

Abstract
Epigenetic changes in gene expression play a role in the development of diabetic complications, including nephropathy. Histone deacetylases (HDACs) are a group of enzymes that exert epigenetic effects by altering the acetylation status of histone and nonhistone proteins. In the current study, we investigated the action of the clinically available HDAC inhibitor vorinostat in a mouse model of diabetic nephropathy, with the following aims: to define its effect on the progression of renal injury and to explore its mechanism of action by focusing on its role in regulating the expression of endothelial nitric oxide synthase (eNOS). Control and streptozotocin-diabetic wild-type and eNOS–/– mice were treated with vorinostat by daily oral dosing for 18 weeks. Without affecting either blood glucose concentration or blood pressure, vorinostat decreased albuminuria, mesangial collagen IV deposition, and oxidative-nitrosative stress in streptozotocin–wild-type mice. These attenuating effects were associated with a >50% reduction in eNOS expression in mouse kidneys and in cultured human umbilical vein endothelial cells. Vorinostat treatment had no effect on albuminuria, glomerular collagen IV concentration, or mesangiolysis in diabetic mice genetically deficient in eNOS. These observations illustrate the therapeutic efficacy of long-term HDAC inhibition in diabetic nephropathy and emphasize the importance of the interplay between eNOS activity and oxidative stress in mediating these effects.
The ability to regulate gene expression is a fundamental attribute of all cells, providing them with the versatility needed to adapt to an ever-changing microenvironment. Although some alterations in gene expression are short-lived, others are far more enduring, providing the transcriptional machinery with access to large regions of DNA for extended periods. Such modifications are frequently semipermanent, involving chemical changes to DNA that include the (de)methylation of cytosine nucleotides and the (de)acetylation of nucleosome histones. Given the persistent and heritable nature of their effects, these “epigenetic” changes have provided mechanistic insights into embryonic development, neoplastic transformation, and, most recently, glycemic memory, affording a plausible explanation for the persistent reduction in diabetic complications long after an earlier phase of intensive glycemic control.1,2
In addition to expanding our understanding of pathogenetic processes, epigenetic phenomena provide new molecular targets that are potentially amenable to therapeutic manipulation. Although the pharmacological modification of the epigenome is still in its infancy, several clinical trials using drugs that target it are in progress, with one agent, the histone deacetylase inhibitor (HDACi), vorinostat (suberoylanilide hydroxamic acid), gaining regulatory approval for the treatment of advanced cutaneous T-cell lymphoma in 2006. Although investigation into the potential beneficial effects of inhibitors of HDACs has primarily focused on neoplastic and neurodegenerative disorders, their ability to modulate the expression of genes involved in the physiological and pathophysiological conditions of the cardiovascular system suggests their broader applicability.
One protein known to be regulated by histone modification is endothelial nitric oxide synthase (eNOS),3 viewed as playing a pivotal, but complex, role in the pathogenesis of diabetic nephropathy. Although eNOS deficiency, through genetic deletion, promotes albuminuria in the diabetic setting,4–7 the enzyme may also contribute to the development of tissue injury through its role in the generation of reactive oxygen species (ROS),8,9 a process commonly referred to as eNOS uncoupling. ROS, generated through increased mitochondrial glycolytic flux10 and impaired antioxidant systems11 in the setting of hyperglycemia, may react with eNOS-derived NO to form peroxynitrite. Oxidization of the eNOS cofactor tetrahydrobiopterin by peroxynitrite may subsequently promote the further generation of superoxide in preference to NO.12,13 Accordingly, in the hyperglycemic milieu, even “normal” levels of eNOS may contribute to oxidative-nitrosative stress.
In this primarily translational study, we sought to explore the effects of HDACi administration in a murine model of diabetic nephropathy, focusing on the potential benefits that might be afforded by this strategy and, more specifically, on the relationship between HDAC inhibition, eNOS activity, and ROS formation.
Materials and Methods
In Vivo Experiments
Male C57BL/6 and eNOS–/– mice (n = 7 to 11 per group) were obtained from Jackson Laboratories (Bar Harbor, ME) at the age of 7 weeks. Diabetes mellitus was induced according to a low-dose streptozotocin (STZ) protocol,14 as previously described.7 Animals were randomized to receive a daily i.p. injection of STZ (50 mg/kg) for 5 consecutive days in 0.1 mol/L sodium citrate buffer (pH 4.5) or citrate buffer alone. Two weeks after the initial STZ injection, diabetes was confirmed by measurement of a fed blood glucose concentration (Accu-check Advantage; Roche, Mississauga, ON); and only animals with a blood glucose concentration >16 mmol/L were considered diabetic. Mice were subsequently randomized to receive either vorinostat (50 mg/kg)15,16 (Exclusive Chemistry Ltd, Obninsk, Russia) or vehicle [0.5% methylcellulose (w/v) and 0.1% Tween 80 (v/v)] by once-daily oral gavage and were observed for 18 weeks.
Systolic blood pressure was measured in anesthetized mice (2% isoflurane) using a noninvasive tail blood pressure system (CODA; Kent Scientific, Torrington, CT). HbA1c was measured using A1cNow+ (Bayer, Sunnyvale, CA). The serum creatinine concentration was determined by the Bartel modification of Jaffe's alkaline picric acid method. Albuminuria was determined after housing mice in metabolic cages for 24 hours after 2 to 3 hours' habituation. Animals continued to have free access to tap water and a standard laboratory diet during this period. After 24 hours in metabolic cages, an aliquot of urine was collected from the 24-hour urine sample and stored at −80°C for subsequent analysis. Urine albumin excretion was determined with a mouse albumin enzyme-linked immunosorbent assay (AssayMax; Assaypro, St Charles, MO). Urine creatinine concentration was determined with a kit (Creatinine Companion; Exocell, Philadelphia, PA). Urinary 8-hydroxydeoxyguanosine (8-OHdG) concentration was determined with a kit (Bioxytech 8-OHdG-EIA; Oxis Health Products Inc., Foster City, CA).
All experimental procedures adhered to the guidelines of the Canadian Council on Animal Care and were approved by St Michael's Hospital Animal Care Committee.
