

Abstract
S100 proteins are small adaptors that regulate the activity of partner proteins by virtue of direct protein interactions. Here, we describe the first small molecule blockers of the interaction between S100A10 and annexin A2. Molecular docking yielded candidate blockers that were screened for competition of the binding of an annexin A2 peptide to S100A10. Several inhibitory clusters were identified with some containing compounds with potency in the lower micromolar range. We chose 3-hydroxy-1-(2-hydroxypropyl)-5-(4-isopropylphenyl)-4-(4-methylbenzoyl)-1H-pyrrol-2(5H)-one (1a) as a starting point for structure−activity studies. These confirmed the hypothetical binding mode from the virtual screen for this series of molecules. Selected compounds disrupted the physiological complex of annexin A2 and S100A10, both in a broken cell preparation and inside MDA-MB-231 breast cancer cells. Thus, this class of compounds has promising properties as inhibitors of the interaction between annexin A2 and S100A10 and may help to elucidate the cellular function of this protein interaction.
Introduction
S100 proteins were first isolated from brain by virtue of their solubility in saturated aqueous ammonium sulfate solution at neutral pH. Since then, over 20 family members have been identified, each of them unique in sequence and tissue distribution. S100 proteins belong to the larger EF hand family of Ca2+-binding proteins and primarily exist as homodimers or heterodimers.1−5 Each S100 protein contains two nonidentical EF hand motifs connected by a linker region. EF hand motifs consist of two α-helices linked by a 12 amino acid long Ca2+ binding loop.(4) Most S100 proteins (dimers) are responsive to Ca2+ by virtue of these EF hand motifs.(6) The exception to this is S100A10, which is permanently locked in a “Ca2+ on” configuration.
The principle function of S100 proteins is the regulation of the localization and activity of other proteins by direct protein−protein interactions. Some of these interactions potentially have therapeutic relevance. For example, the interaction of S100A4 and the coiled coil region of the myosin-IIA heavy chain has been implicated in cell movement in cancer.(7) As such, S100 protein interactions are attracting interest as drug targets.(8) Indeed, progress has been made in the identification of small molecule blockers of S100B and S100A4.9−11
Members of the annexin family of Ca2+/membrane binding proteins are partners of several S100 proteins.(12) Annexin A2 interacts with S100A10 as well as S100A4.2,13 It is highly expressed in endothelial cells and is overexpressed in a number of cancers, including breast and colon cancer, in which it has been implicated in invasive behavior.14−16 Annexin A2 function is crucial for blood vessel formation since its removal by genetic deletion compromises the capacity of endothelial cells to digest the extracellular matrix and to form new networks in the extracellular matrix in the mouse in vivo.(14) Similarly, blocking the cell surface annexin A2 using antibodies reduces the capacity of breast cancer cells to digest and migrate through a collagen matrix.(15)
At the molecular level, the interaction between S100A10 and annexin A2 is very well characterized both by mutagenesis and by crystallography.2,17 Two S100A10 molecules form a dimeric structure that yields two binding pockets, each of which accommodates the 14 amino acid N-terminal region of annexin A2. Biophysical studies suggest that one of the two sites of the S100A10 homodimer must be occupied by the annexin A2 N terminus before the second annexin A2 protein can bind.(18) A synthetic peptide encompassing the N-terminal annexin A2 region can disrupt preformed annexin A2-S100A10 complexes in vitro, suggesting that it has a dominant negative effect on annexin A2-S100A10 complex formation.(19) This same N-terminal peptide inhibited neoangiogenesis into matrigel plugs in a mouse model system,(14) and it also inhibited metastasis of prostate cancer cells into the bone marrow in mice.(20) Thus, complex formation with S100A10 may be the key to regulating several of the processes in which annexin A2 has been implicated.
Detailed inspection of the binding site reveals that the annexin-A2 N terminus buries ca. 660 Å2 of solvent accessible surface area (SAS) in a compact lipophilic pocket of S100A10.(2) Most of the binding energy derives from hydrophobic interactions in the innermost portion of the pocket as well as charge-enhanced H-bonds with the carboxyl groups of E5 and E9 of S100A10.2,17 The main interacting residues at the N terminus (V3, I6, L7, and L10) alone displace ca. 430 Å2 of SAS. The size of the binding pocket is thus within reach of what are commonly considered druglike molecules. Here, we describe the first small molecule blockers of this binding interaction. We used molecular docking to preselect candidate inhibitory molecules and tested these for inhibition of the interaction between S100A10 and a fluorescent annexin A2(1−14) ligand peptide. A pyrrol-2-one hit compound thus identified was explored chemically, and structure−activity relationship (SAR) patterns were observed to be compatible with occupation of the target binding site. Inhibition of the cellular protein interaction was then confirmed using immunoprecipitation. Thus, we identified a novel class of chemical probes suitable for functional exploration of the interaction between S100A10 and annexin A2.
Results and Discussion
Structure-Based Virtual Screening
The N-terminal annexin A2 peptide binding pocket of the S100A10 protein(2) was used as the target to perform a virtual screen of a total of 704511 structurally diverse small molecules. These were selected based on a Mr range between 150 and 600, since modulators of protein−protein interactions are frequently larger than inhibitors of conventional drug targets.21,22 For similar reasons, the number of hydrogen bond donors (up to 7), hydrogen bond acceptors (up to 14), and nonterminal rotatable bonds (up to 12) was allowed to exceed the limits of generally accepted druglikeness rules,23,24 although the calculated physicochemical properties of the majority of compounds (75%) in the screening database conformed to such rules. The predicted partition coefficient of compounds was kept comparatively low (XlogP < 5)(25) to avoid the selection of excessively lipophilic molecules.
Compounds were docked using the genetic optimization for ligand docking (GOLD)26,27 program in virtual screening mode, in which the genetic algorithm (GA) setting was fixed with 10000 operators; hence, search efficiency was limited, to permit faster calculations. Docked ligands were ranked based on the fitness scores (relating to predicted binding affinity)(28) generated from the virtual screening mode, and the top-scoring 1% of hit compounds, whose fitness scores ranged from 40−71, were chosen for further analysis.
The top-ranked 1% of compounds were then rescored using GOLD in standard parameter mode. In this mode, the GA setting was fixed with 100000 operators. As a result, the conformational search efficiency of the ligands was increased. The resulting fitness scores ranged from 41 to 77. On the basis of these scores, 775 top-ranked compounds were selected for binding mode analysis. The suggested binding poses were subjected to visual inspection and ranked based on hit compounds apparently being able (i) to occupy the hydrophobic regions H1, H2, and the hydrophilic pocket H3 (see below); (ii) to form productive hydrogen bonds with the receptor; and (iii) to form electrostatic complementarity with the binding pocket.(29) Furthermore, compounds that required high entropic energy (for instance torsional energy) to fit in the binding pocket were excluded.
On the basis of these criteria, a subset of 394 compounds was selected, and a clustering analysis was performed using a hierarchical clustering algorithm in which 2D fingerprints and atom pairs were taken as metrics to quantify the differences in chemical structures.(30) A total of 27 different clusters were identified in this way as well as three singletons. One distinct cluster contained 79 compounds, and the remaining 26 clusters contained on average 12 compounds.
To evaluate the virtual hit library, the above-prioritized 394 compounds were acquired and screened using a competitive binding assay based on fluorescence resonance energy transfer (FRET) between appropriately labeled S100A10 and an N-terminal annexin A2 peptide.(31) A primary screen was performed in which each compound was tested at a single concentration of 10 μM in quadruplicate. Compounds for which individual replicates showed an inhibitory activity that differed by more than three standard deviations from negative controls were rescreened and half-maximal inhibition concentration (IC50) values determined. This revealed that a total of 29 compounds possessed measurable inhibition potency. These compounds belonged to 10 different clusters (12 compounds from cluster 1, four each from clusters 9 and 19, two each from clusters 5 and 23, and one compound each from clusters 3, 6, 17, 22, and 26). No active compounds were obtained from the remaining clusters. Example compounds representing six clusters are shown in Chart 1.
Chart 1. Clusters of Blockers of the Annexin A2−S100A10 Protein Interaction Identified by GOLD Docking and Biochemical Assay.

Analysis of the Predicted Binding Poses
Analysis of the molecular interaction between the annexin A2 N terminus and the S100A10 protein shows that the annexin recognition site of S100A10 is a comparatively compact, hydrophobic, and concave binding pocket (Figure 1a), composed of residues from both S100A10 molecules in the S100A10 dimer.(2) The annexin A2 N terminus forms an amphipathic helix, the hydrophobic face of which interacts with this binding site. Mutagenesis studies indicate that the hydrophobic annexin A2 residues Val3, Ile6, Leu7, and Leu10 are the main contributors to the protein interaction with S100A10.(17) The N terminus of annexin A2 is posttranslationally modified, and the Ser1 acetyl group is also important for S100A10 binding.(32) The crystal model of the interaction is consistent with these suggestions. Hydrogen bonds are observed from the backbone NH groups of Ser1 and Thr2 to the side chain carboxyl of S100A10 B chain Glu9. Furthermore, the imidazole NH group of annexin A2 His4 hydrogen bonds with the B chain Glu5 carboxylate. Figure 1b shows that the annexin A2 binding site can be divided into three distinct regions: two predominantly hydrophobic pockets (H1 and H2) and a somewhat hydrophilic subsite (H3), which in the native annexin A2−S100A10 complex are occupied mainly by the annexin A2 residue side chains Thr2, Leu7, and AcSer1, respectively, whereas the Val3 side chain is found at the intersection of the three subsites (Figure 1a,b). H1 is composed of Tyr85, Phe86, and Met90 of chain A and Glu9, Phe13, and Met12 of chain B. H2 is formed by Phe38, Pro39, Gly40, Phe41, and Leu78 of chain A and Glu5 and Met8 of chain B. Finally, the S100A10 residues that constitute H3 are Pro1, Ser2, His6, and Glu9 of chain B.
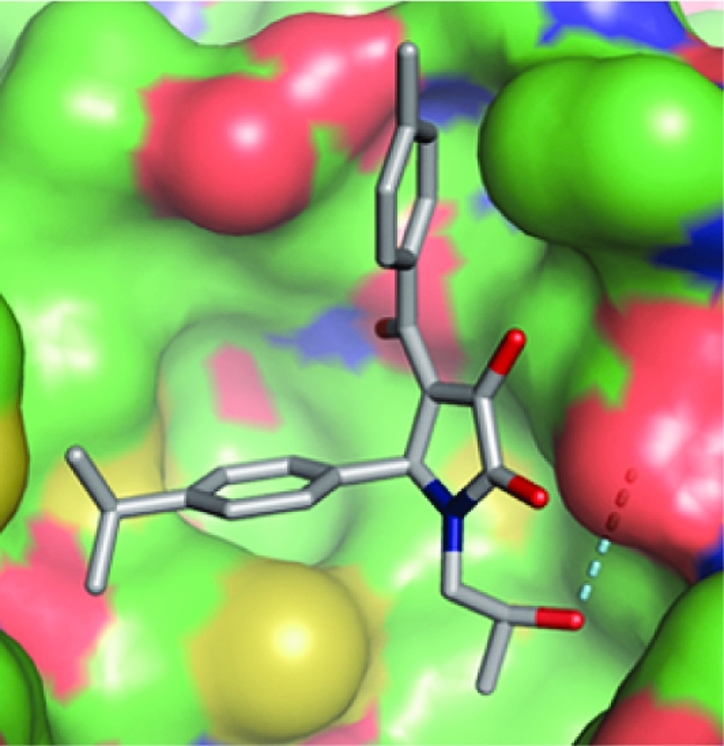
Figure 1.

