

Abstract
Inflammatory immune responses triggered initially to clear West Nile virus (WNV) infection later become detrimental and contribute to the pathological processes such as blood–brain barrier (BBB) disruption and neuronal death, thus complicating WNV-associated encephalitis (WNVE). It has been demonstrated previously that WNV infection in astrocytes results in induction of multiple matrix metalloproteinases (MMPs), which mediate BBB disruption. Cyclooxygenase (COX) enzymes and their product, prostaglandin E2 (PGE2), modulate neuroinflammation and regulate the production of multiple inflammatory molecules including MMPs. Therefore, this study determined and characterized the pathophysiological consequences of the expression of COX enzymes in human brain cortical astrocytes (HBCAs) following WNV infection. Whilst COX-1 mRNA expression did not change, WNV infection significantly induced RNA and protein expression of COX-2 in HBCAs. Similarly, PGE2 production was also enhanced significantly in infected HBCAs and was blocked in the presence of the COX-2-specific inhibitor NS-398, thus suggesting that COX-2, and not COX-1, was the source of the increased PGE2. Treatment of infected HBCAs with NS-398 attenuated the expression of MMP-1, -3 and -9 in a dose-dependent manner. Similarly, expression of interleukin-1β, -6 and -8, which were markedly elevated in infected HBCAs, exhibited a significant reduction in their levels in the presence of NS-398. These results provide direct evidence that WNV-induced COX-2/PGE2 is involved in modulating the expression of multiple neuroinflammatory mediators, thereby directly linking COX-2 with WNV disease pathogenesis. The ability of COX-2 inhibitors to modulate WNV-induced COX-2 and PGE2 signalling warrants further investigation in an animal model as a potential approach for clinical management of neuroinflammation associated with WNVE.
INTRODUCTION
West Nile virus (WNV), a mosquito-borne flavivirus that causes lethal encephalitis, has emerged as a significant cause of viral encephalitis in the USA (Brinton, 2002). In a small subset of cases, WNV targets the central nervous system (CNS), clinically progressing to meningitis, encephalitis or acute flaccid paralysis syndrome, leading to mortality in 10 % of hospitalized patients and complicated neurological sequelae in some who survive (Sejvar et al., 2003). WNV-associated encephalitis (WNVE) in the mouse model is characterized by disruption of the blood–brain barrier (BBB), neuroinflammation, microglial activation and loss of neurons (van Marle et al., 2007; Wang et al., 2004, 2008). Inflammation in the CNS is a major hallmark of WNVE in mice and is associated with a dramatic increase in several pro-inflammatory cytokines such as tumour necrosis factor alpha (TNF-α) and interleukin (IL)-1β and -6 (Garcia-Tapia et al., 2007; Wang et al., 2004) and chemokines such as CCL2 and CXCL10, which regulate leukocyte trafficking into the brain (Glass et al., 2006; Klein et al., 2005; Lim et al., 2006). Although many of these virus-induced cytokines and chemokines play a critical role in the recruitment of virus-specific T cells and virus clearance in the mouse brain, increased production of pro-inflammatory cytokines also contributes to the overall disease pathogenesis.
In the CNS, neurons are the prime target for WNV infection; however, infection of non-neuronal CNS cells such as astrocytes and BBB endothelial cells has been documented (Cheeran et al., 2005; Shrestha et al., 2003; van Marle et al., 2007; Verma et al., 2009). Activation of glial cells along with loss of neurons is considered a key pathogenic feature in WNV infection in humans (Kelley et al., 2003). Although virus infection in human glial cells is not as robust as in neurons, they secrete much higher levels of immune mediators such as chemokines (CXCL10, CCL2 and CCL5) and cytokines (Cheeran et al., 2005; Glass et al., 2005). Furthermore, cytotoxic factors secreted from WNV-infected astrocytes can induce bystander death of naïve neurons (van Marle et al., 2007). Our previous data also demonstrated that WNV infection of human astrocytes results in the induction of multiple matrix metalloproteinases (MMPs), which are capable of degrading the tight junction proteins of human brain microvascular endothelial cells, thereby compromising the integrity of the BBB model (Verma et al., 2010). Thus, although astrocytes are one of the key players in WNV-induced neuroinflammatory responses, the upstream events modulating these inflammatory responses are not well understood.
Prostaglandin E2 (PGE2), the most abundant prostaglandin in the brain, is considered to play an essential role as a local regulator of pathogenic processes in several neurodegenerative diseases (Bazan et al., 2002; Candelario-Jalil & Fiebich, 2008; Hickey et al., 2007). Cyclooxygenase enzymes (COX-1 and -2) catalyse the committed step in the conversion of arachidonic acid to PGE2. COX-1 is expressed ubiquitously and is considered to be an isoform responsible for homeostatic prostaglandin synthesis. By contrast, COX-2 is rapidly induced in many cell types including astrocytes in response to inflammatory stimuli (Bazan, 2001; Bazan et al., 2002; Tzeng et al., 2005). However, in the CNS, COX-2 is also expressed constitutively in the hippocampal neurons (Yang & Chen, 2008). Accumulating evidence suggests that of the two COX isoforms, COX-2, but not COX-1, plays a crucial role in inflammation and disease pathogenesis. PGE2 activates several downstream inflammatory pathways via autocrine or paracrine mechanisms, resulting in the induction of pro-inflammatory mediators (Ferri & Ferguson, 2005; Kyrkanides et al., 2002). Multiple downstream effects of COX-2/PGE2 include induction of chemotactic cytokines, mediators of BBB disruption such as MMPs and plasmin/urokinase plasminogen activator (uPA), apoptotic death and activation of microglia (Bazan, 2001; Bazan et al., 2002; Im et al., 2006). Both MMPs and the plasmin/uPA system belong to the family of multi-domain zinc-containing serine proteases, and their increase in glial cells has been associated with BBB disruption (Conant et al., 2004). Treatment with COX-2 inhibitors such as NS-398 in both in vitro and in vivo model systems can attenuate secretion of cytokines, cell-adhesion molecules, MMP-9 and uPA, and improve overall pathology associated with several neurodegenerative diseases (Im et al., 2006; Iwamoto et al., 2008; Ottino & Bazan, 2001; Pompl et al., 2003; Thomas & Kuhn, 2005). CNS infection of other neurotropic viruses such as human immunodeficiency virus (HIV) and Japanese encephalitis virus (JEV) also results in increased COX-2 and PGE2 production (Flora et al., 2006; Ghoshal et al., 2007).