Tissue Collection
Animals were anesthetized with 2% inhaled isoflurane before cervical dislocation. The right renal artery was clamped and the kidney was removed, decapsulated, sliced transversely, and immersed in 10% neutral-buffered formalin overnight. Tissues were routinely processed, embedded in paraffin, and sectioned. The left kidney was removed, decapsulated, and either snap frozen in liquid nitrogen and stored at −80°C for later molecular biological analysis or cryoembedded in optical cutting temperature compound, flash frozen over dry ice for 15 minutes, and stored at −80°C for subsequent immunofluorescence staining.
Immunohistochemistry
IHC for acetyl-histone H3 (1:60; Cell Signaling, Danvers, MA), collagen IV (1:800; Millipore, Billerica, MA), and nitrotyrosine (1:50; Abcam, Cambridge, MA) was performed as previously described.11,17,18 Incubation with PBS instead of primary antiserum served as the negative control. After incubation with the appropriate horseradish peroxidase–conjugated secondary antibodies, sections were labeled with the liquid diaminobenzidine and substrate chromogen system (DakoCytomation, Glostrup, Denmark) before counterstaining in Mayer's hematoxylin. For determination of histone H3 acetylation in mouse kidneys, the number of positively (brown) staining tubular nuclei was determined in three randomly selected cortical fields (×400 magnification) from each mouse kidney section. For estimation of glomerular collagen IV deposition, slides were scanned with a system (Aperio ScanScope; Aperio Technologies Inc., Vista, CA) and analyzed using ImageScope (Aperio Technologies Inc.), with the proportional glomerular area positive for collagen IV immunostaining determined in 15 randomly selected glomerular profiles from each kidney section. Data are expressed as the fold change relative to that of vehicle-treated nondiabetic animals.
Glomerular Volume
Glomerular volume was calculated on 4-μm PAS-stained kidney sections using the following formula:
where β = 1.38 pertains to the sphere and k = 1.10 is the distribution coefficient.19
Mesangiolysis
The presence of mesangiolysis was determined in 50 glomeruli from eNOS–/– mice after staining kidney sections with silver methenamine.
Electron Microscopy
Small pieces of cortical tissue (1 mm3) were fixed in 2% glutaraldehyde, postfixed in osmium tetroxide, dehydrated in acetone, and embedded in epoxy resin. Ultrathin sections were taken through three randomly selected glomeruli from four mice per group, stained with uranyl acetate and lead citrate, and examined using a transmission electron microscope (Philips CM100; EM Research Services, Newcastle University, Newcastle upon Tyne, UK). Electron micrographs of the whole glomerular profile were taken at ×1100 magnification. Mesangial volume fraction was estimated by superimposing a grid of coarse and fine points (ratio, 1:8) on each image. Fine points landing on mesangial tissue were counted and expressed as a fraction of coarse points landing on the glomerular tuft, the boundary being defined by a minimal–string polygon.20
Confocal Microscopy
Frozen kidney sections, 10-μm thick, were cut on a cryostat (Leica CM1900) and fixed in 4% paraformaldehyde/PBS (pH 7.4) for 5 minutes before incubation with 0.1% Triton X-100, 1% BSA, and normal donkey serum diluted 1:50 in PBS (blocking serum) for 45 minutes. Sections were then exposed to rabbit polyclonal anti-eNOS antibody (1:75; Abcam) in blocking serum at 4°C overnight. Slides were washed and incubated with donkey anti-rabbit IgG (Alexa Fluor 488), diluted 1:100 in PBS for 2 hours (Invitrogen, Carlsbad, CA). Confocal microscopy was performed on a confocal microscope (Leica TCS SL) using a ×20 objective (PlanApo; numerical aperture, 0.7).
In Vitro Experiments
Human umbilical vein endothelial cells were cultured and characterized as previously described, with experiments performed in passage 3 to 4 cells.21 Cells were incubated with either vorinostat at the concentrations described or vehicle (dimethyl sulfoxide) for 24 hours.
RNA and Protein Isolation
RNA was isolated from cultured cell lysates using reagent (TRIzol; Life Technologies, Grand Island, NY). For determination of gene expression in mice kidneys, tissue stored at −80°C was homogenized (Polytron, Kinematica Gmbh, Littau, Switzerland). Total RNA (4 μg) was treated with RQ1 DNase (1 U/μL) (Promega, Madison, WI) to remove genomic DNA. RNA was reverse transcribed with a kit (High-Capacity cDNA Reverse Transcription Kit; Applied Biosystems, Foster City, CA), according to the manufacturer's instructions. Protein was isolated from cultured cell extracts or mouse kidney homogenates (n ≥ 3) after washing in PBS with cell lysis buffer (Cell Signaling) containing 1 mmol/L phenylmethylsulfonyl fluoride. The protein concentration was determined with an assay (Bio-Rad DC Protein Assay; Bio-Rad, Hercules, CA). SDS–polyacrylamide gel electrophoresis was performed after loading gels with 50 μg of protein.
Immunoblotting
Immunoblotting on nitrocellulose membranes was performed with primary antibodies in the following concentrations: acetyl-histone H3 (Lys9), 1:1000 (Cell Signaling); total histone H3, 1:1000 (Cell Signaling); nitrotyrosine, 1:1000 (Abcam); β-actin, 1:5000 (Abcam); and eNOS, 1:5000 (BD Transduction Laboratories, Lexington, KY). After incubation with the appropriate horseradish peroxidase–conjugated secondary antibodies, proteins were detected by the electrochemiluminescence system (Amersham, Buckinghamsire, UK). Nitrotyrosine was determined by densitometry of a prominent approximately 210-kDa nitrated protein band, as previously described.22 Densitometry was performed using software (ImageJ, version 1.39) from the National Institutes of Health (http://rsb.info.nih.gov/ij, last accessed March 9, 2011).
Real-Time PCR
Measurement of eNOS gene expression was performed using SYBR Green on a system (ABI Prism 7900HT Fast PCR System; Applied Biosystems). Sequence-specific primers were designed to span exon-exon boundaries using computer software (Primer Express, v1.5; Applied Biosystems). Primers were obtained from ACGT Corp (Toronto, ON). Primer sequences were as follows: mouse RPL13A, 5′-GCTCTCAAGGTTGTTCGGCTGA-3′ (forward) and 5′-AGATCTGCTTCTTCTTCCGATA-3′ (reverse)23; mouse eNOS, 5′-GGCATCACCAGGAAGAACACC-3′ (forward) and 5′-TCACTCGCTTCGCCATCAC-3′ (reverse); human eNOS, 5′-GGGTCCTGTGTATGGATGAGTATG-3′ (forward) and 5′-GGCCGGACATCTCCATCAG-3′ (reverse); and human RPL32, 5′-CAACATTGGTTATGGAAGCAACA-3′ (forward) and 5′-TGACGTTGTGGACCAGGAACT-3′ (reverse).24 Experiments were performed in triplicate, and data analysis was performed using the comparative CT method (Applied Biosystems).