Structure-based design of annexin A2−S100A10 inhibitors. (a) The interaction between the helical N-terminal annexin A2 peptide (gray CPK coloring) and a binding pocket of S100A10 (green CPK surface); the side chains, including the N-terminal acetyl group of the annexin peptide involved in intimate contact with the receptor through either hydrophobic or hydrogen-bonding (dashed lines) interactions, are shown as solid sticks and are labeled. (b) The hydrophobic (H1 and H2) and hydrophilic (H3) subsites of the S100A10 binding pocket are colored in cyan, yellow, and magenta, respectively. Predicted binding poses of verified virtual screening hits (gray CPK stick models): (c) 1a, (d) 2a, (e) 3, (f) 4, and (g) 5 (refer Chart 1).
The binding modes predicted by the docking studies for all active compounds were analyzed, and the poses of five structurally diverse compounds that showed significant inhibitory activity are illustrated in Figure 1c−g. Compounds 2a (cluster 19) and 4 (cluster 17) interact mainly with the hydrophobic regions H1 and H2 of the binding pocket, whereas compounds 3 (cluster 3), 1a (cluster 9), and 5 (cluster 26) additionally extend toward the hydrophilic pocket H3.
The symmetrical compound 4 mainly occupies the hydrophobic regions of the binding pocket and is involved in good van der Waals and hydrophobic interactions with the surrounding amino acids. In addition, the p-aminophenyl moiety of one-half of the molecule undergoes a π−π stacking interaction with the phenyl ring of Tyr85. Compound 2a hydrogen bonds through one of the protonated piperazine amine groups with a carboxylate oxygen of Glu5. Additional close contacts result from van der Waals interactions with residues Met12 and Phe86.
The amide moiety of 3 extends into the entrance of the hydrophilic H3 site, whereas in the cases of 1a and 5, this site is more fully occupied by aromatic ring substituents. Compound 3 forms hydrophobic interactions with both the H1 and the H2 sites, and its pose is stabilized by a hydrogen bond network involving the ligand primary amide and sulfonyl groups with Glu5 and Glu9 of the receptor. The 4-isopropylphenyl moiety of 1a occupies the H1 site, whereas the 2-hydroxypropyl group binds into the H2 site. The methyl group of the 2-hydroxypropyl substituent undergoes a CH−π stacking interaction with the phenyl ring of Phe41. The hydroxyl moiety donates a strong hydrogen bond to the carboxylate oxygen of Glu5. In addition, the 4-methylbenzoyl group is positioned in the H3 site.
Interestingly, the predicted binding mode of 5 is similar to that of 1a, although the compounds are not similar in structure. The 3,4-dihydropyridine scaffold of 5 is located at the center of the binding pocket, whereas the pyrazole side chain is buried deep within the H2 pocket, making good van der Waals and hydrophobic interactions. Both the methyl and the ethyl ester side chains are located in the hydrophobic region H1. The thiophene side chain extends slightly into H3.
From the above analysis of the binding poses, it appears that the two hydrophobic regions (H1 and H2) play a dominant role in the binding affinity of all of the inhibitors. The proposed hydrogen bond formation of either or both of the Glu5 and the Glu9 receptor residues with the inhibitors (1a, 2a, and 3) appear to mimic the hydrogen bond interaction of Glu5 and Glu9 residues with the annexin A2 N-terminal peptide in the crystal structure. Overall, in view of the chemical nature of the inhibitor compounds analyzed here and the nature of the interactions with the binding pocket, it appears that the hydrogen bond acceptor sites of Glu5 and Glu9 and the two hydrophobic regions (H1 and H2) of the receptor can be considered as the key pharmacophoric features.
Considering cluster SAR analysis, inhibitory potency, druglike properties, and synthetic amenability, we chose cluster 9, and in particular compound 1a, as a medicinal chemistry starting point for the design and development of candidates that target the annexin A2−S100A10 protein−protein interaction. In the following, we describe our initial exploratory hit-to-lead conversion based on the 1-substituted 4-aroyl-3-hydroxy-5-phenyl-1H-pyrrol-2(5H)-one template.
1-Substituted 4-Aroyl-3-hydroxy-5-phenyl-1H-pyrrol-2(5H)-one Analogue Synthesis
The required 1H-pyrrol-2(5H)-one system was prepared using a simple two-step procedure (Scheme 1). In the first step, substituted methyl pyruvates 12 were obtained by Claisen condensation of aryl methyl ketones 10 with dimethyl oxalate 11.33−35 Pyruvates 12 were then subjected in a second step to a three-component coupling reaction with the appropriate amines 7 and aldehydes 8.36−40
Scheme 1. Synthesis of 1-Substituted 4-Aroyl-3-hydroxy-5-phenyl-1H-pyrrol-2(5H)-ones.

Reagents and conditions: (a) 1,4-Dioxane, 15 min. (b) 1,4-Dioxane, overnight reaction. (c) (i) Microwave irradiation, 250 W, 30 °C, 5 min, 2 M NaOMe/MeOH; (ii) H+ (pH 3−4).
For the preparation of methyl pyruvates 12, we developed a rapid protocol for the condensation of acetophenones 10 with dimethyl oxalate 11 using 2 M MeONa in MeOH solution under microwave irradiation (250 W) for 5 min at 30 °C. With most substrates, this reaction gave rise to a precipitate of the sodium enolate Claisen adduct, which upon acidification to pH 3−4 provided the methyl pyruvate products 12 in maximally 52% isolated yields. Alterations to neither base strength nor reaction time were found to result in improved yields. Using this procedure, compounds 12a−k, which exist predominantly in the keto−enol rather than the diketone form, were prepared (Table 1).
Table 1. Chemical Structures and the Keto−Enol Form of Methyl Phenylpyruvates 12a−k.

| no. | R6 | R7 | R8 | X | Y | keto−enol/diketone tautomer ratioa |
|---|---|---|---|---|---|---|
| 12a | H | H | H | C | C | 92:8 |
| 12b | H | H | Me | C | C | 92:8 |
| 12c | H | Me | H | C | C | 93:7 |
| 12d | H | H | OMe | C | C | 93:7 |
| 12e | H | OMe | H | C | C | 90:10 |
| 12f | OEt | H | H | C | C | 91:9 |
| 12g | H | H | Cl | C | C | 89:11 |
| 12h | H | H | CN | C | C | 88:12 |
| 12i | H | Me | N | C | 88:12 | |
| 12j | H | H | N | C | 90:10 | |
| 12k | H | H | C | N | 93:7 |
Tautomer ratios were calculated from the 1H NMR spectra based on the CH signal of the keto−enol form against the CH2 signal of the diketone form.
The three-component reaction of amines 7, aldehydes 8, and methyl phenylpyruvates 12 occurs through initial formation of imines 9, which then undergo cyclocondensation with 12. When 4-isopropyl benzaldehyde was added to 2-hydroxypropylamine in 1,4-dioxane, mass spectromeric analysis after 15 min showed the presence of an ion of m/z [M + H]+ = 206.1537, corresponding to the imine. Pyruvate ester 12b in 1,4-dioxane was then added to the reaction mixture, and precipitation was observed within 1 h of the reaction. Stirring of the mixture was continued overnight to afford authentic compound 1a in 43% isolated yield. This procedure was adopted for the preparation of all of the analogues described here (Tables 2−4).
Table 2. Inhibition of the S100A10 Interaction with Annexin A2 by 5-Substituted 3-Hydroxy-1-(2-hydroxypropyl)-4-(4-methylbenzoyl)-1H-pyrrol-2(5H)-ones and Activity in Counterscreen Assay.

| annexin−S100A10 interaction |
counterscreen | |||||||
|---|---|---|---|---|---|---|---|---|
| no. | type | R2 | R3 | R4 | R5 | pIC50a | IC50 (μM) | % controlb |
| P | 5.99 ± 0.10 | 1 | ||||||
| 1a | I | H | H | iPr | H | 4.85 ± 0.09 | 14 | 101 ± 7 |
| 13 | II | <3 | >1000 | |||||
| 14 | III | 3.89 ± 0.15* | 128 | |||||
| 15 | I | H | H | H | H | 3.60 ± 0.33* | 251 | |
| 16 | I | H | iPr | H | H | 4.94 ± 0.14 | 11 | 100 ± 16 |
| 17 | I | H | H | Me | H | 4.07 ± 0.10* | 85 | |
| 18 | I | H | H | Et | H | 4.80 ± 0.25 | 16 | |
| 19 | I | H | H | nPr | H | 4.74 ± 0.09 | 18 | 97 ± 9 |
| 20 | I | H | H | tBu | H | 4.55 ± 0.05** | 28 | 123 ± 7 |
| 21 | I | H | H | NMe2 | H | 3.56 ± 0.55* | 275 | |
| 22 | I | H | H | OH | H | <3 | >1000 | |
| 23 | I | H | H | OCF3 | H | 5.02 ± 0.11 | 9 | 110 ± 8 |
| 24 | I | H | H | CF3 | H | 5.20 ± 0.36 | 6 | 102 ± 1 |
| 25 | I | H | CF3 | H | H | 5.08 ± 0.09 | 8 | 110 ± 6 |
| 26 | I | CF3 | H | CF3 | H | 5.12 ± 0.09 | 8 | 102 ± 6 |
| 27 | I | H | CF3 | H | CF3 | 5.41 ± 0.12 | 4 | 109 ± 3 |
| 28 | I | H | H | Cl | H | 3.93 ± 0.21* | 117 | |
| 29 | I | H | Cl | H | H | 3.98 ± 0.15* | 105 | |
| 30 | I | H | Cl | H | Cl | 5.16 ± 0.15 | 7 | 119 ± 4 |
| 31 | I | H | CF3 | Cl | H | 5.12 ± 0.06 | 8 | 121 ± 11 |
Data were expressed as % of the specific signal of the nontreated control and analyzed by nonlinear regression using Graphpad Prism. pIC50 ± standard error of the fit (n = 4−8 data points per concentration, 0.4−100 μM). The midpoint of the dose−response curve is given based on a floating fit unless indicated as follows: *, bottom fixed at zero; **, bottom fixed at zero, top fixed at 100%.
Percent remaining signal at 50 μM compound relative to diluent only (mean ± standard error mean of >3 determinations). P = nonlabeled synthetic annexin N terminus peptide.
Table 4. Inhibition of the S100A10 Interaction with Annexin A2 by 1,5-Substituted 4-Aroyl-3-hydroxy-1H-pyrrol-2(5H)-ones and Activity in the Counterscreen Assay.

| annexin−S100A10 interaction |
counterscreen | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| no. | R1 | R3 | R6 | R7 | R8 | X | pIC50a | IC50 (μM) | % controlb |
| 45 | CH2CH=CH2 | H | H | H | N | <3 | >1000 | ||
| 46 | CH2CH=CH2 | H | H | Me | N | 4.55 ± 0.17 | 28 | 102 ± 9 | |
| 47 | CH2CH=CH2 | H | H | H | Cl | C | 5.20 ± 0.06 | 6 | 120 ± 6 |
| 48 | CH2(CH2)2OMe | H | H | H | N | <3 | >1000 | ||
| 49 | CH2(CH2)2OMe | H | H | Me | N | 3.57 ± 0.01* | 269 | ||
| 50 | CH2(CH2)2OMe | F | H | Me | N | 3.74 ± 0.01* | 182 | ||
| 51 | CH2(CH2)2OMe | F | H | H | Cl | C | 3.73 ± 0.42* | 186 | |
| 52 | CH2(CH2)2OMe | H | OEt | H | H | C | 4.97 ± 0.09 | 11 | 117 ± 9 |
| 53 | CH2(CH2)2OMe | F | OEt | H | H | C | 5.31 ± 0.11 | 5 | 127 ± 19 |
Data were expressed as % of the specific signal of the nontreated control and analyzed by nonlinear regression using Graphpad Prism. pIC50 ± standard error of the fit (n = 4−8 data points per concentration, 0.4−100 μM). The midpoint of the dose−response curve is given based on a floating fit unless indicated as follows: *, bottom fixed at zero; **, bottom fixed at zero, top fixed at 100%.