In WNV infection, once the virus enters the brain and triggers inflammation, there is very little that can be done to prevent disease progression. Therefore, understanding the key molecular mechanisms by which WNV modulates inflammation is of utmost importance in the design of effective treatments. The extent of the host innate immune response is critical in determining the end point of WNV infection, i.e. virus clearance versus morbidity. As the COX/PGE2 pathway has been demonstrated to modulate the host innate and inflammatory response in viral infections, a better understanding of this pathway in WNV neuropathogenesis is warranted. Therefore, the purpose of this study was to investigate the role of COX enzymes and their product, PGE2, in WNV infection using primary human brain cortical astrocytes (HBCAs) and to determine the mechanisms by which they contribute to neuroinflammation. Our results indicated that COX-2 and its product, PGE2, are significantly induced in WNV-infected HBCAs and that increased COX-2/PGE2 levels correlate with a peak in the virus titres. Using the COX-2-specific inhibitor NS-398, we further demonstrated that the expression of MMPs and key inflammatory cytokines are tightly regulated by COX-2/PGE2 levels in WNV-infected astrocytes.
RESULTS
WNV infection induces COX-2 expression in HBCAs in a dose- and time-dependent manner
Profound inflammatory reactions including induction of MMPs, cytokines, chemokines and cell-adhesion molecules have been demonstrated in WNV-infected brain cells (Arjona et al., 2007; Glass et al., 2005; Wang et al., 2008). COX-2, an important mediator of inflammation, can modulate inflammatory reactions by inducing cytokines and MMPs. In the brain, astrocytes are an important source of COX-2 and inflammatory mediators in several neurodegenerative diseases. Our previous study demonstrated that WNV infection of primary HBCAs peaked at day 2 post-infection (p.i.) and remained high up to day 4 p.i., following which there was a decline in the virus titre (Verma et al., 2010). Therefore, in this study we analysed the mRNA and protein expression of COX enzymes in HBCAs at the same time points. The global response of HBCAs infected with WNV at an m.o.i. of 5 was determined at days 1 and 3 p.i. by cDNA microarray analysis. As observed in Table 1, WNV infection did not alter the expression profile of the COX-1 gene in HBCAs at either time point, whereas increased expression of the COX-2 gene was observed at day 1 and peaked further at day 3 p.i. An increase in the expression of COX-2 was further validated by qRT-PCR from day 1 to day 4 p.i. in HBCAs infected with WNV at an m.o.i. of 1 and 5. As observed in Table 1, WNV infection at an m.o.i. of 1 induced a time-dependent increase in the mRNA expression of COX-2 from day 1 to day 4 p.i. This increase was first detected at day 1, increased sharply at day 2 and remained high up to day 4 p.i. In concurrence with the microarray data, in HBCAs infected with WNV at m.o.i. of 5, a gradual increase in COX-2 gene expression was observed from day 1 to day 3 p.i. Moreover, infection with UV-inactivated WMV did not induce COX-2 expression (Table 1), suggesting that increased COX-2 expression is a result of active virus replication. Expression of COX-2 protein was characterized by Western blotting. As demonstrated in Fig. 1, although COX-2 protein expression in control HBCAs was minimal, a dramatic increase was observed in infected HBCAs at days 2 and 3 p.i., which coincided with high mRNA transcripts and a peak in virus replication.
Table 1.
mRNA fold changes in COX-1 and -2 levels in WNV-infected HBCAs compared with uninfected controls as determined by cDNA microarray analysis and qRT-PCR
UV-WNV, UV-inactivated WNV; nd, not done.
| Days p.i. | COX-1 | COX-2 | |||
|---|---|---|---|---|---|
| cDNA microarray | cDNA microarray | Real-time qRT-PCR | |||
| m.o.i. 1 | m.o.i. 5 | UV-WNV | |||
| Day 1 | 1.01 | 55 | 27 | 42 | 1.27 |
| Day 2 | nd | nd | 55 | 87 | 2.1 |
| Day 3 | 0.47 | 102 | 83 | 93 | 1.1 |
| Day 4 | nd | nd | 76 | 97 | 1.6 |
Fig. 1.
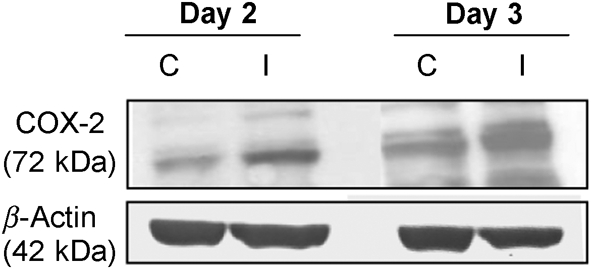
WNV induces COX-2 protein expression in HBCAs. Cell lysates extracted from WNV-infected (m.o.i. 5) (I) and mock-infected control (C) HBCAs at days 2 and 3 p.i. were separated by PAGE, transferred onto PVDF membranes and immunoblotted with antibodies specific to COX-2. Equal loading was confirmed by reprobing with anti-β-actin antibody and the bands were detected by an ECL method.
We next analysed the increase in COX-2 protein expression by immunocytochemical analysis. Whilst mock-infected HBCAs did not exhibit a COX-2 fluorescence signal (Fig. 2a), a strong signal for COX-2 expression was observed at day 2 p.i. in WNV-infected HBCAs (Fig. 2b). Fig. 2(c) demonstrates WNV antigen staining in HBCAs, and, as depicted in Fig. 2(d), increased COX-2 immunostaining was observed mostly in WNV-infected cells. This was further confirmed by counting the number of COX-2- and WNV-positive cells. Based on the counting of approximately 1000 cells from a total of four coverslips, it was found that 79 % of COX-2-positive cells were also positive for WNV antigen staining.
Fig. 2.

Localization of COX-2 in WNV-infected HBCAs using immunofluorescence. Confluent primary HBCAs were infected with WNV at an m.o.i. of 5 and stained with anti-COX-2 (red) or anti-WNV envelope (green) antibody and the nuclei counterstained with DAPI (blue). (a) Mock-infected HBCAs demonstrated faint and diffused COX-2 immunoreactivity. (b) A marked increase in COX-2 immunostaining was observed in infected HBCAs at day 2 p.i. (c) WNV-infected HBCAs stained for WNV antigen. (d) Strong COX-2 staining was observed mostly in WNV-infected HBCAs. (e) HBCAs incubated with secondary antibody only did not demonstrate any immunostaining. The images depicted are representative results of three independent experiments in duplicate. Bars, 10 μm.