Statistics
Data are presented as mean ± SEM, unless otherwise indicated. Statistical significance was determined by one-way analysis of variance with a Newman-Keuls post hoc comparison. All statistical analyses were performed using software (GraphPad Prism version 5.00 for Mac; GraphPad Software, San Diego, CA). P < 0.05 was considered statistically significant.
Results
Vorinostat Attenuates Renal Injury in Diabetic Mice
To determine the effect of long-term HDAC inhibition in diabetic nephropathy, control and STZ-diabetic C57BL/6 mice were treated with vehicle or the HDACi vorinostat by daily oral gavage for 18 weeks (Table 1). In both cultured human umbilical vein endothelial cells and in the kidneys of control and diabetic mice, treatment with vorinostat resulted in an expected increase in acetylation of histone H3 (Figure 1). After 18 weeks, diabetic mice had a reduced b.wt. and an increased kidney size compared with nondiabetic animals (Table 1). Vorinostat treatment was well tolerated in both control and diabetic wild-type mice, with no effect on survival and a nonsignificant reduction in b.wt. (Table 1). At study end, albuminuria, considered as either the 24-hour albumin excretion rate or the albumin/creatinine ratio, was increased approximately 2.5-fold in STZ-C57BL/6 mice compared with controls (Table 1 and Figure 2). Without affecting either blood glucose or blood pressure, vorinostat treatment significantly decreased albuminuria after both 12 and 18 weeks of diabetes (Table 1 and Figure 2). Similarly, kidney size, glomerular volume, and mesangial matrix accumulation (determined by either immunostaining for collagen IV or calculating the mesangial fractional volume by transmission electron microscopy) were each increased in STZ-C57BL/6 mice relative to controls and were reduced with vorinostat (Table 1 and Figure 3).
Table 1.
Functional Characteristics of Control and STZ-Diabetic C57BL/6 Mice Treated with Vehicle or Vorinostat for 18 Weeks
| Treatment | B.wt. (g) | Systolic BP (mm Hg) | HbA1c (%) | Kidney wt. (g) | Kidney wt., b.wt. (%) | Albumin/creatinine ratio (μg/mg) |
|---|---|---|---|---|---|---|
| C57BL/6 + vehicle | 30.1 ± 0.5 | 121 ± 4 | 4.9 ± 0.1 | 0.166 ± 0.005 | 0.55 ± 0.01 | 51.6 ×/÷ 1.1 |
| C57BL/6 + vorinostat | 28.2 ± 0.6 | 121 ± 3 | 4.7 ± 0.1 | 0.151 ± 0.003 | 0.54 ± 0.01 | 57.2 ×/÷ 1.2 |
| STZ-C57BL/6 + vehicle | 22.0 ± 1.3⁎ | 115 ± 4 | 12.8 ± 0.1⁎ | 0.216 ± 0.013⁎ | 0.98 ± 0.02⁎ | 128.5 ×/÷ 1.2† |
| STZ-C57BL/6 + vorinostat | 20.5 ± 1.1 | 109 ± 3 | 12.6 ± 0.3 | 0.185 ± 0.005‡ | 0.91 ± 0.03 | 48.1 ×/÷ 1.3‡ |
Data are given as mean ± SEM.
BP, blood pressure.
P < 0.001 versus C57BL/6 + vehicle.
P < 0.05 versus C57BL/6 + vehicle.
P < 0.05 versus STZ-C57BL/6 + vehicle.
Figure 1.

Effect of vorinostat on histone hyperacetylation in vitro and in vivo. A: Immunoblotting for total and acetylated histone H3 in lysates from human umbilical vein endothelial cells exposed to dimethyl sulfoxide or 10 μmol/L vorinostat for 24 hours. B–E: IHC for acetylated histone H3 with C57BL/6 + vehicle (B), C57BL/6 + vorinostat (C), STZ-C57BL/6 + vehicle (D), and STZ-C57BL/6 + vorinostat (E). F: Quantitation of acetylated histone H3–positive nuclei. *P < 0.01 versus C57BL/6 + vehicle; **P < 0.01 versus STZ-C57BL/6 + vehicle.
Figure 2.
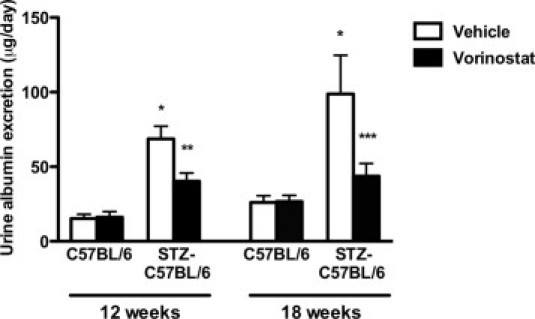
Urine albumin excretion in control and diabetic mice treated with vehicle or vorinostat. *P < 0.001 versus C57BL/6 + vehicle; **P < 0.01 versus STZ-C57BL/6 + vehicle; ***P < 0.05 versus STZ-C57BL/6 + vehicle.
Figure 3.

Renal structure in control and STZ-diabetic C57BL/6 mice treated with vehicle or vorinostat for 18 weeks. A: Glomerular volume. B–E: Glomerular collagen IV immunostaining with C57BL/6 + vehicle (B), C57BL/6 + vorinostat (C), STZ-C57BL/6 + vehicle (D), and STZ-C57BL/6 + vorinostat (E). Original magnification, ×400. F: Quantitation of glomerular collagen IV represented as fold change relative to C57BL/6 + vehicle. G–J: Mesangial fractional volume (Vv Mes/glom) of C57BL/6 + vehicle (G), C57BL/6 + vorinostat (H), STZ-C57BL/6 + vehicle (I), STZ-C57BL/6 + vorinostat (J). K: Vv Mes/glom. *P < 0.001 versus C57BL/6 + vehicle; **P < 0.05 versus STZ-C57BL/6 + vehicle.