Percent remaining signal at 50 μM compound relative to diluent only (mean ± standard error mean of >3 determinations).
SARs
Initially, a number of analogues were investigated in which the substituent at C5 of the 1H-pyrrol-2(5H)-one system present in hits 1a−d was varied (Table 2). Replacement of this substituent with isopropyl (13) abolished the activity completely and with unsubstituted phenyl (15) showed 18-fold decrease in activity, suggesting that the substitution pattern of the phenyl ring was important. We therefore prepared and tested a small set of appropriate analogues 16−31 (Table 2). The facts that the 3- and 4-isopropylphenyl derivatives 1a and 16 were equipotent and that the 2-naphthyl analogue 14 retained some activity show that steric bulk at either the meta- or the para position of the phenyl group, or both, was productive in terms of binding. Alteration of the phenyl para substituent (isopropyl) in 1a from methyl (17) to ethyl (18), n-propyl (19), and t-butyl (20) showed distinct SARs, suggesting that the alkyl chain length and extent of branching in the isopropyl group are optimal. Reference to the modeled binding pose of 1a (Figure 1c), where the 4-isopropylphenyl group fits neatly into the H1 subsite, supports this notion.
Replacement of the lipophilic isopropyl group with the roughly isosteric but more polar dimethylamino group in analogue 21 resulted in loss of activity over 20-fold. Again, this is consistent with the highly hydrophobic nature of the H1 subsite. Similarly, the even more polar 4-phenol derivative 22 was inactive. Whereas the dimethylamino group in 21 is somewhat hydrophobic and electron-donating, the trifluoromethoxy and trifluoromethyl groups, of which the latter is isosteric with dimethylamino and isopropyl in the comparatively more potent analogues 23 and 24, respectively, are also hydrophobic but electron-withdrawing. As with the positional isopropyl isomers 1a and 16, the 3- and 4-trifluoromethylphenyl isomers 24 and 25 displayed similar potency. Interestingly, 3,5-di-CF3 compound 27 was somewhat more active than the corresponding 2,4-isomer 26 and was the most potent compound thus far. A decrease in potency was observed with a chloro substituent at the meta- (29) or para- (28) position of the phenyl ring. However, the 3,5-dichlorophenyl derivative 30 was 15-fold more potent than 28 and 29. Surprisingly, addition of a trifluoromethyl group to the meta position of the phenyl ring in the context of the para-chloro substituent resulted in a comparatively potent compound (31).
We next turned our attention to the 1H-pyrrol-2(5H)-one N1 substituent (32−36, Table 3). In 1a, this is a simple 2-hydroxypropyl group whose alcohol function donates a hydrogen bond to the side chain carboxyl of Glu5 according to our binding hypothesis (Figure 1c). As expected, therefore, replacement of the hydroxyl group with a methyl group (32) resulted in complete loss of activity. Replacement of the 2-hydroxypropyl substituent of 1a with a 2-hydroxyethyl group (33) was well tolerated. However, an unexpected increase in activity was observed with compound 34, where a 2-methoxyethyl substituent, incapable of donating a hydrogen bond, was present. A further gain in potency was observed upon lengthening the substituent to 3-methoxypropyl (35). Introduction of a hydrophobic allyl side chain (36), incapable of participating in hydrogen bonds, also retained activity.
Table 3. Inhibition of the S100A10 Interaction with Annexin A2 by 1-Substituted 4-Aroyl-3-hydroxy-5-(4-isopropylphenyl)-1H-pyrrol-2(5H)-ones and Activity in Counterscreen Assay.

| annexin−S100A10 interaction |
counterscreen | |||||||
|---|---|---|---|---|---|---|---|---|
| no. | R1 | R6 | R7 | R8 | X | pIC50a | IC50 (μM) | % controlb |
| 1a | CH2CHMeOH | H | H | Me | C | 4.85 ± 0.09 | 14 | 101 ± 7 |
| 32 | iBu | H | H | Me | C | <3 | >1000 | |
| 33 | CH2CH2OH | H | H | Me | C | 4.92 ± 0.16 | 12 | 101 ± 6 |
| 34 | CH2CH2OMe | H | H | Me | C | 5.12 ± 0.16 | 8 | 120 ± 11 |
| 35 | CH2(CH2)2OMe | H | H | Me | C | 5.54 ± 0.27 | 3 | 111 ± 19 |
| 36 | CH2CH=CH2 | H | H | Me | C | 5.19 ± 0.05** | 6 | 105 ± 14 |
| 37 | CH2CHMeOH | H | H | H | C | 4.84 ± 0.14 | 14 | |
| 38 | CH2CHMeOH | H | Me | H | C | 4.10 ± 0.10* | 79 | |
| 39 | CH2CHMeOH | H | H | OMe | C | 3.21 ± 0.35* | 616 | |
| 40 | CH2CHMeOH | H | OMe | H | C | 5.16 ± 0.05 | 7 | 110 ± 3 |
| 41 | CH2CHMeOH | OEt | H | H | C | 5.15 ± 0.11 | 7 | 112 ± 11 |
| 42 | CH2CHMeOH | H | - | Me | N | 3.10 ± 0.01* | 794 | |
| 43 | CH2CHMeOH | H | H | CN | C | 4.25 ± 0.31 | 56 | |
| 44 | CH2CHMeOH | H | H | Cl | C | 5.57 ± 0.86 | 3 | 126 ± 5 |
Data were expressed as % of the specific signal of the nontreated control and analyzed by nonlinear regression using Graphpad Prism. pIC50 ± standard error of the fit (n = 4−8 data points per concentration, 0.4−100 μM). The midpoint of the dose−response curve is given based on a floating fit unless indicated as follows: *, bottom fixed at zero; **, bottom fixed at zero, top fixed at 100%.
Percent remaining signal at 50 μM compound relative to diluent only (mean ± standard error mean of >3 determinations).
We finally probed the SARs of the 1H-pyrrol-2(5H)-one 4-aroyl group (37−44, Table 3). Inspection of the predicted binding pose of 1a shows that the 4-methylbenzoyl group occupies the comparatively shallow H3 subsite (Figure 1c) and that substitution of the aromatic ring at the meta or ortho position may result in better contacts. This might be achieved with lipophilic or hydrophilic substituents since the H3 subsite is weakly hydrophilic. Interestingly, switching of the methyl group from the para- (1a) to the meta- (38) position resulted in 5-fold lower activity, whereas removal of the methyl group (37) did not. Surprisingly, replacement of the benzene ring with a more hydrophilic 3-pyridyl ring (42) was poorly tolerated. When the terminal methyl group was replaced with the larger and more polar methoxy group either para (39) or meta (40), this resulted in higher activity in the latter case. As expected from modeling, the ortho-substituted ethoxyphenyl analogue 41 showed slightly better activity than 1a. Replacement of the electron-donating nonpolar 4-methyl group in 1a with the electron-withdrawing polar 4-cyano group (43) resulted in loss of activity, whereas the 4-chlorophenyl derivative 44 retained comparatively better potency.
Because of the somewhat unexpected potency of the 1H-pyrrol-2(5H)-one 3-methoxypropyl (35) and allyl (36) N-substituted analogues, the last phase of our exploratory SAR studies concerned the combination of some of the individual modifications discussed above in the context of these groups (Table 4). Like compound 42, which also contains the 4-methyl-3-pyridyl group, the allyl (46) and 3-methoxypropyl (49 and 50) derivatives were also either less active or inactive, and the corresponding 3-pyridyl analogues (45 and 48) were completely inactive. However, the 4-chlorophenyl derivative 47 with the allyl N-substitution was one of the most potent compound in our series, while a N-(3-methoxypropyl) derivative (51) was comparatively inactive, although the latter compound differs from 47 insofar as it contains an additional 3-fluoro group in the 1H-pyrrol-2(5H)-one C5 phenyl substituent. This difference was not recapitulated in the context of the C5 2-ethoxybenzoyl derivatives 52 and 53, which were both comparatively potent.
Overall, the divergence of the SARs depending on whether the 1H-pyrrol-2(5H)-one N1 substituent is the protic hydroxypropyl or an aprotic (methoxyalkyl or allyl) group suggests that these compounds may have different binding modes. Our modeling studies show that an alternative pose for the methoxyalkyl and allyl compounds (e.g., 35 and 36; Figure 2a,b) may be relevant, where the 4-aroyl group binds in the H2 rather than the H3 site and where the C5-isopropylphenyl occupies the region between H1 and H2, with one of the methyl groups reaching into a small pocket at the base of the binding site in a similar fashion to that observed with the Val3 side chain of the annexin A2 peptide (Figure 1a). A hydrogen bond with the B chain Glu5 carboxylate is maintained but now involves the 1H-pyrrol-2(5H)-one C3-hydroxyl rather than the N1 substituent. This alternative binding pose does not appear to be favored when the C5-phenyl group contains a para-trifluoromethyl group, however, since highly scoring docking poses for, for example, the comparatively potent analogue 47 (Figure 2c) are similar to that of 1a (Figure 1c), except that the alkene function of the N1-allyl group is stacked between the phenyl rings of Phe38 and Phe41 within the H2 subsite.
Figure 2.

Alternative binding poses for N1-methoxyalkyl and allyl 1H-pyrrol-2(5H)-ones. Highly scoring S100A10-docked poses of compounds 35 (a), 36, (b), and 47 (c) are shown.
Comparison of Biological Activity of Inhibitors
We compared the dose response of inhibition of the unlabeled N-terminal annexin A2 peptide with that of our 1H-pyrrol-2(5H)-one compounds (examples are shown in Figure 3). From this analysis, an IC50 value of 1 μM was calculated for the cognate annexin A2 peptide (which compares well with that measured by equilibrium dialysis(31) and isothermal titration calorimetry(18)). The hit compound 1a was about 14-fold less potent (IC50 = 14 μM), and our more potent analogues (e.g., 47) were about 4-fold less potent (IC50 = 6 μM). The apparently steep Hill slopes for both peptide and compounds may be a reflection of the complexity of the underlying binding events as proposed.(18) It is important to note that the addition scheme in the binding assay involves preforming a complex of the annexin A2 N-terminal peptide tracer and the S100A10 dimer prior to exposure to compound; therefore, these compounds are able to disrupt a preformed complex. Importantly, none of the compounds inhibited a nonrelated protein interaction(31) assayed in a comparable fashion (Tables 2−4), suggesting that their inhibitory potential is not due to nonspecific interference with the fluorescence readout or to promiscuous inhibition of protein interactions. The compounds (including inactive compounds) were soluble in aqueous solution at the concentrations tested (Table 5), indicating that the inhibition observed in the biochemical assay is not an artifact of poor solubility.
Figure 3.

Biological activity of peptide and small-molecule annexin A2−S100A10 inhibitors. (A) Dose−response curves for the 14-residue N-terminal annexin A2 peptide (⧫) and compounds 1a (▲), 47 (◼), and 50 (▼) in the FRET-based competitive annexin A2−S100A10 binding assay are shown. Curve fit was performed in Graphpad Prism as described in Table 1. Error bars represent the standard error of the mean of four (compounds) or eight (peptide) observations. (B) As under A except that compound 27 (●; n = 28 ± standard error of the mean) or 22 (○; n = 4 ± standard error of the mean) was analyzed.
Table 5. Aqueous Solubility of Selected Analoguesa.
| no. | aqueous solubility (mM) | no. | aqueous solubility (mM) | no. | aqueous solubility (mM) |
|---|---|---|---|---|---|
| 1a | 0.3 | 24 | 1.5 | 40 | 1.1 |
| 13 | 5.3 | 25 | 4.1 | 42 | 4.9 |
| 15 | 4.1 | 26 | 3.5 | 45 | 2.3 |
| 18 | 0.8 | 27 | 2.3 | 47 | 0.3 |
| 21 | 2.7 | 31 | 2.0 | 48 | 3.4 |
| 22 | 5.8 | 33 | 1.7 | 53 | 3.1 |
Numbers represent averages of two determinations, which differed by less than 5% from the average.