Treatment with COX-2 inhibitor prevents WNV-induced PGE2 production in HBCAs
As PGE2 is the main product of COX-2 and is released extracellularly, it was important to determine its release in the supernatant by ELISA. We observed a four- to fivefold increase in PGE2 levels in the supernatant of WNV-infected HBCAs at day 3 p.i. (P<0.01, Fig. 3). To further confirm the contribution of COX-2 as the main source of PGE2, infected HBCAs were treated with 25 and 50 μM concentrations of the COX-2-specific inhibitor NS-398, and PGE2 levels were assayed at the same time point. As seen in Fig. 3, the level of PGE2 released by infected HBCAs was significantly reduced in the presence of both doses of NS-398 (P<0.01, compared with infected HBCAs without NS-398 treatment). The inhibition was greater in cells treated with 50 μM NS-398, suggesting a dose-dependent effect. HBCAs infected in the presence of vehicle control (0.001 % DMSO) did not exhibit any change in expression levels of COX-2 enzyme (data not shown), thus validating the specificity of NS-398 in blocking the COX-2 enzyme. This observation is similar to previous studies where the vehicle control for NS-398 had no effect on the COX-2 enzyme (Baek et al., 2007; Lu et al., 2008). These findings suggested that the increased levels of PGE2 observed in WNV-infected cells were indeed dependent on COX-2 activation and were not a result of cellular COX-1 activity, which is not inhibited by NS-398. Furthermore, NS-398 treatment did not affect the virus titres in infected HBCAs as measured by plaque assay (data not shown). Taken together, these results demonstrated that WNV infection induces COX-2 and PGE2 production in astrocytes.
Fig. 3.
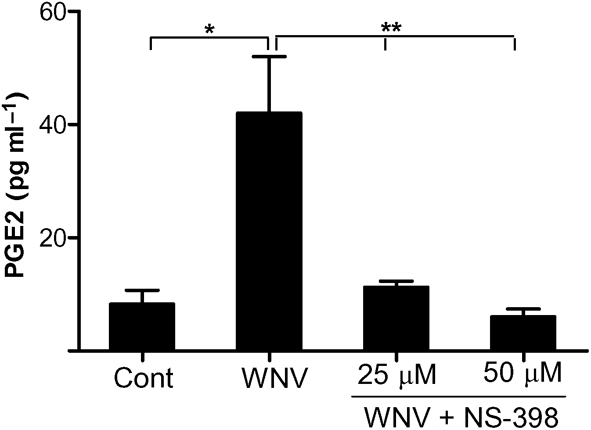
HBCA were infected at an m.o.i. of 5 in the presence or absence of NS-398 and the supernatants collected at day 3 p.i. were assayed by ELISA for PGE2 production. The presence of NS-398 significantly reduced the levels of PGE2 released into the supernatant. The data expressed are the mean concentration±sd (pg ml−1) of the amount of PGE2 secreted into the cell supernatant from at least four independent experiments in duplicate. *, P<0.01 compared with uninfected control; **, P<0.05 compared with infected cells.
WNV-induced COX-2/PGE2 regulates the expression of uPA and MMPs in HBCAs
We have demonstrated previously that WNV infection induces MMP-1, -3 and -9 in HBCAs (Verma et al., 2010). As the COX-2/PGE2 signalling pathway is associated with the induction of MMPs, we assessed the ability of WNV-induced COX-2 and PGE2 to modulate MMP expression in HBCAs. The mRNA expression of MMP-1, -3 and -9 increased in HBCAs at day 3 p.i. (m.o.i. 5), thereby corroborating our previous results. However, treatment of infected HBCAs with NS-398 drastically reduced the expression of MMPs in a dose-dependent manner (Fig. 4a). Compared with WNV-infected cells, treatment of infected HBCAs with NS-398 at a concentration of 50 μM decreased the mRNA expression of MMP-3 by 55 % and of MMP-1 and -9 by 70–75 %. Furthermore, a similar inhibitory effect of NS-398 was also observed in the amount of MMP-3 and -9 secreted into the supernatant of infected HBCAs as assayed by ELISA. As observed in Fig. 4(b), whilst the levels of MMP-3 and -9 increased by five- to sixfold (P<0.005) in the supernatant of infected HBCAs at day 3 p.i., the presence of NS-398 at concentrations of 25 and 50 μM decreased their levels significantly, thereby supporting the qRT-PCR data. These results collectively indicated that COX-2/PGE2 modulates the expression of MMPs in WNV-infected HBCAs. As the activation of uPA/uPA receptor (uPAR) system, which is associated with matrix degradation, is also governed by COX-2, we analysed the response of uPA gene expression to WNV infection. As depicted in Fig. 4(a), the expression of uPA increased by four- to fivefold in WNV-infected HBCAs compared with control cells and treatment of infected HBCAs with NS-398 almost completely blocked any increase in uPA mRNA.
Fig. 4.