Oxidative-Nitrosative Stress Is Increased in Diabetes and Reduced with Vorinostat
The effect of vorinostat on oxidative-nitrosative stress was evaluated using both Western blot analysis and IHC for nitrotyrosine, reflecting peroxynitrite activity, and by determining the urinary excretion of 8-OHdG, a marker of oxidative DNA damage. As an indicator of cellular nitrosylation, we quantitated nitrotyrosine in an approximately 210-kDa heavily nitrated protein band, as previously reported,22 identifying an increase in nitrotyrosine in the kidneys from diabetic mice that was reduced with vorinostat treatment (Figure 4A). By IHC, nitrotyrosine was increased in both tubular and glomerular endothelial compartments and reduced in vorinostat-treated diabetic mice (Figure 4, B–F). Similarly, vorinostat treatment also reduced the 24-hour urinary excretion of 8-OHdG that was increased approximately twofold in STZ-C57BL/6 mice (Figure 4G).
Figure 4.

Oxidative-nitrosative stress in control and STZ-diabetic C57BL/6 mice treated with vehicle or vorinostat for 18 weeks. A and B: Immunoblotting for nitrotyrosine in kidney homogenates revealed the presence of an approximately 210-kDa protein band, the nitrosylation of which was increased with diabetes mellitus and reduced with vorinostat. C–F: Immunostaining for nitrotyrosine with C57BL/6 + vehicle (C), C57BL/6 + vorinostat (D), STZ-C57BL/6 + vehicle (E), and STZ-C57BL/6 + vorinostat (F). G: Urinary 8-OHdG excretion in control and diabetic mice treated with vehicle or vorinostat for 18 weeks. *P < 0.05 versus all other groups; **P < 0.001 versus C57BL/6 + vehicle; ***P < 0.05 versus STZ-C57BL/6 + vehicle. AU indicates arbitrary unit.
Vorinostat Decreases eNOS Expression in the Kidneys of Diabetic Mice and in Cultured Endothelial Cells
After 18 weeks, the magnitude of renal expression of both eNOS mRNA and eNOS protein was equivalent in vehicle-treated diabetic and nondiabetic wild-type mice, with the distribution of the protein being restricted to the endothelium of glomeruli and arterioles (Figure 5, A–F). Real-time PCR of kidney homogenates revealed that eNOS mRNA was reduced by approximately 50% in the kidneys of both control and diabetic mice treated with vorinostat (Figure 5F). In cultured human umbilical vein endothelial cells, vorinostat dose dependently reduced eNOS protein and mRNA, such that with 10 μmol/L vorinostat, eNOS mRNA was reduced by >80% (Figure 5, G and H).
Figure 5.

eNOS protein and mRNA in mouse kidneys and cultured human umbilical vein endothelial cells (HUVECs). A–D: Immunofluorescence for eNOS protein with C57BL/6 + vehicle (A), C57BL/6 + vorinostat (B), STZ-C57BL/6 + vehicle (C), and STZ-C57BL/6 + vorinostat (D). E: Immunoblotting kidney homogenates for eNOS protein. F: Real-time PCR for eNOS in kidney homogenates of control and STZ-diabetic C57BL/6 mice treated with vehicle or vorinostat. G and H: Effect of HDAC inhibition with vorinostat on eNOS expression in cultured HUVECs treated with dimethyl sulfoxide or vorinostat for 24 hours on eNOS protein (G) and eNOS mRNA (H). Data are given for the mean of three experiments. *P < 0.001 versus C57BL/6 + vehicle; **P < 0.05 versus STZ-C57BL/6 + vehicle; ***P < 0.001 versus control; ****P < 0.01 versus 5 μmol/L vorinostat. AU indicates arbitrary unit.
Mice Genetically Deficient in eNOS Are Resistant to the Attenuating Effects of Vorinostat
Finally, to confirm the pivotal role of eNOS repression in mediating the nephroprotective effects of vorinostat, we sought to explore the response to HDAC inhibition in the setting of absolute eNOS deficiency by treating control and STZ-diabetic eNOS–/– mice with either vehicle or vorinostat by daily oral gavage for 18 weeks. Compared with either diabetic wild-type animals or nondiabetic eNOS–/– mice, heavy albuminuria was observed in vehicle-treated STZ-eNOS–/– animals (Figure 6, A and B), without a significant change in 8-OHdG excretion. Urinary 8-OHdG concentrations were as follows: eNOS–/–, 22.7 ± 5.1 ng/24 hours; and STZ-eNOS–/–, 30.3 ± 3.9 ng/24 hours (P = 0.28). Although vorinostat treatment was associated with a reduction in kidney size (Table 2) in STZ-eNOS–/– mice, it had no effect on either albuminuria (Figure 6, A and B) or glomerular collagen IV deposition (Figure 6, C–G). Mesangiolysis was a prominent feature of the glomerular pathological characteristics seen in STZ-eNOS–/– mice, occurring in approximately 10% of glomeruli. As observed with other parameters of renal structure and function, there was no difference in the proportion of glomeruli displaying evidence of mesangiolysis between vehicle- and vorinostat-treated STZ-eNOS–/– mice (Figure 6, H–L).
Figure 6.

Renal structure and function in control and diabetic eNOS–/– mice treated with vehicle or vorinostat for 18 weeks. A: Urine albumin excretion. B: Urine albumin/creatinine ratio. C–F: Glomerular collagen IV immunostaining for eNOS–/– + vehicle (C), eNOS–/– + vorinostat (D), STZ-eNOS–/– + vehicle (E), and STZ-eNOS–/– + vorinostat (F). Original magnification, ×400. G: Quantitation of glomerular collagen IV represented as fold change relative to eNOS–/– + vehicle. H–K: Representative photomicrographs of silver methenamine–stained kidney sections from eNOS–/– + vehicle (H), eNOS–/– + vorinostat (I), STZ-eNOS–/– + vehicle (J), and STZ-eNOS–/– + vorinostat (K), demonstrating mesangiolytic glomeruli in both vehicle- and vorinostat-treated STZ-eNOS–/– mice. Original magnification, ×400. L: Percentage of mesangiolytic glomeruli. *P < 0.01 versus eNOS–/– + vehicle; **P < 0.05 versus eNOS–/– + vehicle. AU indicates arbitrary unit.
Table 2.