The binding assay used for SAR exploration only reports the interaction of the annexin N terminus with S100A10. While there is ample evidence to suggest that this region of annexin A2 represents the sole binding region on S100A10, we sought to confirm the inhibitory activity of this class of compounds on the physiological relevant complex of these two proteins. For this purpose, the physiological complex was obtained by lysing MDA-MB-231 breast cancer cells. The lysate was treated with compound after which S100A10 was immunoprecipitated, and the immunoprecipitates were analyzed by SDS-PAGE and Western blotting for the presence of annexin A2. As can be seen in Figure 4a, annexin A2 is present in the S100A10 immunoprecipitates, indicating that a complex between these proteins exists in the cell lysate. After treatment with 1a or 27, the amount of complex was reduced. Quantification of the coimmunoprecipitated protein showed that at inhibitor concentrations of 50 μM, around 70% of the complex had been lost, which was significantly different from the noninhibitor control (Figure 4b). As expected, the N-terminal annexin A2 peptide also disrupted the S100A10−annexin A2 physiological complex. Thus, these compounds are able to disrupt the complex of S100A10 and full length, intact annexin A2. Interestingly, analogue 22, which is highly soluble (Table 5) but does not inhibit the binding of the annexin N-terminal peptide in the in vitro FRET binding assay (Table 2 and Figure 3b), does not inhibit the complex between the native proteins, suggesting that the SAR patterns observed in the FRET assay translate to the native proteins.
Figure 4.

Analysis of the native complex of S100A10 and annexin A2. (A) MDA-MB-231 cells were lysed and incubated with 50 μM compound as indicated or with the annexin A2 N terminus peptide (P). S100A10 was immunoprecipitated, and the amount of annexin A2 in the immunoprecipitates was analyzed by SDS-PAGE followed by Western blotting using an annexin A2 antibody. Annexin A2 was identified based upon comigration with an annexin A2 reference on the blot. The section of the blot with the annexin A2 signal is shown. (B) MDA-MB-231 cells were lysed and incubated with compound at concentrations as indicated or with the annexin A2 N terminus peptide (P). Western blots were obtained and scanned, and scans were quantified using Scion Image software. Quantified data were expressed as % of nontreated control. Error bars represent the standard error of the mean (n = 3 from three separate lysates). (C) As in B, except that MDA-MB-231 cells were treated with compound for 6 h after which the cells were lysed and washed, and immunoprecipitations were performed as above. Error bars represent the standard error of the mean (n = 3 from three separate lysates). *p < 0.05 (nonpaired t test).
We also investigated the ability of selected analogues from this series of compounds to inhibit the physiological complex inside the cell. Intact MDA-MB-231 cells were pretreated with different concentrations of 1a or 27 after which the cells were lysed and immunoprecipitation assays were performed. It can be seen in Figure 4b that pretreatment of the cells with 27 leads to a statistically significant disruption of the interaction between S100A10 and cellular annexin A2. The extent of inhibition at 100 μM is similar to that observed at 10 μM in the broken cell immunoprecipitation assay, suggesting a 10-fold drop-off in potency, possibly due to limited cell penetration of the compound. Compound 1a apparently inhibits the cellular interaction, but this inhibition was not statistically significant from the control treatment. Solubility may affect the assayability of 1a at these higher concentrations (Table 5). Importantly, in contrast to peptide blockers, 27 can penetrate the cell and can therefore be used to evaluate the cellular function of this protein interaction.
Conclusion
In this study, we have employed a structure-based virtual screening approach to identify nonpeptide inhibitors of the annexin A2−S100A10 protein interaction. Systematic alteration of the 3-hydroxy-1H-pyrrol-2(5H)-one hit compound 1a resulted in distinct SARs that are in fairly good agreement with our hypothetical binding mode for the series. Importantly, the SAR translates to the native proteins, and the compounds are potentially useful for exploration of the physiological importance of the protein interaction. So far, peptides have been used to compete with the interaction between these proteins. While these peptides have generated some understanding of the function of the complex at the cell surface, their use is limited. The blockers described here provide a template to develop inhibitors for wider exploration of the function of this complex. The current inhibitors may themselves be useful as probes, and the fact that we have identified active as well as inactive analogues in this series indicates this may be an option. However, further potency gains are desirable, and a better understanding of the solubility vs membrane penetration is required to allow this class of compounds to become useful as probes of the intracellular complex.
Experimental Section
Preparation of the Annexin A2 N-Terminal Peptide−S100A10 Complex Coordinates
The complex between the S100A10 protein and the annexin A2 N-terminal peptide (PDB ID: 1BT6) was modified as follows: The chain termini were charged, and a total of 1664 hydrogen atoms were added, including water molecules. AMBER7 FF99 charges were assigned to the complex. Side chain amides of Gln residues were reoriented to maximize hydrogen bonding. Then, the complex was subjected to energy minimization in three steps using AMBER7 FF99 parameters. Minimization method, Powell; initial optimization, none; termination criteria, gradient 0.5 kcal/mol; and maximum iterations, 5000. The annexin A2 binding site of S100A10 was defined as a cavity by a set of residues within 7 Å of the annexin A2 ligand peptide from the energy-minimized complex. A centroid for the binding site was calculated using a shell of all atoms within 5 Å of the ligand peptide involving 283 atoms, and the peptide was then extracted from the complex. There were five water molecules within the binding site, and these were also deleted.
Compound Database Preparation
Compounds were selected using the ZINC database (release 6, 2006)(41) entries from the vendors Asinex and Chembridge. A web-based query tool in ZINC was used to specify molecular property constraints, and 3D structures of the selected compounds were generated using the program Concord (Tripos). Structures with inappropriate isomeric specifications and those containing disallowed atoms in terms of valence or with errors in ring definitions were excluded. A database was generated from the remaining 704511 3D-encoded compounds.
Virtual Screening
Docking was performed using GOLD V3.0.1. Ten docking runs were performed for each molecule in this database, allowing early termination of the docking runs if the top three solutions were within 1.5 Å rmsd of each other. A binding site radius of 12 Å was found to be optimal. Flipping was not allowed for those ligands that have ring-NHR and ring-NR1R2 groups to avoid the addition of huge torsional energy penalties to the total fitness scores.
To optimize the fitness score, GOLD uses a GA, a search technique that mimics the process of natural evolution. An initial population of ligand conformations is transformed via GA operations (mutations, crossovers, and migrations) into a final population. Individual ligand conformations are known as chromosomes. Genes represent the flexible torsion angles of the ligand. Each chromosome is assigned a fitness score based on its predicted binding affinity, and the chromosomes within the population are ranked according to the fitness score. The selection of parent chromosomes is biased toward fitter members of the population, that is, chromosomes corresponding to ligand dockings with good fitness scores. Each population in GA is called an island. Chromosomes can migrate between adjacent islands using the migration operator. Mutation is an intrachromosomal operator that introduces new genetic material into the population. Crossover selects the genes from the parent chromosomes and creates new offspring. The number of GA operations applied over the course of a GA run determines the search efficiency of the GOLD run.
During the run in virtual screening mode, 10000 GA operations were performed on a single population (single island) of 100 individuals (default value); hence, there cannot be any migration. Operator weights for the crossover and mutation were set at default (100, 100, respectively). A cutoff value of 2.0 Å was applied for the hydrogen bonds to avoid the selection of poor hydrogen bonds during the GA run. Similarly, a cutoff value of 10 Å was applied for the van der Waals interactions, tolerating a few bad bumps during the GA run. Rescoring was then performed using standard GA settings with the number of operators increased to 100000. Five different populations (islands) each consisting of 100 chromosomes were maintained during the entire GA run. Ten migrations were allowed between the islands. For every crossover, one mutation was applied. Cut-off values of 2.5 Å for hydrogen bonds and 4 Å for van der Waals interactions were applied. The parameters active site radius, early termination criteria, and ligand flexibility remained unchanged during the rescoring of selected compounds.
Cluster Analysis
For cluster analysis, the Selector module of SYBYL was used, and analysis was performed as described.(30) Two-dimensional fingerprints and atom pairs were selected as diverse metrics. All of the compounds were processed using the hierarchical clustering method with a weight metric of 1.00. A total of 27 clustering classes were generated, and the diversity of these clusters was analyzed based on the dendrogram obtained, in which nodes of the dendrogram correspond to the common core structure.
Solubility Assay
Compounds were dissolved at 10 mM in phosphate buffer (pH 7.4) containing 2.7 mM potassium chloride and 137 mM sodium chloride, by stirring for 27 h at room temperature. The resulting solution was filtered using GHP Acrodisc 25 mm syringe filters with 0.2 μm GHP membrane. Compound concentrations were measured by absorption on a Perkin-Elmer Envision plate reader, at the wavelength showing maximum absorption in a scan between 250 and 700 nm, determined separately for each compound. The absorbance of each filtrate was measured, and the concentration was determined by comparison with a standard curve of compound in 5% aqueous DMSO.
Fluorescence Screening Assay
Routine assessment of compound activity was performed as described in Li et al.(31) Briefly, a Cy5-labeled S100A10 tracer was developed, and binding of a Cy3-labeled annexin A2(1−14) peptide ligand was assessed using a FRET readout. Assays were carried out in Nunc black nontreated 384-well plates at 20 °C in 50 μL of buffer C. All incubations were performed in quadruplicate. Compounds, peptide, and buffer controls were added to the wells in a 10 μL volume in 5% DMSO. Cy5-labeled S100A10 tracer (407 nM) and Cy3-labeled annexin A2(1−14) peptide ligand (1.33 μM) were preincubated for 5 min at 20 °C, and 40 μL of the preformed complex was then added to the wells and mixed for 10 s to yield a final DMSO concentration of 1%. After 5 min of incubation at 20 °C, readings were taken on a Perkin-Elmer Envision plate reader by excitation at 488 nm and emission at 695 nm. Each screening plate contained as controls the individual labeled proteins, as well as the two protein partners with and without a 12.8 μM concentration of the nonlabeled AA2(1−14) competitor (all in quadruplicate). FRET was calculated by subtracting the sum of the fluorescence emission of S100A10-Cy5 and AA2(1−14)-Cy3 individually from that measured for the coincubated partners. A counterscreen assay(31) that measured the FRET signal from a Cy3-conjugated donkey antigoat IgG (4 μg/mL) onto a Cy5-labeled goat IgG (3 μg/mL) was used in parallel to assess nonspecific interference with the fluorescence readout. Compound binding was calculated as percentage of nontreated control, and data were analyzed by nonlinear regression (dose response−variable slope) using Graphpad Prism Software to determine the IC50 of binding (primary assay) or expressed as a percentage of nontreated controls measured at the 50 μM concentration (counterscreen assay).