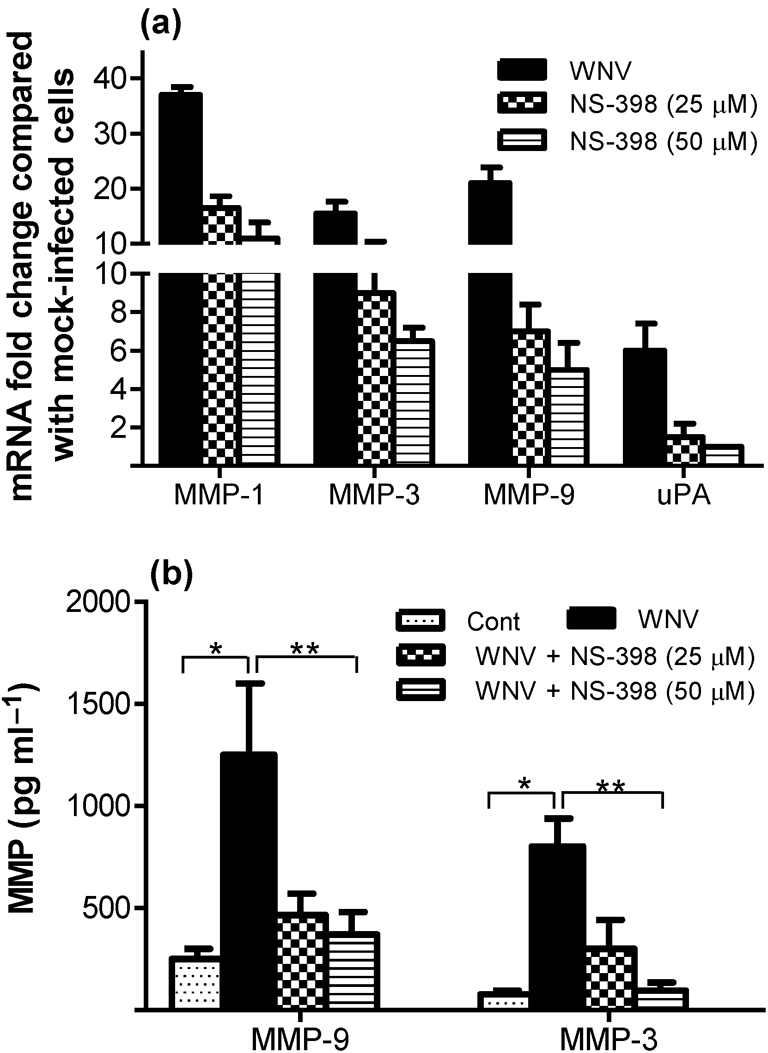
COX-2 inhibitor attenuates the expression of MMPs and uPA. (a) qRT-PCR was conducted on RNA extracted from HBCAs treated with or without NS-398 at day 3 p.i. (m.o.i. 5) to determine the fold change in MMP-1, -3 and -9 gene expression. Changes in the levels of each gene were first normalized to the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene and the fold change in infected cells compared with corresponding control cells was calculated. Data represent the means±sd of at least four independent experiments conducted in duplicate. (b) The supernatant medium from WNV- and mock-infected HBCAs with or without NS-398 treatment was analysed for total MMP-3 and -9 levels by ELISA at day 3 p.i. WNV-induced MMP-3 and -9 were significantly reduced in the presence of 50 μM NS-398. Results are expressed as means±sd (pg ml−1) of the amount of MMP-3 and -9 secreted into the cell supernatant in duplicate from at least three independent experiments. Mean comparisons were based on extrapolated confidence intervals for at least four data points. *, P<0.005 compared with uninfected control; **, P<0.05 compared with infected cells.
Expression of inflammatory cytokines is governed by COX-2-derived PGE2
Increased production of inflammatory cytokines is another major neuropathological event associated with WNV infection. Therefore, we next investigated the possibility of COX-2 as one of the cellular factors responsible for modulating cytokine production in WNV-infected HBCAs. Increases in pro-inflammatory cytokines such as IL-1β and -6 has been documented recently in WNV-infected astrocytes (van Marle et al., 2007). Here, we measured both mRNA and protein levels of key inflammatory cytokines such as IL-1β, -6 and -8 induced by WNV in HBCAs in the presence or absence of NS-398. At an m.o.i. of 5, the levels of mRNA expression of IL-1β, -6 and -8 increased by 5−11-fold at 24 h p.i. (data not shown), followed by a dramatic increase at day 2, which peaked further at day 3 p.i. (Table 2). This sharp increase in IL-1β, -6 and -8 occurred at days 2 and 3 p.i. when the expression of COX-2 was also high (Fig. 1) and coincided with the peak in virus titres. However, as depicted in Table 2, treatment of HBCAs with NS-398 led to a 60–90 % reduction in mRNA expression of these cytokines at both time points. This decrease was more prominent in HBCAs treated with 50 μM NS-398 compared with treatment with 25 μM NS-398, suggesting that the reduction in cytokine expression is dose dependent. As cytokines are secretory proteins, their release into the supernatant of infected HBCAs treated with or without NS-398 was confirmed by ELISA. A significant increase in the levels of IL-1β, -6 and -8 was observed in the supernatant from WNV-infected HBCAs at day 3 p.i. (P<0.005), which decreased significantly in the presence of NS-398 (P<0.05, Fig. 5). These data suggested that COX-2 has a significant regulatory effect on the induction of inflammatory cytokines in WNV-infected cells and that blocking of COX-2 alone was sufficient to reduce the levels of key inflammatory cytokines released from the infected cells.
Table 2.
mRNA fold changes in cytokine levels as determined by qRT-PCR in WNV-infected HBCAs in the presence or absence of the COX-2-specific inhibitor NS-398
| Gene | Day 2 | Day 3 | ||||
|---|---|---|---|---|---|---|
| WNV | WNV+NS-398 | WNV | WNV+NS-398 | |||
| 25 μM | 50 μM | 25 μM | 50 μM | |||
| IL-1β | 81 | 18 | 9 | 187 | 68 | 32 |
| IL-6 | 76 | 15 | 8 | 108 | 37 | 21 |
| IL-8 | 48 | 8 | 3 | 137 | 22 | 11 |
Fig. 5.