Functional Characteristics of eNOS–/– Mice Treated with Vehicle or Vorinostat for 18 Weeks
| Treatment | B.wt. (g) | Systolic BP (mm Hg) | Serum creatinine ×10–1 (mg/dL) | HbA1c (%) | Kidney wt. (g) | Kidney wt., b.wt. (%) |
|---|---|---|---|---|---|---|
| eNOS–/– + vehicle | 27.3 ± 0.6 | 133 ± 3 | 0.91 ± 0.08 | 4.9 ± 0.1 | 0.138 ± 0.004 | 0.50 ± 0.01 |
| eNOS–/– + vorinostat | 26.4 ± 0.6 | 135 ± 3 | 1.31 ± 0.11 | 4.7 ± 0.1 | 0.131 ± 0.003 | 0.50 ± 0.01 |
| STZ-eNOS–/– + vehicle | 20.4 ± 0.5⁎ | 110 ± 4⁎ | 2.50 ± 0.51† | 12.5 ± 0.4⁎ | 0.186 ± 0.006⁎ | 0.91 ± 0.02⁎ |
| STZ-eNOS–/– + vorinostat | 18.8 ± 1.3 | 110 ± 3 | 2.27 ± 0.30 | 12.2 ± 0.4 | 0.152 ± 0.012‡ | 0.83 ± 0.04§ |
Data are given as mean ± SEM.
BP, blood pressure.
P < 0.001 versus eNOS–/– + vehicle.
P < 0.01 versus eNOS–/– + vehicle.
P < 0.01 versus STZ-eNOS–/– + vehicle.
P < 0.05 versus STZ-eNOS–/– + vehicle.
Discussion
During recent years, HDAC inhibitors have emerged as a class of agents with therapeutic potential for the treatment of complex chronic diseases, including diabetes. In the current study, we explored the efficacy of long-term HDAC inhibition using the orally bioavailable broad-spectrum HDACi, vorinostat. Treatment of diabetic mice with vorinostat resulted in a persistent attenuation of oxidative stress, albuminuria, and mesangial matrix accumulation. Given the wide range of target genes that are regulated by acetylation of histone proteins at their promoter regions, HDAC inhibition is likely to affect multiple injurious pathways in diabetes. However, the findings of the current study indicate a pivotal role for eNOS down-regulation in mediating the effects of HDAC inhibition, as evidenced by the reduction in kidney and endothelial cell eNOS mRNA after vorinostat administration and the absence of an effect in the setting of eNOS gene deletion. These findings, combined with the known role that eNOS uncoupling plays in diabetes-associated ROS generation, extend the mechanisms of action of HDAC inhibitors in chronic kidney disease beyond previously described antifibrotic effects.
Originally developed for their antiproliferative properties in transformed cells,25,26 inhibitors of HDACs have more recently been applied in a spectrum of experimental models of nonneoplastic disease.27 The first evidence for an antialbuminuric action of broad-spectrum HDAC inhibition was in a murine model of lupus nephritis.28 Unfortunately, the agent used in the initial study28 and in several subsequent studies,29–31 trichostatin A, is limited by its low oral bioavailability and relatively short half-life.32 Accordingly, we explored the properties of the orally bioavailable and clinically applicable HDACi, vorinostat. Similarly, although the studies with trichostatin A in kidney disease have primarily focused on the agent's actions in antagonizing transforming growth factor-β and its prosclerotic effects,29–31 we set out to examine alternative mechanisms of action, using a diabetic mouse model that is characterized by the development of albuminuria and modest mesangial expansion but that is relatively resistant to late-stage fibrotic disease.33 Consistent with observations in other mouse strains,33 we observed a reduction in systolic blood pressure in both wild-type and eNOS–/– mice in the context of STZ-induced diabetes. Without affecting either blood pressure or blood glucose concentration, vorinostat decreased albuminuria, glomerular hypertrophy, and glomerular collagen IV deposition, the chief collagenous component of mesangial matrix that is increased in diabetes,34 suggesting the potential for this class of agent to exert a beneficial effect in diabetic nephropathy as an adjunct to antihyperglycemic and antihypertensive strategies.
In neoplasia, the ability to promote ROS accumulation is a major mechanism underlying the cytotoxic effects of HDAC inhibitors.35,36 However, one of the most remarkable properties of these agents is their cytotoxic selectivity for transformed cells.37 In fact, low doses of HDAC inhibitors have been repeatedly cytoprotective in neurodegenerative diseases,38,39 with specific inhibition of HDAC6 attenuating oxidative stress–induced neuronal injury.40 Conversely, hydrogen peroxide increases HDAC activity in cultured tubular epithelial cells,41 indicating a bidirectional relationship between the oxidation-reduction and histone acetylation status of the cell. Our observations, and studies in other models of kidney disease, illustrate a persistent benefit of HDAC inhibition despite the apparent potential for this class of agent to promote oxidative stress. One mechanism for the antioxidant effect of vorinostat in diabetes is the reduction in eNOS mRNA and protein levels observed in vitro and in vivo. Although this decrease in eNOS expression did not appear to affect renal function in normoglycemic animals, it was associated with a reduction in oxidative-nitrosative stress in diabetic mice, consistent with the known actions of eNOS in mediating the formation of superoxide and peroxynitrite in the setting of reduced substrate bioavailability and high ambient glucose concentrations.8,9
The transcriptional regulation of eNOS is an archetypal example of a cell-specific histone code. In endothelial cells, the eNOS promoter is highly enriched with acetylated histones.42 In previous experiments, HDAC inhibition with trichostatin A decreased eNOS mRNA while paradoxically increasing the activity of its promoter, suggesting the induction of an eNOS mRNA-destabilizing factor.3 This factor has subsequently been identified as the antisense mRNA, sONE.43 Because vorinostat nonselectively inhibits the actions of class I (HDACs 1 to 3 and 8) and class II (HDACs 4 to 7, 9, and 10) HDAC enzymes and potentially increases the lysine acetylation status of more than 1700 nonhistone proteins,44 we did not seek to precisely define which HDACs are implicated in the pathogenesis of diabetic nephropathy. Instead, we took a translational approach to determine the long-term efficacy of a clinically available HDACi.