Immunoprecipitation Assay
Human MDA-MB-231 breast cancer cells were extracted in 50 mM Tris-HCl (pH 8.0) buffer containing 150 mM sodium chloride and 1% (v/v) NP-40. Lysates were centrifuged at 17000g and incubated at 4 °C for 16 h with 10 μL of S100A10 antibody (BD transduction Laboratories) and 50 μL of protein A/G agarose (Alpha Diagnostic International Inc.). Protein A/G agarose was then recovered by centrifugation at 2400g, washed three times with 10 mM phosphate buffer (pH 7.4) containing 2.7 mM potassium chloride and 137 mM sodium chloride, resuspended in 50 μL of 160 mM Tris-HCl (pH 6.8) buffer containing 4% (w/v) sodium lauryl sulfate, 20% (v/v) glycerol, 0.04% (w/v) bromophenol blue, and 10% (v/v) 2-mercaptoethanol, boiled at 99 °C for 10 min, and centrifuged at 17000g for 10 min. The supernatant was then analyzed by sodium lauryl sulfate polyacrylamide gel electrophoresis after which the gel was transferred to a nitrocellulose filter. The filter was incubated with an annexin A2 monoclonal antibody (1:3000; BD Transduction Laboratories) followed by incubation with an antimouse horseradish peroxidase IgG conjugate (1:5000; GE Healthcare) and then developed using the ECL detection reagent (GE Healthcare). Where indicated, test compounds were incubated for 1 h with cell lysate prior to immunoprecipitation. To determine inhibition of the intracellular complex, MDA-MB-231 cells were pretreated with compound for 6 h after which the cells were washed three times with 10 mM phosphate buffer (pH 7.4) containing 2.7 mM potassium chloride and 137 mM sodium chloride, prior to extraction and immunoprecipitation as described above.
Synthesis
All reagents were purchased directly from commercial sources and used as supplied unless otherwise stated. Melting points were measured using a Gallenkamp melting point apparatus and are uncorrected. Accurate mass and nominal mass measurements were performed using a Waters 2795-Micromass LCT electrospray mass spectrometer. Infrared spectra were recorded using Avatar 360 FT-IR system. Samples were prepared as KBr discs and scanned from 4000 to 500 cm−1. All NMR spectra were recorded in deutero-DMSO in 5 mm tubes, with tetramethylsilane as an internal standard, using a Bruker ACS-120 instrument at 400 (1H NMR) and 100.6 MHz (13C NMR). Thin-layer chromatography was performed using aluminum-backed silica gel 60 plates (0.20 mm layer), and the ascending technique was used with a variety of solvents. Visualization was by UV light at either 254 or 365 nm. HPLC was carried out using two different methods. Method 1: Kromasil (250 mm × 4.6 mm, 5 μM particle size) column with MeCN/H2O (5−95% organic over 20 min at 1 mL/min). Method 2: Onyx Monolith C18 (100 mm × 4.6 mm, 5 μM particle size) column with MeOH/H2O (5−95% organic over 10 min at 3 mL/min). In both of the methods, detection was at 260 nm. All compounds were isolated in >95% purity unless otherwise stated (refer to the Supporting Information).
Step 1: Synthesis of Methyl Phenylpyruvates
Sodium (184 mg, 8 mmol) was dissolved in dry methanol (4 mL) at 0 °C to produce a 2.00 M solution of sodium methoxide. Dimethyl oxalate (590 mg, 5.0 mmol, 1.0 equiv) was added, and the solution was stirred for 15 min. The appropriate aromatic ketone (5.0 mmol, 1.0 equiv) was then added. A microwave-assisted reaction was carried out for 5 min at 250 W, 250 P, and 30 °C.
Method A
After 5 min of the microwave reaction, for some products, precipitation was observed immediately (12a,d,e,g−k). For other products (12b,c), precipitation occurred after standing the solution for 15 min. The precipitate was collected by filtration and was washed with Et2O. The resulting solid was dissolved in H2O (volume depending on amount of precipitate obtained) and stirred for 1 h at room temperature. The solution was acidified by addition of glacial AcOH to pH 3−4 and kept at 0 °C for 1 h. The precipitate was filtered, washed with distilled H2O, and dried in a freeze dryer.
Method B
No precipitate was observed at the end of 5 min of the microwave reaction (product 12f). The reaction mixture was poured into 50 mL of ice-cold H2O and acidified with glacial AcOH to pH 3−4. Precipitation was observed, and the reaction mixture was kept at 0 °C for 1 h. The precipitate was filtered, washed with distilled H2O, and dried in a freeze dryer.
2-Hydroxy-4-oxo-4-phenyl-but-2-enoic Acid Methyl Ester (12a)
The product was isolated as a white powder (52% yield); mp 60−62 °C. m/z (ES): found 205.0300 (C11H9O4 [M − H]−) requires 205.0579.
2-Hydroxy-4-oxo-4-p-tolyl-but-2-enoic Acid Methyl Ester (12b)
The product was isolated as a white powder (49% yield); mp 84−86 °C. m/z (ES): found 221.0838 (C12H13O4 [M + H]+) requires 221.0736.
2-Hydroxy-4-oxo-4-m-tolyl-but-2-enoic Acid Methyl Ester (12c)
The product was isolated as a white powder (37% yield); mp 69−71 °C. m/z (ES): found 219.0389 (C12H11O4 [M − H]−) requires 219.0736.
2-Hydroxy-4-(4-methoxy-phenyl)-4-oxo-but-2-enoic Acid Methyl Ester (12d)
The product was isolated as a pale white powder (47% yield); mp 94−96 °C. m/z (ES): found 235.0806 (C12H11O5 [M − H]−) requires 235.0685.
2-Hydroxy-4-(3-methoxy-phenyl)-4-oxo-but-2-enoic Acid Methyl Ester (12e)
The product was isolated as a white powder (26% yield); mp 90−92 °C. m/z (ES): found 235.0647 (C12H11O5 [M − H]−) requires 235.0685.
4-(2-Ethoxy-phenyl)-2-hydroxy-4-oxo-but-2-enoic Acid Methyl Ester (12f)
The product was isolated as a yellow powder (46% yield); mp 51−53 °C. m/z (ES): found 249.0685 (C13H13O5 [M − H]−) requires 249.0841.
4-(4-Chloro-phenyl)-2-hydroxy-4-oxo-but-2-enoic Acid Methyl Ester (12g)
The product was isolated as a white powder (48% yield); mp 109−111 °C. m/z (ES): found 239.0037 (C11H8ClO4 [M − H]−) requires 239.0189.
4-(4-Cyano-phenyl)-2-hydroxy-4-oxo-but-2-enoic Acid Methyl Ester (12h)
The product was isolated as a pale white powder (30% yield); mp 161−163 °C. m/z (ES): found 230.0474 (C12H8NO4 [M − H]−) requires 230.0532.
2-Hydroxy-4-(6-methyl-pyridin-3-yl)-4-oxo-but-2-enoic Acid Methyl Ester (12i)
The product was isolated as a pale yellow powder (16% yield); mp 100−102 °C. m/z (ES): found 222.0764 (C11H12NO4 [M + H]+) requires 222.0688.
2-Hydroxy-4-oxo-4-pyridin-3-yl-but-2-enoic Acid Methyl Ester (12j)
The product was isolated as a pale yellow powder (40% yield); mp 121−123 °C. m/z (ES): found 208.0641 (C10H10NO4 [M + H]+) requires 208.0532.
2-Hydroxy-4-oxo-4-pyridin-4-yl-but-2-enoic Acid Methyl Ester (12k)
The product was isolated as a yellow powder (12% yield); mp 99−101 °C. m/z (ES): found 206.0398 (C10H8NO4 [M − H]−) requires 206.0532.
Step 2: Synthesis of 1,5-Disubstituted-4-substituted Aroyl-3-hydroxy-5-phenyl-1H-pyrrol-2(5H)-ones
3-Hydroxy-1-(2-hydroxy-propyl)-5-(4-isopropyl-phenyl)-4-(4-methyl-benzoyl)-1,5-dihydro-pyrrol-2-one (1a)
4-Isopropylbenzaldehyde (308 μL, 2.0 mmol, 1.0 equiv) and 1-amino-2-propanol (166 μL, 2.0 mmol, 1.0 equiv) were stirred in 1,4-dioxane (5 mL) for 15 min. To this was added a solution of 2-hydroxy-4-oxo-4-p-tolyl-but-2-enoic acid methyl ester (12b) (440 mg in 5.0 mL of 1,4-dioxane, 2.0 mmol, 1.0 equiv), and the reaction mixture was allowed to stir at room temperature overnight. The yellow precipitate formed was filtered, washed with Et2O, followed by EtOH, and was then dried in vacuo. The desired product 1a was obtained as a white powder (335 mg, 43% yield); mp 249−251 °C. m/z (ES): found 394.2076 (C24H28NO4 [M + H]+) requires 394.1940.
3-Hydroxy-1-(2-hydroxy-propyl)-5-isopropyl-4-(4-methyl-benzoyl)-1,5-dihydro-pyrrol-2-one (13)
The procedure was similar to the procedure for 1a except that isobutyraldehyde (227 μL, 2.5 mmol, 1.0 equiv) was used and the reaction mixture was allowed to stir at room temperature for 24 h. The precipitate obtained was washed only with Et2O and gave the desired product 13 as a white powder (14% yield); mp 214−216 °C. m/z (ES): found 318.1652 (C18H24NO4 [M + H]+) requires 318.1627.
3-Hydroxy-1-(2-hydroxy-propyl)-4-(4-methyl-benzoyl)-5-naphthalen-2-yl-1,5-dihydro-pyrrol-2-one (14)
The procedure was similar to the procedure for 1a except that 2-naphthaldehyde (398 mg, 2.5 mmol, 1.0 equiv) was used. Product 14 was isolated as a white powder (54% yield); mp 247−249 °C. m/z (ES): found 402.1804 (C25H24NO4 [M + H]+) requires 402.1627.
3-Hydroxy-1-(2-hydroxy-propyl)-4-(4-methyl-benzoyl)-5-phenyl-1,5-dihydro-pyrrol-2-one (15)
The procedure was similar to the procedure for 1a except that benzaldehyde (305 μL, 3.0 mmol, 1.0 equiv) was used, and the reaction mixture was allowed to stir at room temperature for only 3 h. Product 15 was isolated as a white powder (80% yield); mp 251−253 °C. m/z (ES): found 352.1584 (C21H22NO4 [M + H]+) requires 352.1471.
3-Hydroxy-1-(2-hydroxy-propyl)-5-(3-isopropyl-phenyl)-4-(4-methyl-benzoyl)-1,5-dihydro-pyrrol-2-one (16)
The procedure was similar to the procedure for 1a except that 3-isopropyl benzaldehyde (371 μL, 2.5 mmol, 1.0 equiv) was used. No precipitation was observed after the overnight reaction, and the reaction mixture was poured into cold H2O (25 mL). The resulting solid was filtered and washed with EtOH/petroleum ether 1:1 to afford 16 as a white powder (62% yield); mp 176−178 °C. m/z (ES): found 394.2066 (C24H28NO4 [M + H]+) requires 394.1940.
3-Hydroxy-1-(2-hydroxy-propyl)-4-(4-methyl-benzoyl)-5-p-tolyl-1,5-dihydro-pyrrol-2-one (17)
The procedure was similar to the procedure for 1a except that p-tolualdehyde (370 μL, 3.0 mmol, 1.0 equiv) was used and the reaction mixture was allowed to stir at room temperature for only 3 h. The precipitate obtained was washed with EtOH gave product 17 as a white powder (4% yield); mp 233−235 °C. m/z (ES): found 366.2106 (C22H24NO4 [M + H]+) requires 366.1627.
5-(4-Ethyl-phenyl)-3-hydroxy-1-(2-hydroxy-propyl)-4-(4-methyl-benzoyl)-1,5-dihydro-pyrrol-2-one (18)
The procedure was similar to the procedure for 1a except that 4-ethyl benzaldehyde (420 μL, 3.0 mmol, 1.0 equiv) was used and the reaction mixture was allowed to stir at room temperature for only 3 h. The precipitate obtained was washed with EtOH and gave product 18 as a white powder (5% yield); mp 243−245 °C. m/z (ES): found 380.1794 (C23H26NO4 [M + H]+) requires 380.1784.
