi: 10.1097/01.asn.0000095248.91994.d3. [DOI] [PubMed] [Google Scholar]
- 28.Nakamura T., Nawa K., Ichihara A. Partial purification and characterization of hepatocyte growth factor from serum of hepatectomized rats. Biochem Biophys Res Commun. 1984;122:1450–1459. doi: 10.1016/0006-291x(84)91253-1. [DOI] [PubMed] [Google Scholar]
- 29.Russell W.E., McGowan J.A., Bucher N.L. Partial characterization of a hepatocyte growth factor from rat platelets. J Cell Physiol. 1984;119:183–192. doi: 10.1002/jcp.1041190207. [DOI] [PubMed] [Google Scholar]
- 30.Nakamura T., Nawa K., Ichihara A., Kaise N., Nishino T. Purification and subunit structure of hepatocyte growth factor from rat platelets. FEBS Lett. 1987;224:311–316. doi: 10.1016/0014-5793(87)80475-1. [DOI] [PubMed] [Google Scholar]
- 31.Miyazawa K., Tsubouchi H., Naka D., Takahashi K., Okigaki M., Arakaki N., Nakayama H., Hirono S., Sakiyama O., Takahashi K., Gohda E., Daikuhara Y., Kitamura N. Molecular cloning and sequence analysis of cDNA for human hepatocyte growth factor. Biochem Biophys Res Commun. 1989;163:967–973. doi: 10.1016/0006-291x(89)92316-4. [DOI] [PubMed] [Google Scholar]
- 32.Nakamura T., Nishizawa T., Hagiya M., Seki T., Shimonishi M., Sugimura A., Tashiro K., Shimizu S. Molecular cloning and expression of human hepatocyte growth factor. Nature. 1989;342:440–443. doi: 10.1038/342440a0. [DOI] [PubMed] [Google Scholar]
- 33.Ishibashi K., Sasaki S., Sakamoto H., Nakamura Y., Hata T., Nakamura T., Marumo F. Hepatocyte growth factor is a paracrine factor for renal epithelial cells: stimulation of DNA synthesis and NA,K-ATPase activity. Biochem Biophys Res Commun. 1992;182:960–965. doi: 10.1016/0006-291x(92)91825-b. [DOI] [PubMed] [Google Scholar]
- 34.Matsumoto K., Nakamura T. Hepatocyte growth factor: renotropic role and potential therapeutics for renal diseases. Kidney Int. 2001;59:2023–2038. doi: 10.1046/j.1523-1755.2001.00717.x. [DOI] [PubMed] [Google Scholar]
- 35.Igawa T., Matsumoto K., Kanda S., Saito Y., Nakamura T. Hepatocyte growth factor may function as a renotropic factor for regeneration in rats with acute renal injury. Am J Physiol. 1993;265:F61–F69. doi: 10.1152/ajprenal.1993.265.1.F61. [DOI] [PubMed] [Google Scholar]
- 36.Longati P., Albero D., Comoglio P.M. Hepatocyte growth factor is a pleiotropic factor protecting epithelial cells from apoptosis. Cell Death Differ. 1996;3:23–28. [PubMed] [Google Scholar]
- 37.Yamasaki N., Nagano T., Mori-Kudo I., Tsuchida A., Kawamura T., Seki H., Taiji M., Noguchi H. Hepatocyte growth factor protects functional and histological disorders of HgCl(2)-induced acute renal failure mice. Nephron. 2002;90:195–205. doi: 10.1159/000049042. [DOI] [PubMed] [Google Scholar]
- 38.Yang J., Liu Y. Blockage of tubular epithelial to myofibroblast transition by hepatocyte growth factor prevents renal interstitial fibrosis. J Am Soc Nephrol. 2002;13:96–107. doi: 10.1681/ASN.V13196. [DOI] [PubMed] [Google Scholar]