As with many factors implicated in the pathogenesis of diabetic complications, the role that eNOS plays in nephropathy is dependent on its relative expression levels, with detrimental effects reported in the setting of both enhanced and reduced eNOS activity. Although absolute eNOS deficiency, through genetic knockout, markedly augments albuminuria in diabetic mice,4–7 increased NO generation has been implicated in the pathogenesis of glomerular hypertrophy, hyperfiltration, and microalbuminuria in early diabetes.45,46 As has been previously described,4–7 we observed heavy albuminuria in diabetic eNOS–/– mice in the absence of an increase in oxidative injury,7 illustrating the critical role that eNOS plays in both the regulation of macromolecular flow across the filtration barrier and the formation of oxygen radicals in the presence of hyperglycemia. In contrast to the pathological effects of genetic eNOS deletion, incomplete down-regulation of eNOS, by HDAC inhibition, was renoprotective in diabetes by decreasing oxidative-nitrosative stress.
The failure to demonstrate an antialbuminuric effect of vorinostat in STZ-eNOS–/– mice highlights the limitations of knockout models for the study of novel therapies, in which the deleted gene is implicated in the pathogenetic process. Many knockout mouse models are derived on the C57BL/6 genetic background strain that, in contrast to some other mouse strains, is relatively nephropathy resistant.47 Accordingly, both a strength and a weakness of the current study is that the renal injury observed in STZ-C57BL/6 mice was relatively mild. For example, we did not identify a significant worsening in tubulointerstitial fibrosis in these animals (data not shown), relative to normoglycemic mice, enabling the elucidation of a novel renoprotective mechanism for HDAC inhibition (eNOS down-regulation) beyond previously identified antifibrotic effects.31 Although the current study entailed a daily gavage treatment for 18 weeks, it remains to be determined whether even longer-term treatment with vorinostat would attenuate later-stage renal fibrosis in diabetes.
In summary, long-term treatment with vorinostat improved albuminuria and mesangial matrix accumulation in diabetic mice through an eNOS-dependent mechanism. These findings provide further support to a growing body of evidence attesting to the potential utility of HDAC inhibitors for the treatment of cardiorenal diseases.27
Acknowledgments
We thank Bailey Stead, Golam Kabir, M.D., Kodie Lee, and Christine Botelho, RLAT (Toronto, ON, Canada), for their excellent technical assistance.
Footnotes
Supported by grants from the Canadian Diabetes Association (OG-3-10-2949-AA), the J. P. Bickell Foundation, a Servier Vascular Research Award (A.A.), a KRESCENT postdoctoral fellowship (D.A.Y.), a Clinician Scientist Award from the Heart and Stroke Foundation of Ontario (K.A.C.), a Canadian Institutes of Health Research team grant (R.E.G.), and in part by the Canadian Diabetes Association and the Canada Research Chair Program. A.A. is a Canadian Diabetes Association Clinician Scientist, and R.E.G. is a Canada Research Chair in Diabetes Complications.
References
- 1.Nathan D.M., Cleary P.A., Backlund J.Y., Genuth S.M., Lachin J.M., Orchard T.J., Raskin P., Zinman B. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med. 2005;353:2643–2653. doi: 10.1056/NEJMoa052187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Holman R.R., Paul S.K., Bethel M.A., Matthews D.R., Neil H.A. 10-Year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med. 2008;359:1577–1589. doi: 10.1056/NEJMoa0806470. [DOI] [PubMed] [Google Scholar]
- 3.Rossig L., Li H., Fisslthaler B., Urbich C., Fleming I., Forstermann U., Zeiher A.M., Dimmeler S. Inhibitors of histone deacetylation downregulate the expression of endothelial nitric oxide synthase and compromise endothelial cell function in vasorelaxation and angiogenesis. Circ Res. 2002;91:837–844. doi: 10.1161/01.res.0000037983.07158.b1. [DOI] [PubMed] [Google Scholar]
- 4.Mohan S., Reddick R.L., Musi N., Horn D.A., Yan B., Prihoda T.J., Natarajan M., Abboud-Werner S.L. Diabetic eNOS knockout mice develop distinct macro- and microvascular complications. Lab Invest. 2008;88:515–528. doi: 10.1038/labinvest.2008.23. [DOI] [PubMed] [Google Scholar]
- 5.Zhao H.J., Wang S., Cheng H., Zhang M.Z., Takahashi T., Fogo A.B., Breyer M.D., Harris R.C. Endothelial nitric oxide synthase deficiency produces accelerated nephropathy in diabetic mice. J Am Soc Nephrol. 2006;17:2664–2669. doi: 10.1681/ASN.2006070798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nakagawa T., Sato W., Glushakova O., Heinig M., Clarke T., Campbell-Thompson M., Yuzawa Y., Atkinson M.A., Johnson R.J., Croker B. Diabetic endothelial nitric oxide synthase knockout mice develop advanced diabetic nephropathy. J Am Soc Nephrol. 2007;18:539–550. doi: 10.1681/ASN.2006050459. [DOI] [PubMed] [Google Scholar]
- 7.Kanetsuna Y., Takahashi K., Nagata M., Gannon M.A., Breyer M.D., Harris R.C., Takahashi T. Deficiency of endothelial nitric-oxide synthase confers susceptibility to diabetic nephropathy in nephropathy-resistant inbred mice. Am J Pathol. 2007;170:1473–1484. doi: 10.2353/ajpath.2007.060481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xia Y., Tsai A.L., Berka V., Zweier J.L. Superoxide generation from endothelial nitric-oxide synthase: a Ca2+/calmodulin-dependent and tetrahydrobiopterin regulatory process. J Biol Chem. 1998;273:25804–25808. doi: 10.1074/jbc.273.40.25804. [DOI] [PubMed] [Google Scholar]