3-Hydroxy-1-(2-hydroxy-propyl)-4-(4-methyl-benzoyl)-5-(4-propyl-phenyl)-1,5-dihydro-pyrrol-2-one (19)
The procedure was similar to the procedure for 1a except that 4-propyl benzaldehyde (380 μL, 2.5 mmol, 1.0 equiv) was used and the reaction mixture was allowed to stir at room temperature for only 3 h. The precipitate obtained was washed with EtOH and gave product 19 as a white powder (52% yield); mp 248−250 °C. m/z (ES): found 394.2096 (C24H28NO4 [M + H]+) requires 394.1940.
5-(4-tert-Butyl-phenyl)-3-hydroxy-1-(2-hydroxy-propyl)-4-(4-methyl-benzoyl)-1,5-dihydro-pyrrol-2-one (20)
The procedure was similar to the procedure for 1a except that 4-tert-butyl benzaldehyde (431 μL, 2.5 mmol, 1.0 equiv) was used and the reaction mixture was allowed to stir at room temperature for only 3 h. Product 20 was isolated as a white powder (62% yield); mp 263−265 °C. m/z (ES): found 408.2273 (C25H30NO4 [M + H]+) requires 408.2097.
5-(4-Dimethylamino-phenyl)-3-hydroxy-1-(2-hydroxy-propyl)-4-(4-methyl-benzoyl)-1,5-dihydro-pyrrol-2-one (21)
The procedure was similar to the procedure for 1a except that 4-dimethyl amino benzaldehyde (448 mg, 3.0 mmol, 1.0 equiv) was used and the reaction mixture was allowed to stir at room temperature for only 3 h. Product 21 was isolated as a yellow powder (32% yield); mp 242−244 °C. m/z (ES): found 395.2034 (C23H27N2O4 [M + H]+) requires 395.1893.
3-Hydroxy-5-(4-hydroxy-phenyl)-1-(2-hydroxy-propyl)-4-(4-methyl-benzoyl)-1,5-dihydro-pyrrol-2-one (22)
The procedure was similar to the procedure for 1a except that 4-hydroxy benzaldehyde (249 mg, 2.0 mmol, 1.0 equiv) was used. Product 22 was isolated as a pale yellow powder (15% yield); mp 225−227 °C. m/z (ES): found 368.1541 (C21H22NO5 [M + H]+) requires 368.1420.
3-Hydroxy-1-(2-hydroxy-propyl)-4-(4-methyl-benzoyl)-5-(4-trifluoromethoxy-phenyl)-1,5-dihydro-pyrrol-2-one (23)
The procedure was similar to the procedure for 1a except that 4-(trifluoromethoxy)-benzaldehyde (372 μL, 2.5 mmol, 1.0 equiv) was used and the reaction mixture was allowed to stir at room temperature for only 3 h. The precipitate obtained was washed with EtOH and gave product 23 as a white powder (32% yield); mp 246−248 °C. m/z (ES): found 436.1570 (C22H21F3NO5 [M + H]+) requires 436.1294.
3-Hydroxy-1-(2-hydroxy-propyl)-4-(4-methyl-benzoyl)-5-(4-trifluoromethyl-phenyl)-1,5-dihydro-pyrrol-2-one (24)
The procedure was similar to the procedure for 1a except that trifluoro-p-tolualdehyde (409 μL, 3.0 mmol, 1.0 equiv) was used and the reaction mixture was allowed to stir at room temperature for only 3 h. The precipitate obtained was washed with EtOH and gave product 24 as a white powder (19% yield); mp 256−258 °C. m/z (ES): found 420.1533 (C22H21F3NO4 [M + H]+) requires 420.1344.
3-Hydroxy-1-(2-hydroxy-propyl)-4-(4-methyl-benzoyl)-5-(3-trifluoromethyl-phenyl)-1,5-dihydro-pyrrol-2-one (25)
The procedure was similar to the procedure for 1a except that 3-trifluoromethyl benzaldehyde (276 μL, 2.0 mmol, 1.0 equiv) was used. No precipitation was observed after the overnight reaction, and the reaction mixture was poured into cold H2O (25 mL). The resulting solid was filtered and washed with distilled H2O followed by recrystallization from MeOH. Product 25 was isolated as a white powder (9% yield); mp 205−207 °C. m/z (ES): found 420.1573 (C22H21F3NO4 [M + H]+) requires 420.1344.
5-(2,4-Bis-trifluoromethyl-phenyl)-3-hydroxy-1-(2-hydroxy-propyl)-4-(4-methyl-benzoyl)-1,5-dihydro-pyrrol-2-one (26)
The procedure was similar to the procedure for 1a except that 2,4-bis-(trifluoromethyl) benzaldehyde (417 μL, 2.5 mmol, 1.0 equiv) was used. The precipitate obtained was washed only with Et2O and gave the desired product 26 as a white powder (21% yield); mp 204−206 °C. m/z (ES): found 488.1429 (C23H20F6NO4 [M + H]+) requires 488.1218.
5-(3,5-Bis-trifluoromethyl-phenyl)-3-hydroxy-1-(2-hydroxy-propyl)-4-(4-methyl-benzoyl)-1,5-dihydro-pyrrol-2-one (27)
1-Amino-2-propanol (416 μL, 5.00 mmol) and 3,5-bis-(trifluoromethyl) benzaldehyde (850 μL, 5.00 mmol) were stirred in 1,4-dioxane (10 mL) for 15 min. To this was added 12b (1.101 g in 10 mL of 1,4-dioxane, 5.00 mmol), and the reaction mixture was stirred at room temperature for 22 h. The precipitate that formed was filtered, washed with Et2O, and then dried in vacuo to give 27. To recover more of the compound 27, the filtrate was then poured into cold H2O (50 mL) and allowed to stand for 6 h. The resulting yellow precipitate was filtered, washed with water, and dried in vacuo. To the dried yellow precipitate was added Et2O (120 mL), and the mixture was stirred for 4 h. The white precipitate that formed was filtered and dried in vacuo to give 27. The total amount of 27 obtained from both steps was 1.021 g (42% yield); mp 234−236 °C. m/z (ES): found 488.1485 (C23H20F6NO4 [M + H]+) requires 488.1218.
5-(4-Chloro-phenyl)-3-hydroxy-1-(2-hydroxy-propyl)-4-(4-methyl-benzoyl)-1,5-dihydro-pyrrol-2-one (28)
The procedure was similar to the procedure for 1a except that 4-chloro benzaldehyde (362 mg, 2.5 mmol, 1.0 equiv) was used and the reaction mixture was allowed to stir at room temperature for only 3 h. Product 28 was isolated as a white powder (28% yield); mp 238−240 °C. m/z (ES): found 386.1226 (C21H21ClNO4 [M + H]+) requires 386.1081.
5-(3-Chloro-phenyl)-3-hydroxy-1-(2-hydroxy-propyl)-4-(4-methyl-benzoyl)-1,5-dihydro-pyrrol-2-one (29)
The procedure was similar to the procedure for 1a except that 3-chloro benzaldehyde (293 μL, 2.5 mmol, 1.0 equiv) was used. Product 29 was isolated as a white powder (47% yield); mp 235−237 °C. m/z (ES): found 386.1232 (C21H21ClNO4 [M + H]+) requires 386.1081.
5-(3,5-Dichloro-phenyl)-3-hydroxy-1-(2-hydroxy-propyl)-4-(4-methyl-benzoyl)-1,5-dihydro-pyrrol-2-one (30)
The procedure was similar to the procedure for 1a except that 3,5-dichloro benzaldehyde (451 mg, 2.5 mmol, 1.0 equiv) was used. No precipitate was observed after the overnight reaction, and the reaction mixture was heated under reflux for 10 h to complete the reaction. As precipitation was not observed on standing, the reaction mixture was poured onto crushed ice (50 mL). A brown precipitate formed that was filtered and recrystallized from MeOH. Product 30 was isolated as a white powder (18% yield); mp 245−247 °C. m/z (ES): found 420.0893 (C21H20Cl2NO4 [M + H]+) requires 420.0691.
5-(4-Chloro-3-trifluoromethyl-phenyl)-3-hydroxy-1-(2-hydroxy-propyl)-4-(4-methyl-benzoyl)-1,5-dihydro-pyrrol-2-one (31)
The procedure was similar to the procedure for 1a except that 4-chloro-3-trifluoromethyl benzaldehyde (532 μL, 2.5 mmol, 1.0 equiv) was used and the reaction mixture was allowed to stir at room temperature for only 3 h. The precipitate obtained was washed with EtOH and gave product 31 as a white powder (45% yield); mp 225−227 °C. m/z (ES): found 454.1246 (C22H20ClF3NO4 [M + H]+) requires 454.0955.
3-Hydroxy-1-isobutyl-5-(4-isopropyl-phenyl)-4-(4-methyl-benzoyl)-1,5-dihydro-pyrrol-2-one (32)
The procedure was similar to the procedure for 1a except that isobutyl amine (201 μL, 2.0 mmol, 1.0 equiv) was used and the reaction mixture was allowed to stir at room temperature for only 3 h. The precipitate obtained was washed with Et2O followed by MeOH and gave product 32 as a white powder (57% yield); mp 262−264 °C. m/z (ES): found 392.2286 (C25H30NO3 [M + H]+) requires 392.2147.
3-Hydroxy-1-(2-hydroxy-ethyl)-5-(4-isopropyl-phenyl)-4-(4-methyl-benzoyl)-1,5-dihydro-pyrrol-2-one (33)
The procedure was similar to the procedure for 1a except that ethanolamine (151 μL, 2.5 mmol, 1.0 equiv) was used and the reaction mixture was allowed to stir at room temperature for only 3 h. The precipitate obtained was washed with Et2O followed by MeOH and gave product 33 as a white powder (74% yield); mp 236−238 °C. m/z (ES): found 380.1957 (C23H26NO4 [M + H]+) requires 380.1784.
3-Hydroxy-5-(4-isopropyl-phenyl)-1-(2-methoxy-ethyl)-4-(4-methyl-benzoyl)-1,5-dihydro-pyrrol-2-one (34)
The procedure was similar to the procedure for 1a except that 2-methoxy ethylamine (218 μL, 2.5 mmol, 1.0 equiv) was used and the reaction mixture was allowed to stir at room temperature for only 3 h. The precipitate obtained was washed with Et2O followed by MeOH and gave product 34 as a white powder (68% yield); mp 247−249 °C. m/z (ES): found 394.2099 (C24H28NO4 [M + H]+) requires 394.1940.
3-Hydroxy-5-(4-isopropyl-phenyl)-1-(3-methoxy-propyl)-4-(4-methyl-benzoyl)-1,5-dihydro-pyrrol-2-one (35)
The procedure was similar to the procedure for 1a except that 3-methoxy propylamine (256 μL, 2.5 mmol, 1.0 equiv) was used and the reaction mixture was allowed to stir at room temperature for only 3 h. The precipitate obtained was washed with Et2O followed by MeOH and gave product 35 as a white powder (47% yield); mp 252−254 °C. m/z (ES): found 406.2099 (C25H28NO4 [M - H]−) requires 406. 2097.
1-Allyl-3-hydroxy-5-(4-isopropyl-phenyl)-4-(4-methyl-benzoyl)-1,5-dihydro-pyrrol-2-one (36)
The procedure was similar to the procedure for 1a except that allyl amine (189 μL, 2.5 mmol, 1.0 equiv) was used and the reaction mixture was allowed to stir at room temperature for only 3 h. The precipitate obtained was washed with Et2O followed by MeOH and gave product 36 as a white powder (52% yield); mp 249−251 °C. m/z (ES): found 376.1973 (C24H26NO3 [M + H]+) requires 376.1834.
4-Benzoyl-3-hydroxy-1-(2-hydroxy-propyl)-5-(4-isopropyl-phenyl)-1,5-dihydro-pyrrol-2-one (37)
The procedure was similar to the procedure for 1a except that 12a (618 mg, 3.0 mmol, 1.0 equiv) was used and the reaction mixture was allowed to stir at room temperature for only 3 h. The precipitate obtained was washed with EtOH and gave product 37 as a white powder (50% yield); mp 257−259 °C. m/z (ES): found 380.1749 (C23H26NO4 [M + H]+) requires 380.1784.