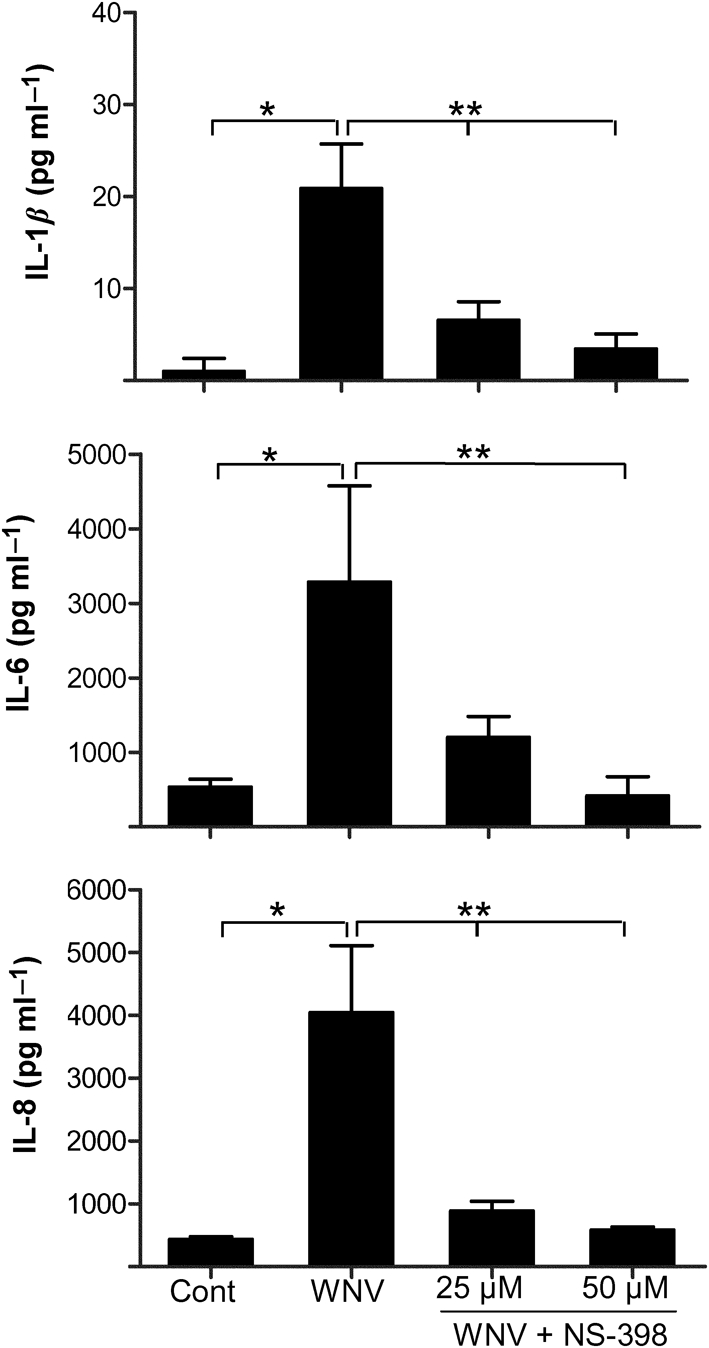
COX-2 inhibitor limits the expression of cytokines in WNV-infected HBCAs. Supernatants collected from HBCAs infected in the presence or absence of NS-398 at day 3 p.i. were analysed by ELISA for levels of IL-1β, -6 and -8. The data are expressed as mean concentration±sd (pg ml−1) of the amount of protein secreted into the supernatant from at least three independent experiments in duplicate. *, P<0.005 compared with uninfected control; **, P<0.05 compared with infected cells.
DISCUSSION
Neuroinflammation and disruption of the BBB are the common pathological features of WNVE (Sejvar et al., 2003; Wang et al., 2004, 2008), but the upstream signalling pathways modulating neuroinflammation are unclear. In the present study, we investigated the upstream mechanisms by which WNV modulates the expression of MMPs as well as key inflammatory cytokines. Our results not only highlight the fact that astrocytes respond to WNV infection by significantly increasing COX-2 expression and production of its substrate, PGE2, but also link its increase to downstream pathogenic events such as induction of matrix-degrading enzymes and inflammatory cytokines.
The COX-2 enzyme is specifically implicated as being important in host responses to infection, and the ability of PGE2 to modulate inflammation and the immune response is well documented (Bazan, 2001; Candelario-Jalil & Fiebich, 2008). COX-2 enzymic processes not only generate PGE2 but also promote the production and release of reactive oxygen intermediates such as oxygen radicals and nitric oxide (Gebicke-Haerter, 2001) and are a major target of widely prescribed non-steroidal anti-inflammatory drugs and newer COX-2-specific inhibitors such as celecoxib. We have demonstrated for the first time that the COX-2 enzyme is induced at both the mRNA and protein level in human astrocytes (Fig. 1). This increase coincided with a peak in virus titres, and infection with UV-inactivated WNV failed to induce COX-2 in HBCAs, suggesting that induction of COX-2 is a result of active virus replication. The increase in COX-derived PGE2 as observed in Fig. 3 also correlated with COX-2 induction, and its dramatic decrease in the presence of the COX-2-specific inhibitor NS-398 clearly indicated that COX-2, rather than COX-1, was the source of PGE2 in HBCAs. It is likely that COX-2-derived PGE2 acts as an autocrine or paracrine molecule, thereby mediating cellular events involved in WNV-induced neuroinflammatory responses. Although our in vitro data identified astrocytes as one of the sources of COX-2 in infected brain, it is possible that in vivo other cell types such as microglia and infiltrating monocytes may also contribute to the increased COX-2/PGE2 levels. A recent study by Alvarez et al. (2008) demonstrated that COX-2 induced by HIV-2 in human astrocytes modulated their activation status, such as expression of glial fibrillary acidic protein, and that this effect was blocked by NS-398 treatment. Increased COX-2/PGE2 has been linked to pathogenesis associated with other viruses such as HIV (Flora et al., 2006), hepatitis C virus (Lu et al., 2008), Epstein–Barr virus (Murono et al., 2001), Theiler's murine encephalomyelitis virus (Molina-Holgado et al., 2002), vesicular stomatitis virus (VSV) (Chen et al., 2002) and respiratory syncytial virus (Radi et al., 2010). Recent findings also report COX-2 induction and microglial activation following infection with JEV, another flavivirus (Ghoshal et al., 2007).