- 9.Vasquez-Vivar J., Kalyanaraman B., Martasek P., Hogg N., Masters B.S., Karoui H., Tordo P., Pritchard K.A., Jr Superoxide generation by endothelial nitric oxide synthase: the influence of cofactors. Proc Natl Acad Sci U S A. 1998;95:9220–9225. doi: 10.1073/pnas.95.16.9220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 11.Advani A., Gilbert R.E., Thai K., Gow R.M., Langham R.G., Cox A.J., Connelly K.A., Zhang Y., Herzenberg A.M., Christensen P.K., Pollock C.A., Qi W., Tan S.M., Parving H.H., Kelly D.J. Expression, localization, and function of the thioredoxin system in diabetic nephropathy. J Am Soc Nephrol. 2009;20:730–741. doi: 10.1681/ASN.2008020142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Santilli F., Cipollone F., Mezzetti A., Chiarelli F. The role of nitric oxide in the development of diabetic angiopathy. Horm Metab Res. 2004;36:319–335. doi: 10.1055/s-2004-814489. [DOI] [PubMed] [Google Scholar]
- 13.Forstermann U., Munzel T. Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation. 2006;113:1708–1714. doi: 10.1161/CIRCULATIONAHA.105.602532. [DOI] [PubMed] [Google Scholar]
- 14.Brosius F.C., 3rd, Alpers C.E., Bottinger E.P., Breyer M.D., Coffman T.M., Gurley S.B., Harris R.C., Kakoki M., Kretzler M., Leiter E.H., Levi M., McIndoe R.A., Sharma K., Smithies O., Susztak K., Takahashi N., Takahashi T. Mouse models of diabetic nephropathy. J Am Soc Nephrol. 2009;20:2503–2512. doi: 10.1681/ASN.2009070721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leoni F., Zaliani A., Bertolini G., Porro G., Pagani P., Pozzi P., Dona G., Fossati G., Sozzani S., Azam T., Bufler P., Fantuzzi G., Goncharov I., Kim S.H., Pomerantz B.J., Reznikov L.L., Siegmund B., Dinarello C.A., Mascagni P. The antitumor histone deacetylase inhibitor suberoylanilide hydroxamic acid exhibits antiinflammatory properties via suppression of cytokines. Proc Natl Acad Sci U S A. 2002;99:2995–3000. doi: 10.1073/pnas.052702999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kulp S.K., Chen C.S., Wang D.S., Chen C.Y., Chen C.S. Antitumor effects of a novel phenylbutyrate-based histone deacetylase inhibitor, (S)-HDAC-42, in prostate cancer. Clin Cancer Res. 2006;12:5199–5206. doi: 10.1158/1078-0432.CCR-06-0429. [DOI] [PubMed] [Google Scholar]
- 17.Advani A., Kelly D.J., Advani S.L., Cox A.J., Thai K., Zhang Y., White K.E., Gow R.M., Marshall S.M., Steer B.M., Marsden P.A., Rakoczy P.E., Gilbert R.E. Role of VEGF in maintaining renal structure and function under normotensive and hypertensive conditions. Proc Natl Acad Sci U S A. 2007;104:14448–14453. doi: 10.1073/pnas.0703577104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Advani A., Kelly D.J., Cox A.J., White K.E., Advani S.L., Thai K., Connelly K.A., Yuen D., Trogadis J., Herzenberg A.M., Kuliszewski M.A., Leong-Poi H., Gilbert R.E. The (pro)renin receptor: site-specific and functional linkage to the vacuolar H+-ATPase in the kidney. Hypertension. 2009;54:261–269. doi: 10.1161/HYPERTENSIONAHA.109.128645. [DOI] [PubMed] [Google Scholar]
- 19.Hirose K., Osterby R., Nozawa M., Gundersen H.J. Development of glomerular lesions in experimental long-term diabetes in the rat. Kidney Int. 1982;21:689–695. doi: 10.1038/ki.1982.82. [DOI] [PubMed] [Google Scholar]
- 20.Osterby R., Gundersen H.J., Nyberg G., Aurell M. Advanced diabetic glomerulopathy: quantitative structural characterization of nonoccluded glomeruli. Diabetes. 1987;36:612–619. doi: 10.2337/diab.36.5.612. [DOI] [PubMed] [Google Scholar]
- 21.Fish J.E., Yan M.S., Matouk C.C., St Bernard R., Ho J.J., Gavryushova A., Srivastava D., Marsden P.A. Hypoxic repression of endothelial nitric-oxide synthase transcription is coupled with eviction of promoter histones. J Biol Chem. 2010;285:810–826. doi: 10.1074/jbc.M109.067868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haorah J., Knipe B., Leibhart J., Ghorpade A., Persidsky Y. Alcohol-induced oxidative stress in brain endothelial cells causes blood-brain barrier dysfunction. J Leukoc Biol. 2005;78:1223–1232. doi: 10.1189/jlb.0605340. [DOI] [PubMed] [Google Scholar]
- 23.Siddens L.K., Henderson M.C., Vandyke J.E., Williams D.E., Krueger S.K. Characterization of mouse flavin-containing monooxygenase transcript levels in lung and liver, and activity of expressed isoforms. Biochem Pharmacol. 2008;75:570–579. doi: 10.1016/j.bcp.2007.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ahn K., Huh J.W., Park S.J., Kim D.S., Ha H.S., Kim Y.J., Lee J.R., Chang K.T., Kim H.S. Selection of internal reference genes for SYBR green qRT-PCR studies of rhesus monkey (Macaca mulatta) tissues. BMC Mol Biol. 2008;9:78. doi: 10.1186/1471-2199-9-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marks P.A. Discovery and development of SAHA as an anticancer agent. Oncogene. 2007;26:1351–1356. doi: 10.1038/sj.onc.1210204. [DOI] [PubMed] [Google Scholar]
- 26.Marks P.A., Breslow R. Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nature Biotechnol. 2007;25:84–90. doi: 10.1038/nbt1272. [DOI] [PubMed] [Google Scholar]
- 27.Bush E.W., McKinsey T.A. Protein acetylation in the cardiorenal axis: the promise of histone deacetylase inhibitors. Circ Res. 2010;106:272–284. doi: 10.1161/CIRCRESAHA.109.209338. [DOI] [PubMed] [Google Scholar]
- 28.Mishra N., Reilly C.M., Brown D.R., Ruiz P., Gilkeson G.S. Histone deacetylase inhibitors modulate renal disease in the MRL-lpr/lpr mouse. J Clin Invest. 2003;111:539–552. doi: 10.1172/JCI16153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marumo T., Hishikawa K., Yoshikawa M., Hirahashi J., Kawachi S., Fujita T. Histone deacetylase modulates the proinflammatory and -fibrotic changes in tubulointerstitial injury. Am J Physiol Renal Physiol. 2010;298:F133–F141. doi: 10.1152/ajprenal.00400.2009. [DOI] [PubMed] [Google Scholar]