3-Hydroxy-1-(2-hydroxy-propyl)-5-(4-isopropyl-phenyl)-4-(3-methyl-benzoyl)-1,5-dihydro-pyrrol-2-one (38)
The procedure was similar to the procedure for 1a except that 12c (661 mg, 3.0 mmol, 1.0 equiv) was used and the reaction mixture was allowed to stir at room temperature for only 3 h. Product 38 was isolated as a pale white powder (17% yield); mp 221−223 °C. m/z (ES): found 394.2122 (C24H28NO4 [M + H]+) requires 394.1940.
3-Hydroxy-1-(2-hydroxy-propyl)-5-(4-isopropyl-phenyl)-4-(4-methoxy-benzoyl)-1,5-dihydro-pyrrol-2-one (39)
The procedure was similar to the procedure for 1a except that 12d (591 mg, 2.5 mmol, 1.0 equiv) was used. Product 39 was isolated as a white powder (29% yield); mp 238−240 °C. m/z (ES): found 410.2090 (C24H28NO5 [M + H]+) requires 410.1889.
3-Hydroxy-1-(2-hydroxy-propyl)-5-(4-isopropyl-phenyl)-4-(3-methoxy-benzoyl)-1,5-dihydro-pyrrol-2-one (40)
The procedure was similar to the procedure for 1a except that 12e (709 mg, 3.0 mmol, 1.0 equiv) was used. Product 40 was isolated as a white powder (12% yield); mp 227−229 °C. m/z (ES): found 410.2022 (C24H28NO5 [M + H]+) requires 410.1889.
4-(2-Ethoxy-benzoyl)-3-hydroxy-1-(2-hydroxy-propyl)-5-(4-isopropyl-phenyl)-1,5-dihydro-pyrrol-2-one (41)
The procedure was similar to the procedure for 1a except that 12f (500.5 mg, 2.0 mmol, 1.0 equiv) was used. No precipitation was observed after the overnight reaction, and the reaction mixture was poured into cold H2O (25 mL). The resulting solid was filtered and washed with distilled H2O to afford product 41 as a whitish brown powder (44% yield); mp 128−130 °C. m/z (ES): found 424.2231 (C25H30NO5 [M + H]+) requires 424.2046. The purity was obtained 91% (see the Supporting Information).
3-Hydroxy-1-(2-hydroxy-propyl)-5-(4-isopropyl-phenyl)-4-(6-methyl-pyridine-3-carbonyl)-1,5-dihydro-pyrrol-2-one (42)
The procedure was similar to the procedure for 1a except that 12i (331.8 mg, 1.5 mmol, 1.0 equiv) was used. The precipitate obtained was washed only with Et2O and gave the desired product 42 as a pale yellow powder (46% yield); mp 234−236 °C. m/z (ES): found 395.1867 (C23H27N2O4 [M + H]+) requires 395.1893.
4-[4-Hydroxy-1-(2-hydroxy-propyl)-2-(4-isopropyl-phenyl)-5-oxo-2,5-dihydro-1H-pyrrole-3-carbonyl]-benzonitrile (43)
The procedure was similar to the procedure for 1a except that 12h (462.4 mg, 2.0 mmol, 1.0 equiv) was used. The reaction was carried out overnight at 70 °C. As precipitation was not observed on standing, the reaction mixture was poured onto crushed ice (40 mL). The precipitate formed was filtered and washed with Et2O, followed by recrystallization from MeOH. Product 43 was isolated as a white powder (12% yield); mp 234−236 °C. m/z (ES): found 405.1912 (C24H25N2O4 [M + H]+) requires 405.1736.
4-(4-Chloro-benzoyl)-3-hydroxy-1-(2-hydroxy-propyl)-5-(4-isopropyl-phenyl)-1,5-dihydro-pyrrol-2-one (44)
The procedure was similar to the procedure for 1a except that 12g (481.3 mg, 2.0 mmol, 1.0 equiv). The precipitate obtained was washed only with Et2O and gave the desired product 44 as a white powder (56% yield); mp 256−258 °C. m/z (ES): found 414.1546 (C23H25ClNO4 [M + H]+) requires 414.1394.
1-Allyl-3-hydroxy-4-(pyridine-3-carbonyl)-5-(4-trifluoromethyl-phenyl)-1,5-dihydro-pyrrol-2-one (45)
4-Trifluoromethyl benzaldehyde (273 μL, 2.0 mmol, 1.0 equiv) and allyl amine (152 μL, 2.0 mmol, 1.0 equiv) were stirred in 1,4-dioxane (5 mL) for 15 min. To this was added a warm solution of 12j (414 mg in 5.0 mL of 1,4-dioxane, 2.0 mmol, 1.0 equiv), and the reaction mixture was allowed to stir at room temperature overnight. The yellow precipitate formed was filtered, washed with diethyl ether followed by ethanol, and then dried in vacuo. Product 45 was isolated as a white powder (84% yield); mp 265−267 °C. m/z (ES): found 389.1143 (C20H16F3N2O3 [M + H]+) requires 389.1035.
1-Allyl-3-hydroxy-4-(6-methyl-pyridine-3-carbonyl)-5-(4-trifluoromethyl-phenyl)-1,5-dihydro-pyrrol-2-one (46)
4-Trifluoromethyl benzaldehyde (273 μL, 2.0 mmol, 1.0 equiv) and allyl amine (152 μL, 2.0 mmol, 1.0 equiv) were stirred in 1,4-dioxane (5 mL) for 15 min. To this was added a solution of 12i (442 mg in 5.0 mL of 1,4-dioxane, 2.0 mmol, 1.0 equiv), and the reaction mixture was allowed to stir at room temperature for 2.30 h. The yellow precipitate obtained was washed only with Et2O and gave the desired product 46 as a pale yellow powder (18% yield); mp 237−239 °C decomp. m/z (ES): found 403.1359 (C21H18F3N2O3 [M + H]+) requires 403.1191.
1-Allyl-4-(4-chloro-benzoyl)-3-hydroxy-5-(4-trifluoromethyl-phenyl)-1,5-dihydro-pyrrol-2-one (47)
4-Trifluoromethyl benzaldehyde (273 μL, 2.0 mmol, 1.0 equiv) and allyl amine (152 μL, 2.0 mmol, 1.0 equiv) were stirred in 1,4-dioxane (5 mL) for 15 min. To this was added a solution of 12g (481.3 mg in 5.0 mL of 1,4-dioxane, 2.0 mmol, 1.0 equiv), and the reaction mixture was allowed to stir at room temperature overnight. The yellow precipitate obtained was washed only with Et2O and gave the desired product 47 as a white powder (59% yield); mp 264−266 °C. m/z (ES): found 422.0850 (C21H16ClF3NO3 [M + H]+) requires 422.0693.
3-Hydroxy-1-(3-methoxy-propyl)-4-(pyridine-3-carbonyl)-5-(4-trifluoromethyl-phenyl)-1,5-dihydro-pyrrol-2-one (48)
4-Trifluoromethyl benzaldehyde (273 μL, 2.0 mmol, 1.0 equiv) and 3-methoxy propylamine (205 μL, 2.0 mmol, 1.0 equiv) were stirred in 1,4-dioxane (5 mL) for 15 min. To this was added a solution of 12j (414 mg in 5.0 mL of 1,4-dioxane, 2.0 mmol, 1.0 equiv), and the reaction mixture was allowed to stir at room temperature overnight. The yellow precipitate obtained was washed only with Et2O and gave the desired product 48 as a white powder (59% yield); mp 253−255 °C. m/z (ES): found 421.1439 (C21H20F3N2O4 [M + H]+) requires 421.1297.
3-Hydroxy-1-(3-methoxy-propyl)-4-(6-methyl-pyridine-3-carbonyl)-5-(4-trifluoromethyl-phenyl)-1,5-dihydro-pyrrol-2-one (49)
4-Trifluoromethyl benzaldehyde (273 μL, 2.0 mmol, 1.0 equiv) and 3-methoxy propylamine (205 μL, 2.0 mmol, 1.0 equiv) were stirred in 1,4-dioxane (5 mL) for 15 min. To this was added a solution of 12i (442.4 mg in 5.0 mL of 1,4-dioxane, 2.0 mmol, 1.0 equiv), and the reaction mixture was allowed to stir at room temperature for 3 h. The yellow precipitate obtained was washed only with Et2O and gave the desired product 49 as a white powder (57% yield); mp 261−263 °C. m/z (ES): found 435.1588 (C22H22F3N2O4 [M + H]+) requires 435.1453.
5-(3-Fluoro-4-trifluoromethyl-phenyl)-3-hydroxy- 1-(3-methoxy-propyl)-4-(6-methyl-pyridine-3-carbonyl)-1,5-dihydro-pyrrol-2-one (50)
3-Fluoro-4-trifluoromethyl benzaldehyde (205 μL, 1.5 mmol, 1.0 equiv) and 3-methoxy propylamine (154 μL, 1.5 mmol, 1.0 equiv) were stirred in 1,4-dioxane (5 mL) for 15 min. To this was added a solution of 12i (332 mg in 5.0 mL of 1,4-dioxane, 1.5 mmol, 1.0 equiv), and the reaction mixture was allowed to stir at room temperature overnight. The yellow precipitate obtained was washed only with Et2O and gave the desired product 50 as a white powder (65% yield); mp 257−259 °C. m/z (ES): found 453.1475 (C22H21F4N2O4 [M + H]+) requires 453.1359.
4-(4-Chloro-benzoyl)-5-(3-fluoro-4-trifluoromethyl-phenyl)-3-hydroxy-1-(3-methoxy-propyl)-1,5-dihydro-pyrrol-2-one (51)
3-Fluoro-4-trifluoromethyl benzaldehyde (273 μL, 2.0 mmol, 1.0 equiv) and 3-methoxy propylamine (205 μL, 2.0 mmol, 1.0 equiv) were stirred in 1,4-dioxane (5 mL) for 15 min. To this was added a solution of 12g (481 mg in 5.0 mL of 1,4-dioxane, 2.0 mmol, 1.0 equiv), and the reaction mixture was allowed to stir at room temperature overnight. The yellow precipitate formed was filtered, washed with diethyl ether followed by ethanol, and then dried in vacuo. Product 51 was isolated as a white powder (76% yield); mp 252−254 °C. m/z (ES): found 470.0819 (C22H17ClF4NO4 [M − H]−) requires 470.0860.
4-(2-Ethoxy-benzoyl)-3-hydroxy-1-(3-methoxy-propyl)-5-(4-trifluoromethyl-phenyl)-1,5-dihydro-pyrrol-2-one (52)
4-Trifluoromethyl benzaldehyde (279 μL, 2.0 mmol, 1.0 equiv) and 3-methoxy propylamine (205 μL, 2.0 mmol, 1.0 equiv) were stirred in 1,4-dioxane (5 mL) for 15 min. To this was added a solution of 12f (501 mg in 5.0 mL of 1,4-dioxane, 2.0 mmol, 1.0 equiv), and the reaction mixture was allowed to stir at room temperature overnight. No precipitation was observed, and the reaction mixture was poured onto cold water (25 mL). The resulting solid was filtered and washed with distilled H2O followed by Et2O to afford product 52 as a white powder (6% yield); mp 138−140 °C. m/z (ES): found 462.1536 (C24H23F3NO5 [M − H]−) requires 462.1607.