- 39.Ma P.C., Maulik G., Christensen J., Salgia R. c-Met: structure, functions and potential for therapeutic inhibition. Cancer Metastasis Rev. 2003;22:309–325. doi: 10.1023/a:1023768811842. [DOI] [PubMed] [Google Scholar]
- 40.Fornoni A., Li H., Foschi A., Striker G.E., Striker L.J. Hepatocyte growth factor, but not insulin-like growth factor I, protects podocytes against cyclosporin A-induced apoptosis. Am J Pathol. 2001;158:275–280. doi: 10.1016/S0002-9440(10)63966-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu Y. Hepatocyte growth factor in kidney fibrosis: therapeutic potential and mechanisms of action. Am J Physiol Renal Physiol. 2004;287:F7–F16. doi: 10.1152/ajprenal.00451.2003. [DOI] [PubMed] [Google Scholar]
- 42.Khwaja A., Lehmann K., Marte B.M., Downward J. phosphoinositide 3-kinase induces scattering and tubulogenesis in epithelial cells through a novel pathway. J Biol Chem. 1998;273:18793–18801. doi: 10.1074/jbc.273.30.18793. [DOI] [PubMed] [Google Scholar]
- 43.Besser D., Bardelli A., Didichenko S., Thelen M., Comoglio P.M., Ponzetto C., Nagamine Y. Regulation of the urokinase-type plasminogen activator gene by the oncogene Tpr-Met involves GRB2. Oncogene. 1997;14:705–711. doi: 10.1038/sj.onc.1200879. [DOI] [PubMed] [Google Scholar]
- 44.Manning B.D., Cantley L.C. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schiffer M., Bitzer M., Roberts I.S., Kopp J.B., ten Dijke P., Mundel P., Bottinger E.P. Apoptosis in podocytes induced by TGF-beta and Smad7. J Clin Invest. 2001;108:807–816. doi: 10.1172/JCI12367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dai C., Saleem M.A., Holzman L.B., Mathieson P., Liu Y. Hepatocyte growth factor signaling ameliorates podocyte injury and proteinuria. Kidney Int. 2010;77:962–973. doi: 10.1038/ki.2010.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sanden S.K., Wiggins J.E., Goyal M., Riggs L.K., Wiggins R.C. Evaluation of a thick and thin section method for estimation of podocyte number, glomerular volume, and glomerular volume per podocyte in rat kidney with Wilms' tumor-1 protein used as a podocyte nuclear marker. J Am Soc Nephrol. 2003;14:2484–2493. doi: 10.1097/01.asn.0000089829.45296.7c. [DOI] [PubMed] [Google Scholar]
- 48.Racusen L.C., Colvin R.B., Solez K., Mihatsch M.J., Halloran P.F., Campbell P.M., Cecka M.J., Cosyns J.P., Demetris A.J., Fishbein M.C., Fogo A., Furness P., Gibson I.W., Glotz D., Hayry P., Hunsickern L., Kashgarian M., Kerman R., Magil A.J., Montgomery R., Morozumi K., Nickeleit V., Randhawa P., Regele H., Seron D., Seshan S., Sund S., Trpkov K. Antibody-mediated rejection criteria: an addition to the Banff 97 classification of renal allograft rejection. Am J Transplant. 2003;3:708–714. doi: 10.1034/j.1600-6143.2003.00072.x. [DOI] [PubMed] [Google Scholar]
- 49.Racusen L.C., Halloran P.F., Solez K. Banff 2003 meeting report: new diagnostic insights and standards. Am J Transplant. 2004;4:1562–1566. doi: 10.1111/j.1600-6143.2004.00585.x. [DOI] [PubMed] [Google Scholar]