The integrity of the BBB in neuroinflammation is highly dependent on endothelial cells, production of cytokines and matrix-degrading enzymes, and recruitment of circulating inflammatory cells such as monocytes. An increase in the levels of MMPs, key matrix-degrading enzymes, has been linked to a compromised BBB and tight junction protein degradation in infections with neurotropic viruses such as HIV, herpes simplex virus and WNV (Keogh et al., 2003; Sellner et al., 2006; Wang et al., 2008). Our previous results also demonstrated that increased MMPs produced by WNV-infected HBCAs are capable of digesting tight junction proteins of endothelial cells (Verma et al., 2010). Here, we further demonstrated that uPA is induced by WNV in HBCAs. Increased uPA observed in neurodegenerative diseases and infection with pathogens (Conant et al., 2004; East et al., 2005; Paul et al., 2005) is believed to activate plasmin, which can degrade most components of basal lamina either directly or through activation of pro-collagenases such as MMP-3 (Iwamoto et al., 2008; Zhao et al., 2008). These data suggest that several matrix-degrading components such as MMPs and uPA are induced in the astrocytes in response to WNV infection, which collectively might contribute to the BBB disruption in vivo. Upstream signalling mechanisms that govern expression of these matrix-degrading enzymes include COX-2 and cytokines such as IL-1β and TNF-α (Aid et al., 2010; Johnatty et al., 1997). Inhibition of COX-2 limits lipopolysaccharide-induced MMP-3 and -9 levels in both in vitro and in vivo mouse models (Aid et al., 2010; Candelario-Jalil et al., 2007). Similarly, in infections with pathogens such as Helicobacter pylori, the COX-2/PGE2 pathway is involved in the induction of the uPA/uPAR signalling pathway (Iwamoto et al., 2008). Our results demonstrating a significant reduction in the levels of MMPs and uPA in the presence of NS-398, as depicted in Fig. 4, suggest that WNV-induced COX-2 and its product, PGE2, are one of the key upstream signals that govern the activity of these matrix-degrading enzymes. However, these studies do not address the question of whether these MMPs are induced directly by PGE2 or by PGE2-modulated cytokines such as IL-1β. Treatment with TNF-α results in the induction of MMPs and BBB disruption in mice, and the use of COX-2-specific inhibitors can eliminate TNF-α-induced MMP activity and reverse BBB disruption, thereby supporting the role of COX-2 in governing MMP activities (Candelario-Jalil et al., 2007). Similarly, post-ischaemic treatment with the COX-2 inhibitor nimesulide reduces BBB disruption and leukocyte infiltration following transient focal cerebral ischaemia in rats (Candelario-Jalil et al., 2005).
Pro-inflammatory cytokines such as TNF-α and IL-1 have been reported as potent inducers of neuronal injury in several neurodegenerative diseases such as cerebral ischaemia, spinal cord injury, multiple sclerosis and viral infections including HIV and JEV (Brabers & Nottet, 2006; Ghoshal et al., 2007; McColl et al., 2008). WNV infection also results in dramatic increases of several pro-inflammatory cytokines and chemokines such as IL-1β and CXCL10 in both human brain cells and mouse brain (Garcia-Tapia et al., 2007; Kumar et al., 2010). Our data demonstrating an increase in IL-1β in infected astrocytes is consistent with a previous observation reported by van Marle et al. (2007) where they not only documented increased production of IL-1β by WNV-infected astrocytes but also established the role of astrocyte-derived neurotoxic mediators in inducing the death of naïve neurons (van Marle et al., 2007). Expression of IL-6 and -8, which are endogenous pyrogens exerting multiple downstream inflammatory signalling pathways, is elevated in many CNS disorders including infection with JEV. The upstream mechanisms that control the production of different cytokines are complex and include COX-2-derived PGE2, intracellular potassium levels and reactive oxygen species (Aktas et al., 2007; Flora et al., 2006; Salminen et al., 2008). Cytokines such as TNF-α and IL-1β can stimulate the expression of several secondary cytokines and COX-2. In contrast, it is also well established that COX-2-derived PGE2 can amplify the signalling cascade and production of cytokines, MMPs and cell-adhesion molecules (Kyrkanides et al., 2002; Mark et al., 2001; Tzeng et al., 2005). Similarly, PGE2 mediates the cellular effects of IL-1β on parvocellular neurons in the hypothalamus, and the presence of NS-398 can abolish that effect (Ferri & Ferguson, 2005). Virus infections can induce multiple signalling pathways, which may cross-talk with each other or converge on common downstream effectors. In WNV infection, the initial stimulus for cytokine production in the brain is not clear. In the present study, our data demonstrating the ability of NS-398 to significantly block the production and release of IL-1β, -6 and -8 (Fig. 5) indicate that COX-2/PGE2 is one of the pathways through which expression of key inflammatory cytokines is modulated in WNV-infected HBCAs. This finding is particularly important because it suggests that targeting COX-2 alone is likely to reduce multiple neuroinflammatory downstream molecules that contribute to complex pathological events such as chemoattraction of peripheral immune cells into the CNS, disruption of the BBB and neuronal injury.
NS-398 administration to modulate inflammation has been employed successfully in several neurodegenerative disease models and significantly attenuates levels of several inflammatory mediators (Bazan et al., 2002; Candelario-Jalil & Fiebich, 2008), thus establishing a complex feedback regulation. Furthermore, it is COX-2 activity, but not COX-1 activity, that contributes to the progression of focal ischaemic brain injury, and the beneficial effects observed with non-selective COX inhibitors are associated with COX-2 rather than COX-1 inhibition (Candelario-Jalil et al., 2003). Although the data are limited, a few studies have characterized the relative effects of COX-1 and COX-2 in modulating the host immune response (Aid et al., 2010; Candelario-Jalil et al., 2003). In influenza virus infection, important but opposite effects of the COX-1 and COX-2 enzymes have been documented, wherein COX-1 deficiency is detrimental to the host whilst COX-2 deficiency is beneficial (Carey et al., 2005). Similarly, selective inhibition of COX-2 also suppresses replication of VSV (Chen et al., 2000, 2002).
It has been suggested that it is the overwhelming inflammatory response that contributes to the severity of WNV disease, blocking of which not only improves disease parameters but also reduces virus titres in the brain (Wang et al., 2004, 2008). Several of these cytokines act synergistically, resulting in cell death; therefore, it is important to identify upstream mechanism(s) by which the production of inflammatory molecules is modulated in WNV infection. Extrapolation of our results to animal models and testing the effect of COX-2 inhibitors on WNV titres, BBB disruption and CNS inflammation ultimately resulting in improved WNV disease outcome are warranted. In conclusion, our findings establish a critical role for COX-2 and PGE2 in modulating the inflammatory response in the CNS to WNV infection.
METHODS
Cells and virus.
Primary HBCAs were obtained from the Applied Cell Biology Research Institute (Kirkland, WA, USA) and propagated as described previously (Verma et al., 2010). All experiments were performed with cells between passage 6 and 10. Infections with WNV (lineage I, WNV strain NY99 originally isolated from crow brain and passaged once in Vero cells) and UV-inactivated WNV were conducted as described previously (Verma et al., 2008, 2009) and cells were harvested at various time points p.i.
Treatment of HBCAs with COX-2 inhibitor.