- 30.Pang M., Kothapally J., Mao H., Tolbert E., Ponnusamy M., Chin Y.E., Zhuang S. Inhibition of histone deacetylase activity attenuates renal fibroblast activation and interstitial fibrosis in obstructive nephropathy. Am J Physiol Renal Physiol. 2009;297:F996–F1005. doi: 10.1152/ajprenal.00282.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yoshikawa M., Hishikawa K., Marumo T., Fujita T. Inhibition of histone deacetylase activity suppresses epithelial-to-mesenchymal transition induced by TGF-beta1 in human renal epithelial cells. J Am Soc Nephrol. 2007;18:58–65. doi: 10.1681/ASN.2005111187. [DOI] [PubMed] [Google Scholar]
- 32.Tischler J.L., Abuaita B., Cuthpert S.C., Fage C., Murphy K., Saxe A., Furr E.B., Hedrick J., Meyers J., Snare D., Zand A.R. Simple inhibitors of histone deacetylase activity that combine features of short-chain fatty acid and hydroxamic acid inhibitors. J Enzyme Inhib Med Chem. 2008;23:549–555. doi: 10.1080/14756360701715703. [DOI] [PubMed] [Google Scholar]
- 33.Breyer M.D., Bottinger E., Brosius F.C., 3rd, Coffman T.M., Harris R.C., Heilig C.W., Sharma K. Mouse models of diabetic nephropathy. J Am Soc Nephrol. 2005;16:27–45. doi: 10.1681/ASN.2004080648. [DOI] [PubMed] [Google Scholar]
- 34.Yagame M., Kim Y., Zhu D., Suzuki D., Eguchi K., Nomoto Y., Sakai H., Groppoli T., Steffes M.W., Mauer S.M. Differential distribution of type IV collagen chains in patients with diabetic nephropathy in non-insulin-dependent diabetes mellitus. Nephron. 1995;70:42–48. doi: 10.1159/000188542. [DOI] [PubMed] [Google Scholar]
- 35.Dai Y., Rahmani M., Dent P., Grant S. Blockade of histone deacetylase inhibitor-induced RelA/p65 acetylation and NF-kappaB activation potentiates apoptosis in leukemia cells through a process mediated by oxidative damage: XIAP downregulation, and c-Jun N-terminal kinase 1 activation. Mol Cell Biol. 2005;25:5429–5444. doi: 10.1128/MCB.25.13.5429-5444.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Portanova P., Russo T., Pellerito O., Calvaruso G., Giuliano M., Vento R., Tesoriere G. The role of oxidative stress in apoptosis induced by the histone deacetylase inhibitor suberoylanilide hydroxamic acid in human colon adenocarcinoma HT-29 cells. Int J Oncol. 2008;33:325–331. [PubMed] [Google Scholar]
- 37.Bolden J.E., Peart M.J., Johnstone R.W. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 38.Shein N.A., Grigoriadis N., Alexandrovich A.G., Simeonidou C., Lourbopoulos A., Polyzoidou E., Trembovler V., Mascagni P., Dinarello C.A., Shohami E. Histone deacetylase inhibitor ITF2357 is neuroprotective, improves functional recovery, and induces glial apoptosis following experimental traumatic brain injury. FASEB J. 2009;23:4266–4275. doi: 10.1096/fj.09-134700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leng Y., Chuang D.M. Endogenous alpha-synuclein is induced by valproic acid through histone deacetylase inhibition and participates in neuroprotection against glutamate-induced excitotoxicity. J Neurosci. 2006;26:7502–7512. doi: 10.1523/JNEUROSCI.0096-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rivieccio M.A., Brochier C., Willis D.E., Walker B.A., D'Annibale M.A., McLaughlin K., Siddiq A., Kozikowski A.P., Jaffrey S.R., Twiss J.L., Ratan R.R., Langley B. HDAC6 is a target for protection and regeneration following injury in the nervous system. Proc Natl Acad Sci U S A. 2009;106:19599–19604. doi: 10.1073/pnas.0907935106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Noh H., Oh E.Y., Seo J.Y., Yu M.R., Kim Y.O., Ha H., Lee H.B. Histone deacetylase-2 is a key regulator of diabetes- and transforming growth factor-beta1-induced renal injury. Am J Physiol Renal Physiol. 2009;297:F729–F739. doi: 10.1152/ajprenal.00086.2009. [DOI] [PubMed] [Google Scholar]
- 42.Fish J.E., Matouk C.C., Rachlis A., Lin S., Tai S.C., D'Abreo C., Marsden P.A. The expression of endothelial nitric-oxide synthase is controlled by a cell-specific histone code. J Biol Chem. 2005;280:24824–24838. doi: 10.1074/jbc.M502115200. [DOI] [PubMed] [Google Scholar]
- 43.Robb G.B., Carson A.R., Tai S.C., Fish J.E., Singh S., Yamada T., Scherer S.W., Nakabayashi K., Marsden P.A. Post-transcriptional regulation of endothelial nitric-oxide synthase by an overlapping antisense mRNA transcript. J Biol Chem. 2004;279:37982–37996. doi: 10.1074/jbc.M400271200. [DOI] [PubMed] [Google Scholar]
- 44.Choudhary C., Kumar C., Gnad F., Nielsen M.L., Rehman M., Walther T.C., Olsen J.V., Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 45.Tolins J.P., Shultz P.J., Raij L., Brown D.M., Mauer S.M. Abnormal renal hemodynamic response to reduced renal perfusion pressure in diabetic rats: role of NO. Am J Physiol. 1993;265:F886–F895. doi: 10.1152/ajprenal.1993.265.6.F886. [DOI] [PubMed] [Google Scholar]
- 46.Sugimoto H., Shikata K., Matsuda M., Kushiro M., Hayashi Y., Hiragushi K., Wada J., Makino H. Increased expression of endothelial cell nitric oxide synthase (ecNOS) in afferent and glomerular endothelial cells is involved in glomerular hyperfiltration of diabetic nephropathy. Diabetologia. 1998;41:1426–1434. doi: 10.1007/s001250051088. [DOI] [PubMed] [Google Scholar]
- 47.Qi Z., Fujita H., Jin J., Davis L.S., Wang Y., Fogo A.B., Breyer M.D. Characterization of susceptibility of inbred mouse strains to diabetic nephropathy. Diabetes. 2005;54:2628–2637. doi: 10.2337/diabetes.54.9.2628. [DOI] [PubMed] [Google Scholar]