- 50.Solez K., Colvin R.B., Racusen L.C., Haas M., Sis B., Mengel M., Halloran P.F., Baldwin W., Banfi G., Collins A.B., Cosio F., David D.S., Drachenberg C., Einecke G., Fogo A.B., Gibson I.W., Glotz D., Iskandar S.S., Kraus E., Lerut E., Mannon R.B., Mihatsch M., Nankivell B.J., Nickeleit V., Papadimitriou J.C., Randhawa P., Regele H., Renaudin K., Roberts I., Seron D., Smith R.N., Valente M. Banff 07 classification of renal allograft pathology: updates and future directions. Am J Transplant. 2008;8:753–760. doi: 10.1111/j.1600-6143.2008.02159.x. [DOI] [PubMed] [Google Scholar]
- 51.Cen L., Arnoczky K.J., Hsieh F.C., Lin H.J., Qualman S.J., Yu S., Xiang H., Lin J. Phosphorylation profiles of protein kinases in alveolar and embryonal rhabdomyosarcoma. Mod Pathol. 2007;20:936–946. doi: 10.1038/modpathol.3800834. [DOI] [PubMed] [Google Scholar]
- 52.Benvenuti S., Comoglio P.M. The MET receptor tyrosine kinase in invasion and metastasis. J Cell Physiol. 2007;213:316–325. doi: 10.1002/jcp.21183. [DOI] [PubMed] [Google Scholar]
- 53.Eder J.P., Vande Woude G.F., Boerner S.A., LoRusso P.M. Novel therapeutic inhibitors of the c-Met signaling pathway in cancer. Clin Cancer Res. 2009;15:2207–2214. doi: 10.1158/1078-0432.CCR-08-1306. [DOI] [PubMed] [Google Scholar]
- 54.Mundel P., Kriz W. Cell culture of podocytes. Exp Nephrol. 1996;4:263–266. [PubMed] [Google Scholar]
- 55.Deveraux Q.L., Takahashi R., Salvesen G.S., Reed J.C. X-linked IAP is a direct inhibitor of cell-death proteases. Nature. 1997;388:300–304. doi: 10.1038/40901. [DOI] [PubMed] [Google Scholar]
- 56.Deveraux Q.L., Roy N., Stennicke H.R., Van Arsdale T., Zhou Q., Srinivasula S.M., Alnemri E.S., Salvesen G.S., Reed J.C. IAPs block apoptotic events induced by caspase-8 and cytochrome c by direct inhibition of distinct caspases. EMBO J. 1998;17:2215–2223. doi: 10.1093/emboj/17.8.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.World Medical Association . World Medical Association; Ferney-Voltaire, France: 2008. WMA Declaration of Helsinki: Ethical Principles for Medical Research Involving Human Subjects.http://www.wma.net/en/30publications/10policies/b3/ [Google Scholar]
- 58.Kriz W., LeHir M. Pathways to nephron loss starting from glomerular diseases: insights from animal models. Kidney Int. 2005;67:404–419. doi: 10.1111/j.1523-1755.2005.67097.x. [DOI] [PubMed] [Google Scholar]
- 59.Kriz W. The pathogenesis of ‘classic’ focal segmental glomerulosclerosis: lessons from rat models. Nephrol Dial Transplant. 2003;18(Suppl 6):vi39–vi44. doi: 10.1093/ndt/gfg1064. [DOI] [PubMed] [Google Scholar]
- 60.Osterby R., Nyberg G., Karlberg I., Svalander C. Glomerular volume in kidneys transplanted into diabetic and non-diabetic patients. Diabet Med. 1992;9:144–149. doi: 10.1111/j.1464-5491.1992.tb01751.x. [DOI] [PubMed] [Google Scholar]
- 61.Schurek H.J., Neumann K.H., Jesinghaus W.P., Aeikens B., Wonigeit K. Influence of cyclosporine A on adaptive hypertrophy after unilateral nephrectomy in the rat. Clin Nephrol. 1986;25(Suppl 1):S144–S147. [PubMed] [Google Scholar]