The toxicity of the COX-2-specific inhibitor NS-398 (Cayman Chemical) was tested by cultivating HBCAs in medium in the presence of NS-398 in a concentration range of 10–80 μM using a CellTiter 96 AQueous One Solution Cell Proliferation Assay kit (Promega) (Verma et al., 2008). None of these concentrations of NS-398, or use of vehicle only (0.001 % DMSO), was found to be toxic to HBCAs. For all inhibitor experiments, HBCAs were treated with 25 or 50 μM NS-398 at 12 h p.i. with a final concentration of DMSO of 0.001 %.
qRT-PCR.
cDNA prepared from total RNA extracted from HBCAs was used for qRT-PCR as described previously (Verma et al., 2010). The primer sequences and annealing temperatures used for the amplification of GAPDH and MMPs have been described previously (Verma et al., 2009), and the primers for COX and inflammatory cytokines are described in Supplementary Table S1 (available in JGV Online). For all in vitro fold change analyses, the housekeeping gene GAPDH was used as an internal baseline reference to normalize each gene of interest, and the mRNA fold change of the gene of interest was determined at each time point compared with its corresponding infected control. All experiments were performed on samples from at least three independent infections in duplicate.
Western immunoblot analysis of COX-2.
Total protein from HBCAs was extracted as described previously (Verma et al., 2008, 2009, 2010). The nitrocellulose membranes were incubated with rabbit polyclonal anti-COX-2 antibody (1 mg ml−1; Santa Cruz Biotechnologies) followed by HRP-conjugated secondary antibody and developed using an ECL detection kit (Pierce Technologies).
Immunocytochemical analysis.
Fixed HBCAs were immunostained using anti-3.67G WNV envelope mAb (diluted 1 : 800; a generous gift from Dr D. Gubler, Duke-NUS Graduate Medical School, Singapore; originally produced by Dr R. H. Hall, University of Queensland, St Lucia, Australia) or anti-COX-2 (diluted 1 : 100) mAb followed by secondary antibodies conjugated to Alexa Fluor 488 or 546 (Invitrogen) and examined using a Zeiss confocal Pascal microscope equipped with a Zeiss Axiovert 200, as described previously (Verma et al., 2009).
Analysis of PGE2, MMPs and cytokines by ELISA.
At day 4 p.i., the supernatant medium from WNV- and mock-infected HBCAs in the presence or absence of NS-398 was collected to assay the levels of PGE2, MMPs and cytokines. All ELISAs were performed using undiluted supernatant medium in Biosafety Level 3 laboratories without any UV treatment. PGE2 was measured using a PGE2 Parameter kit (R&D Systems) according to the manufacturer's instructions. ELISAs for total MMP-3 and -9 in the cell supernatant were conducted using Quantikine human MMP-3 and -9 ELISA kits, respectively (R&D systems), as described previously (Verma et al., 2010). IL-6 and -8 levels were measured using Quantikine ELISA kits, whilst the IL-1β level was measured using a Quantikine High Sensitivity kit according to the manufacturer's instructions. The ELISA plates were analysed using a Victor 3 microtitre reader equipped with Workout version 1.5 software (Perkin Elmer).
Statistical analysis.
All in vitro data are reported as the mean±sd of at least three independent experiments in duplicate. An unpaired Student t-test using GraphPad Prism software version 5.0 was used to compare the levels of cytokines and MMPs assayed by ELISA. Differences where P<0.05 were considered statistically significant.
Supplementary Material
Acknowledgments
This work was partly supported by grants from the Hawaii Community Foundation (20050405), Research Centers in Minority Institutions Program (G12RR003061) and Centers of Biomedical Research Excellence (P20RR018727), National Center for Research Resources, National Institutes of Health and institutional funds. We thank Ms Janet Meeks and Ms Kelsey Roe for valuable technical assistance, and Ms Esther Volper for microarray analysis.
Footnotes
Primer sequences are available with the online version of this paper.
References
- Aid, S., Silva, A. C., Candelario-Jalil, E., Choi, S. H., Rosenberg, G. A. & Bosetti, F. (2010). Cyclooxygenase-1 and -2 differentially modulate lipopolysaccharide-induced blood−brain barrier disruption through matrix metalloproteinase activity. J Cereb Blood Flow Metab 30, 370–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aktas, O., Ullrich, O., Infante-Duarte, C., Nitsch, R. & Zipp, F. (2007). Neuronal damage in brain inflammation. Arch Neurol 64, 185–189. [DOI] [PubMed] [Google Scholar]
- Alvarez, S., Blanco, A., Kern, F., Fresno, M. & Munoz-Fernandez, M. A. (2008). HIV-2 induces NF-κB activation and cyclooxygenase-2 expression in human astroglial cells. Virology 380, 144–151. [DOI] [PubMed] [Google Scholar]
- Arjona, A., Foellmer, H. G., Town, T., Leng, L., McDonald, C., Wang, T., Wong, S. J., Montgomery, R. R., Fikrig, E. & Bucala, R. (2007). Abrogation of macrophage migration inhibitory factor decreases West Nile virus lethality by limiting viral neuroinvasion. J Clin Invest 117, 3059–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek, J. Y., Hur, W., Wang, J. S., Bae, S. H. & Yoon, S. K. (2007). Selective COX-2 inhibitor, NS-398, suppresses cellular proliferation in human hepatocellular carcinoma cell lines via cell cycle arrest. World J Gastroenterol 13, 1175–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazan, N. G. (2001). COX-2 as a multifunctional neuronal modulator. Nat Med 7, 414–415. [DOI] [PubMed] [Google Scholar]
- Bazan, N. G., Colangelo, V. & Lukiw, W. J. (2002). Prostaglandins and other lipid mediators in Alzheimer's disease. Prostaglandins Other Lipid Mediat 68–69, 197–210. [DOI] [PubMed] [Google Scholar]