4-(2-Ethoxy-benzoyl)-5-(3-fluoro-4-trifluoromethyl-phenyl)-3-hydroxy-1-(3-methoxy-propyl)-1,5-dihydro-pyrrol-2-one (53)
The procedure was similar as for 52 except that 3-fluoro-4-trifluoromethyl benzaldehyde (273 μL, 2.0 mmol, 1.0 equiv) was used. Product 53 was isolated as a white powder (9% yield); mp 140−142 °C. m/z (ES): found 480.1387 (C24H22F4NO5 [M − H]−) requires 480.1512.
Acknowledgments
We thank Wen Yuen Lim and Catherine Pereira for experimental help at various stages of this research and Dr. Cristina de Matteis for virtual screening software. This research was supported by grants from Cancer Research UK. H.K.M. was funded by a Biotechnology and Biological Sciences Research Council studentship.
Glossary
Abbreviations Used
- FRET
fluorescence resonance energy transfer
- GA
genetic algorithm
- GOLD
genetic optimization for ligand docking
- SAS
solvent accessible surface area
Supporting Information Available
IR, 1H and 13C NMR spectroscopic data, and assessment of compound purity by HPLC. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- Donato R. Functional roles of S100 proteins, calcium-binding proteins of the EF-hand type. Biochim. Biophys. Acta, Mol. Cell Res. 1999, 1450, 191–231. [DOI] [PubMed] [Google Scholar]
- Rety S.; Sopkova J.; Renouard M.; Osterloh D.; Gerke V.; Tabaries S.; Russo-Marie F.; Lewit-Bentley A. The crystal structure of a complex of p11 with the annexin II N-terminal peptide. Nat. Struct. Biol. 1999, 6, 89–95. [DOI] [PubMed] [Google Scholar]
- Garrett S. C.; Varney K. M.; Weber D. J.; Bresnick A. R. S100A4, a mediator of metastasis. J. Biol. Chem. 2006, 281, 677–680. [DOI] [PubMed] [Google Scholar]
- Santamaria-Kisiel L.; Rintala-Dempsey A. C.; Shaw G. S. Calcium-dependent and -independent interactions of the S100 protein family. Biochem. J. 2006, 396, 201–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salama I.; Malone P. S.; Mihaimeed F.; Jones J. L. A review of the S100 proteins in cancer. Eur. J. Surg. Oncol. 2008, 34, 357–364. [DOI] [PubMed] [Google Scholar]
- Pathuri P.; Vogeley L.; Luecke H. Crystal Structure of Metastasis-Associated Protein S100A4 in the Active Calcium-Bound Form. J. Mol. Biol. 2008, 383, 62–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z.-H.; Bresnick A. R. The S100A4 metastasis factor regulates cellular motility via a direct interaction with myosin-IIA. Cancer Res. 2006, 66, 5173–5180. [DOI] [PubMed] [Google Scholar]
- Donato R. Intracellular and extracellular roles of S100 proteins. Microsc. Res. Tech. 2003, 60, 540–551. [DOI] [PubMed] [Google Scholar]
- Markowitz J.; Chen I.; Gitti R.; Baldisseri D. M.; Pan Y.; Udan R.; Carrier F.; MacKerell A. D. Jr.; Weber D. J. Identification and Characterization of Small Molecule Inhibitors of the Calcium-Dependent S100B-p53 Tumor Suppressor Interaction. J. Med. Chem. 2004, 47, 5085–5093. [DOI] [PubMed] [Google Scholar]
- Garrett S. C.; Hodgson L.; Rybin A.; Toutchkine A.; Hahn K. M.; Lawrence D. S.; Bresnick A. R. A Biosensor of S100A4 Metastasis Factor Activation: Inhibitor Screening and Cellular Activation Dynamics. Biochemistry 2008, 47, 986–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malashkevich V. N.; Dulyaninova N. G.; Ramagopal U. A.; Liriano M. A.; Varney K. M.; Knight D.; Brenowitz M.; Weber D. J.; Almo S. C.; Bresnick A. R. Phenothiazines inhibit S100A4 function by inducing protein oligomerization. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 8605–8610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerke V.; Creutz C. E.; Moss S. E. Annexins: Linking Ca2+ signalling to membrane dynamics. Nat. Rev. Mol. Cell. Biol. 2005, 6, 449–461. [DOI] [PubMed] [Google Scholar]
- Semov A.; Moreno M. J.; Onichtchenko A.; Abulrob A.; Ball M.; Ekiel I.; Pietrzynski G.; Stanimirovic D.; Alakhov V. Metastasis-associated protein S100A4 induces angiogenesis through interaction with Annexin II and accelerated plasmin formation. J. Biol. Chem. 2005, 280, 20833–20841. [DOI] [PubMed] [Google Scholar]
- Ling Q.; Jacovina A. T.; Deora A.; Febbraio M.; Simantov R.; Silverstein R. L.; Hempstead B.; Mark W. H.; Hajjar K. A. Annexin II regulates fibrin homeostasis and neoangiogenesis in vivo. J. Clin. Invest. 2004, 113, 38–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma M. R.; Koltowski L.; Ownbey R. T.; Tuszynski G. P.; Sharma M. C. Angiogenesis-associated protein annexin II in breast cancer: selective expression in invasive breast cancer and contribution to tumor invasion and progression. Exp. Mol. Pathol. 2006, 81, 146–156. [DOI] [PubMed] [Google Scholar]
- Emoto K.; Yamada Y.; Sawada H.; Fujimoto H.; Ueno M.; Takayama T.; Kamada K.; Naito A.; Hirao S.; Nakajima Y. Annexin II overexpression correlates with stromal tenascin-C overexpression: A prognostic marker in colorectal carcinoma. Cancer 2001, 92, 1419–1426. [DOI] [PubMed] [Google Scholar]
- Becker T.; Weber K.; Johnsson N. Protein-protein recognition via short amphiphilic helixes; a mutational analysis of the binding site of annexin II for p11. EMBO J. 1990, 9, 4207–4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streicher W. W.; Lopez M. M.; Makhatadze G. I. Annexin I and Annexin II N-Terminal Peptides Binding to S100 Protein Family Members: Specificity and Thermodynamic Characterization. Biochemistry 2009, 48, 2788–2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konig J.; Prenen J.; Nilius B.; Gerke V. The annexin II-p11 complex is involved in regulated exocytosis in bovine pulmonary artery endothelial cells. J. Biol. Chem. 1998, 273, 19679–19684. [DOI] [PubMed] [Google Scholar]
- Shiozawa Y.; Havens A. M.; Jung Y.; Ziegler A. M.; Pedersen E. A.; Wang J.; Wang J.; Lu G.; Roodman G. D.; Loberg R. D.; Pienta K. J.; Taichman R. S. Annexin II/Annexin II receptor axis regulates adhesion, migration, homing, and growth of prostate cancer. J. Cell. Biochem. 2008, 105, 370–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg T. Small-molecule inhibitors of protein-protein interactions. Curr. Opin. Drug Discovery Dev. 2008, 11, 666–674. [PubMed] [Google Scholar]
- Fischer P. M. Protein-protein interactions in drug discovery. Drug Des. Rev. Online 2005, 2, 179–207. [Google Scholar]
- Lipinski C. A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discovery Today 2004, 1, 337–341. [DOI] [PubMed] [Google Scholar]
- Veber D. F.; Johnson S. R.; Cheng H.-Y.; Smith B. R.; Ward K. W.; Kopple K. D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [DOI] [PubMed] [Google Scholar]
- Wang R.; Fu Y.; Lai L. A New Atom-Additive Method for Calculating Partition Coefficients. J. Chem. Inf. Comput. Sci. 1997, 37, 615–621. [Google Scholar]
- Verdonk M. L.; Cole J. C.; Hartshorn M. J.; Murray C. W.; Taylor R. D. Improved protein-ligand docking using GOLD. Proteins 2003, 52, 609–623. [DOI] [PubMed] [Google Scholar]
- Jones G.; Willett P.; Glen R. C.; Leach A. R.; Taylor R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [DOI] [PubMed] [Google Scholar]
- Kontoyianni M.; Sokol G. S.; McClellan L. M. Evaluation of library ranking efficacy in virtual screening. J. Comput. Chem. 2005, 26, 11–22. [DOI] [PubMed] [Google Scholar]
- SYBYL MOLCAD, Electrostatic Potential, Version 7.3; Tripos, Inc.: St. Louis, MO, 2006. [Google Scholar]
- SYBYL Molecular Diversity Selector, Version 7.3; Tripos, Inc.: St. Louis, MO, 2006. [Google Scholar]
- Li C.; Reddy T. R. K.; Fischer P. M.; Dekker L. V. A Cy5-labeled S100A10 tracer used to identify inhibitors of the protein interaction with annexin A2. Assay Drug Dev. Technol. 2010, 8, 85–95. [DOI] [PubMed] [Google Scholar]
- Johnsson N.; Marriott G.; Weber K. p36, the major cytoplasmic substrate of src tyrosine protein kinase, binds to its p11 regulatory subunit via a short amino-terminal amphiphatic helix. EMBO J. 1988, 7, 2435–2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurin C.; Bailly F.; Cotelle P. Improved preparation and structural investigation of 4-aryl-4-oxo-2-hydroxy-2-butenoic acids and methyl esters. Tetrahedron Lett. 2004, 60, 6479–6486. [Google Scholar]
- Drysdale M. J.; Hind S. L.; Jansen M.; Reinhard J. F. Jr. Synthesis and SAR of 4-Aryl-2-hydroxy-4-oxobut-2-enoic Acids and Esters and 2-Amino-4-aryl-4-oxobut-2-enoic Acids and Esters: Potent Inhibitors of Kynurenine-3-hydroxylase as Potential Neuroprotective Agents. J. Med. Chem. 2000, 43, 123–127. [DOI] [PubMed] [Google Scholar]
- Sechi M.; Bacchi A.; Carcelli M.; Compari C.; Duce E.; Fisicaro E.; Rogolino D.; Gates P.; Derudas M.; Al-Mawsawi L. Q.; Neamati N. From Ligand to Complexes: Inhibition of Human Immunodeficiency Virus Type 1 Integrase by Î2-Diketo Acid Metal Complexes. J. Med. Chem. 2006, 49, 4248–4260. [DOI] [PubMed] [Google Scholar]
- Gein V. L.; Kasimova N. N. Three-component condensation of methyl acylpyruvates with aromatic aldehydes and ethylenediamine. Chemical properties of the products. Russ. J. General Chem. 2005, 75, 254–260. [Google Scholar]
- Gein V. L.; Kasimova N. N.; Potemkin K. D. Simple Three-Component Synthesis of 4-Acyl-1-(2-aminoethyl)-5-aryl-3-hydroxy-2,5-dihydropyrrol-2(1H)-ones. Russ. J. Gen. Chem. 2002, 72, 1150–1151. [Google Scholar]
- Gein V. L.; Kasimova N. N.; Voronina E. V.; Gein L. F. Synthesis and antimicrobial activity of 5-aryl-4-acyl-1-(N,N-dimethylaminoethyl)-3-hydroxy-3-pyrrolin-2-ones. Pharm. Chem. J. 2001, 35, 151–154. [Google Scholar]
- Silina T. A.; Gein V. L.; Gein L. F.; Voronina E. V. Synthesis and Antimicrobial Activity of 3-Hydroxy- and 3-Arylamino-5-aryl-4-acyl-1-(pyridyl)-3-pyrrolin-2-ones. Pharm. Chem. J. 2003, 37, 585–587. [Google Scholar]
- Silina T. A.; Pulina N. A.; Gein L. F.; Gein V. L. Simple three-component synthesis of 4-acyl-5-phenyl-1-(2-heteroaryl)-3-hydroxy-3-pyrrolin-2-ones. Chem. Heterocycl. Compd. 1998, 34, 739. [Google Scholar]
- Irwin J. J.; Shoichet B. K. ZINC—A Free Database of Commercially Available Compounds for Virtual Screening. J. Chem. Inf. Comput. Sci. 2005, 45, 177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.