- 62.Brenner B.M., Meyer T.W., Hostetter T.H. Dietary protein intake and the progressive nature of kidney disease: the role of hemodynamically mediated glomerular injury in the pathogenesis of progressive glomerular sclerosis in aging, renal ablation, and intrinsic renal disease. N Engl J Med. 1982;307:652–659. doi: 10.1056/NEJM198209093071104. [DOI] [PubMed] [Google Scholar]
- 63.Brenner B.M. Nephron adaptation to renal injury or ablation. Am J Physiol. 1985;249:F324–F337. doi: 10.1152/ajprenal.1985.249.3.F324. [DOI] [PubMed] [Google Scholar]
- 64.Terasaki P.I., Koyama H., Cecka J.M., Gjertson D.W. The hyperfiltration hypothesis in human renal transplantation. Transplantation. 1994;57:1450–1454. [PubMed] [Google Scholar]
- 65.Sola R., Paredes D., Antonijoan R.M., Estorch M., Vila L.P., Guirado L.L., Diaz J.M., Gich I., Barbanoj M.J. Glomerular hyperfiltration, intrarenal hemodynamics, and chronic allograft nephropathy: physiopathology of chronic allograft nephropathy. Transplant Proc. 2002;34:340–342. doi: 10.1016/s0041-1345(01)02790-7. [DOI] [PubMed] [Google Scholar]
- 66.Garovic V.D., Wagner S.J., Turner S.T., Rosenthal D.W., Watson W.J., Brost B.C., Rose C.H., Gavrilova L., Craigo P., Bailey K.R., Achenbach J., Schiffer M., Grande J.P. Urinary podocyte excretion as a marker for preeclampsia. Am J Obstet Gynecol. 2007;196:320. doi: 10.1016/j.ajog.2007.02.007. e321–e327. [DOI] [PubMed] [Google Scholar]
- 67.Kretzler M. Regulation of adhesive interaction between podocytes and glomerular basement membrane. Microsc Res Tech. 2002;57:247–253. doi: 10.1002/jemt.10083. [DOI] [PubMed] [Google Scholar]
- 68.Trusolino L., Cavassa S., Angelini P., Ando M., Bertotti A., Comoglio P.M., Boccaccio C. HGF/scatter factor selectively promotes cell invasion by increasing integrin avidity. FASEB J. 2000;14:1629–1640. doi: 10.1096/fj.14.11.1629. [DOI] [PubMed] [Google Scholar]
- 69.Skoberne A., Konieczny A., Schiffer M. Glomerular epithelial cells in the urine: what has to be done to make them worthwhile? Am J Physiol Renal Physiol. 2009;296:F230–F241. doi: 10.1152/ajprenal.90507.2008. [DOI] [PubMed] [Google Scholar]
- 70.Eremina V., Baelde H.J., Quaggin S.E. Role of the VEGF—a signaling pathway in the glomerulus: evidence for crosstalk between components of the glomerular filtration barrier. Nephron Physiol. 2007;106:32–37. doi: 10.1159/000101798. [DOI] [PubMed] [Google Scholar]
- 71.Eremina V., Cui S., Gerber H., Ferrara N., Haigh J., Nagy A., Ema M., Rossant J., Jothy S., Miner J.H., Quaggin S.E. Vascular endothelial growth factor A signaling in the podocyte-endothelial compartment is required for mesangial cell migration and survival. J Am Soc Nephrol. 2006;17:724–735. doi: 10.1681/ASN.2005080810. [DOI] [PubMed] [Google Scholar]
- 72.Eremina V., Quaggin S.E. The role of VEGF-A in glomerular development and function. Curr Opin Nephrol Hypertens. 2004;13:9–15. doi: 10.1097/00041552-200401000-00002. [DOI] [PubMed] [Google Scholar]
- 73.Schumacher V.A., Jeruschke S., Eitner F., Becker J.U., Pitschke G., Ince Y., Miner J.H., Leuschner I., Engers R., Everding A.S., Bulla M., Royer-Pokora B. Impaired glomerular maturation and lack of VEGF165b in Denys-Drash syndrome. J Am Soc Nephrol. 2007;18:719–729. doi: 10.1681/ASN.2006020124. [DOI] [PubMed] [Google Scholar]
- 74.Florquin S., Rouschop K.M. Reciprocal functions of hepatocyte growth factor and transforming growth factor-beta1 in the progression of renal diseases: a role for CD44? Kidney Int Suppl. 2003:S15–S20. doi: 10.1046/j.1523-1755.64.s86.4.x. [DOI] [PubMed] [Google Scholar]
- 75.Border W.A., Noble N.A., Yamamoto T., Harper J.R., Yamaguchi Y., Pierschbacher M.D., Ruoslahti E. Natural inhibitor of transforming growth factor-beta protects against scarring in experimental kidney disease. Nature. 1992;360:361–364. doi: 10.1038/360361a0. [DOI] [PubMed] [Google Scholar]
- 76.Sato M., Markiewicz M., Yamanaka M., Bielawska A., Mao C., Obeid L.M., Hannun Y.A., Trojanowska M. Modulation of transforming growth factor-beta (TGF-beta) signaling by endogenous sphingolipid mediators. J Biol Chem. 2003;278:9276–9282. doi: 10.1074/jbc.M211529200. [DOI] [PubMed] [Google Scholar]
- 77.Gaciong Z., Koziak K., Religa P., Lisiecka A., Morzycka-Michalik M., Rell K., Kozlowska-Boszko B., Lao M. Increased expression of growth factors during chronic rejection of human kidney allograft. Transplant Proc. 1995;27:928–929. [PubMed] [Google Scholar]
- 78.Shihab F.S., Yamamoto T., Nast C.C., Cohen A.H., Noble N.A., Gold L.I., Border W.A. Transforming growth factor-beta and matrix protein expression in acute and chronic rejection of human renal allografts. J Am Soc Nephrol. 1995;6:286–294. doi: 10.1681/ASN.V62286. [DOI] [PubMed] [Google Scholar]
- 79.Mauiyyedi S., Crespo M., Collins A.B., Schneeberger E.E., Pascual M.A., Saidman S.L., Tolkoff-Rubin N.E., Williams W.W., Delmonico F.L., Cosimi A.B., Colvin R.B. Acute humoral rejection in kidney transplantation: II: Morphology, immunopathology, and pathologic classification. J Am Soc Nephrol. 2002;13:779–787. doi: 10.1681/ASN.V133779. [DOI] [PubMed] [Google Scholar]
- 80.Palomar R., Lopez-Hoyos M., Pastor J.M., Fernandez-Fresnedo G., Rodrigo E., Ruiz J.C., Cotorruelo J.G., Valero R., Castaneda O., San Segundo D., Arias M. Impact of HLA antibodies on transplant glomerulopathy. Transplant Proc. 2005;37:3830–3832. doi: 10.1016/j.transproceed.2005.10.077. [DOI] [PubMed] [Google Scholar]
- 81.Rampino T., Gregorini M., Camussi G., Conaldi P.G., Soccio G., Maggio M., Bottelli A., Dal Canton A. Hepatocyte growth factor and its receptor Met are induced in crescentic glomerulonephritis. Nephrol Dial Transplant. 2005;20:1066–1074. doi: 10.1093/ndt/gfh740. [DOI] [PubMed] [Google Scholar]
- 82.Kusumoto K., Ido A., Moriuchi A., Katsura T., Kim I., Takahama Y., Numata M., Kodama M., Hasuike S., Nagata K., Uto H., Inui K., Tsubouchi H. Repeated intravenous injection of recombinant human hepatocyte growth factor ameliorates liver cirrhosis but causes albuminuria in rats. Int J Mol Med. 2006;17:503–509. [PubMed] [Google Scholar]
- 83.Takano Y., Yamauchi K., Hiramatsu N., Kasai A., Hayakawa K., Yokouchi M., Yao J., Kitamura M. Recovery and maintenance of nephrin expression in cultured podocytes and identification of HGF as a repressor of nephrin. Am J Physiol Renal Physiol. 2007;292:F1573–F1582. doi: 10.1152/ajprenal.00423.2006. [DOI] [PubMed] [Google Scholar]
- 84.Goldstein D., Leavitt J. Expression of neoplasia-related proteins of chemically transformed HuT fibroblasts in human osteosarcoma HOS fibroblasts and modulation of actin expression upon elevation of tumorigenic potential. Cancer Res. 1985;45:3256–3261. [PubMed] [Google Scholar]
- 85.Schmidt L., Duh F.M., Chen F., Kishida T., Glenn G., Choyke P., Scherer S.W., Zhuang Z., Lubensky I., Dean M., Allikmets R., Chidambaram A., Bergerheim U.R., Feltis J.T., Casadevall C., Zamarron A., Bernues M., Richard S., Lips C.J., Walther M.M., Tsui L.C., Geil L., Orcutt M.L., Stackhouse T., Lipan J., Slife L., Brauch H., Decker J., Niehans G., Hughson M.D., Moch H., Storkel S., Lerman M.I., Linehan W.M., Zbar B. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat Genet. 1997;16:68–73. doi: 10.1038/ng0597-68. [DOI] [PubMed] [Google Scholar]
- 86.Mizuno S., Kurosawa T., Matsumoto K., Mizuno-Horikawa Y., Okamoto M., Nakamura T. Hepatocyte growth factor prevents renal fibrosis and dysfunction in a mouse model of chronic renal disease. J Clin Invest. 1998;101:1827–1834. doi: 10.1172/JCI1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dworkin L.D., Gong R., Tolbert E., Centracchio J., Yano N., Zanabli A.R., Esparza A., Rifai A. Hepatocyte growth factor ameliorates progression of interstitial fibrosis in rats with established renal injury. Kidney Int. 2004;65:409–419. doi: 10.1111/j.1523-1755.2004.00417.x. [DOI] [PubMed] [Google Scholar]
- 88.Yazawa K., Isaka Y., Takahara S., Imai E., Ichimaru N., Shi Y., Namba Y., Okuyama A. Direct transfer of hepatocyte growth factor gene into kidney suppresses cyclosporin A nephrotoxicity in rats. Nephrol Dial Transplant. 2004;19:812–816. doi: 10.1093/ndt/gfh064. [DOI] [PubMed] [Google Scholar]
- 89.Mizui M., Isaka Y., Takabatake Y., Mizuno S., Nakamura T., Ito T., Imai E., Hori M. Electroporation-mediated HGF gene transfer ameliorated cyclosporine nephrotoxicity. Kidney Int. 2004;65:2041–2053. doi: 10.1111/j.1523-1755.2004.00625.x. [DOI] [PubMed] [Google Scholar]
- 90.Azuma H., Takahara S., Matsumoto K., Ichimaru N., Wang J.D., Moriyama T., Waaga A.M., Kitamura M., Otsuki Y., Okuyama A., Katsuoka Y., Chandraker A., Sayegh M.H., Nakamura T. Hepatocyte growth factor prevents the development of chronic allograft nephropathy in rats. J Am Soc Nephrol. 2001;12:1280–1292. doi: 10.1681/ASN.V1261280. [DOI] [PubMed] [Google Scholar]
- 91.Herrero-Fresneda I., Torras J., Franquesa M., Vidal A., Cruzado J.M., Lloberas N., Fillat C., Grinyo J.M. HGF gene therapy attenuates renal allograft scarring by preventing the profibrotic inflammatory-induced mechanisms. Kidney Int. 2006;70:265–274. doi: 10.1038/sj.ki.5001510. [DOI] [PubMed] [Google Scholar]