- Brabers, N. A. & Nottet, H. S. (2006). Role of the pro-inflammatory cytokines TNF-α and IL-1β in HIV-associated dementia. Eur J Clin Invest 36, 447–458. [DOI] [PubMed] [Google Scholar]
- Brinton, M. A. (2002). The molecular biology of West Nile virus: a new invader of the western hemisphere. Annu Rev Microbiol 56, 371–402. [DOI] [PubMed] [Google Scholar]
- Candelario-Jalil, E. & Fiebich, B. L. (2008). Cyclooxygenase inhibition in ischemic brain injury. Curr Pharm Des 14, 1401–1418. [DOI] [PubMed] [Google Scholar]
- Candelario-Jalil, E., González-Falcón, A., García-Cabrera, M., Álvarez, D., Al-Dalain, S., Martínez, G., León, O. S. & Springer, J. E. (2003). Assessment of the relative contribution of COX-1 and COX-2 isoforms to ischemia-induced oxidative damage and neurodegeneration following transient global cerebral ischemia. J Neurochem 86, 545–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candelario-Jalil, E., Mhadu, N. H., González-Falcón, A., García-Cabrera, M., Muñoz, E., León, O. S. & Fiebich, B. L. (2005). Effects of the cyclooxygenase-2 inhibitor nimesulide on cerebral infarction and neurological deficits induced by permanent middle cerebral artery occlusion in the rat. J Neuroinflammation 2, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candelario-Jalil, E., Taheri, S., Yang, Y., Sood, R., Grossetete, M., Estrada, E. Y., Fiebich, B. L. & Rosenberg, G. A. (2007). Cyclooxygenase inhibition limits blood−brain barrier disruption following intracerebral injection of tumor necrosis factor-α in the rat. J Pharmacol Exp Ther 323, 488–498. [DOI] [PubMed] [Google Scholar]
- Carey, M. A., Bradbury, J. A., Seubert, J. M., Langenbach, R., Zeldin, D. C. & Germolec, D. R. (2005). Contrasting effects of cyclooxygenase-1 (COX-1) and COX-2 deficiency on the host response to influenza A viral infection. J Immunol 175, 6878–6884. [DOI] [PubMed] [Google Scholar]
- Cheeran, M. C., Hu, S., Sheng, W. S., Rashid, A., Peterson, P. K. & Lokensgard, J. R. (2005). Differential responses of human brain cells to West Nile virus infection. J Neurovirol 11, 512–524. [DOI] [PubMed] [Google Scholar]
- Chen, N., Warner, J. L. & Reiss, C. S. (2000). NSAID treatment suppresses VSV propagation in mouse CNS. Virology 276, 44–51. [DOI] [PubMed] [Google Scholar]
- Chen, N., Restivo, A. & Reiss, C. S. (2002). Selective inhibition of COX-2 is beneficial to mice infected intranasally with VSV. Prostaglandins Other Lipid Mediat 67, 143–155. [DOI] [PubMed] [Google Scholar]
- Conant, K., St Hillaire, C., Anderson, C., Galey, D., Wang, J. & Nath, A. (2004). Human immunodeficiency virus type 1 Tat and methamphetamine affect the release and activation of matrix-degrading proteinases. J Neurovirol 10, 21–28. [DOI] [PubMed] [Google Scholar]
- East, E., Baker, D., Pryce, G., Lijnen, H. R., Cuzner, M. L. & Gveric, D. (2005). A role for the plasminogen activator system in inflammation and neurodegeneration in the central nervous system during experimental allergic encephalomyelitis. Am J Pathol 167, 545–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferri, C. C. & Ferguson, A. V. (2005). Prostaglandin E2 mediates cellular effects of interleukin-1β on parvocellular neurones in the paraventricular nucleus of the hypothalamus. J Neuroendocrinol 17, 498–508. [DOI] [PubMed] [Google Scholar]
- Flora, G., Pu, H., Hennig, B. & Toborek, M. (2006). Cyclooxygenase-2 is involved in HIV-1 Tat-induced inflammatory responses in the brain. Neuromolecular Med 8, 337–352. [DOI] [PubMed] [Google Scholar]
- Garcia-Tapia, D., Hassett, D. E., Mitchell, W. J., Jr, Johnson, G. C. & Kleiboeker, S. B. (2007). West Nile virus encephalitis: sequential histopathological and immunological events in a murine model of infection. J Neurovirol 13, 130–138. [DOI] [PubMed] [Google Scholar]
- Gebicke-Haerter, P. J. (2001). Microglia in neurodegeneration: molecular aspects. Microsc Res Tech 54, 47–58. [DOI] [PubMed] [Google Scholar]
- Ghoshal, A., Das, S., Ghosh, S., Mishra, M. K., Sharma, V., Koli, P., Sen, E. & Basu, A. (2007). Proinflammatory mediators released by activated microglia induces neuronal death in Japanese encephalitis. Glia 55, 483–496. [DOI] [PubMed] [Google Scholar]
- Glass, W. G., Lim, J. K., Cholera, R., Pletnev, A. G., Gao, J. L. & Murphy, P. M. (2005). Chemokine receptor CCR5 promotes leukocyte trafficking to the brain and survival in West Nile virus infection. J Exp Med 202, 1087–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass, W. G., McDermott, D. H., Lim, J. K., Lekhong, S., Yu, S. F., Frank, W. A., Pape, J., Cheshier, R. C. & Murphy, P. M. (2006). CCR5 deficiency increases risk of symptomatic West Nile virus infection. J Exp Med 203, 35–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickey, R. W., Adelson, P. D., Johnnides, M. J., Davis, D. S., Yu, Z., Rose, M. E., Chang, Y. F. & Graham, S. H. (2007). Cyclooxygenase-2 activity following traumatic brain injury in the developing rat. Pediatr Res 62, 271–276. [DOI] [PubMed] [Google Scholar]
- Im, J. Y., Kim, D., Paik, S. G. & Han, P. L. (2006). Cyclooxygenase-2-dependent neuronal death proceeds via superoxide anion generation. Free Radic Biol Med 41, 960–972. [DOI] [PubMed] [Google Scholar]
- Iwamoto, J., Mizokami, Y., Takahashi, K., Matsuoka, T. & Matsuzaki, Y. (2008). The effects of cyclooxygenase2−prostaglandinE2 pathway on Helicobacter pylori-induced urokinase-type plasminogen activator system in the gastric cancer cells. Helicobacter 13, 174–182. [DOI] [PubMed] [Google Scholar]
- Johnatty, R. N., Taub, D. D., Reeder, S. P., Turcovski-Corrales, S. M., Cottam, D. W., Stephenson, T. J. & Rees, R. C. (1997). Cytokine and chemokine regulation of proMMP-9 and TIMP-1 production by human peripheral blood lymphocytes. J Immunol 158, 2327–2333. [PubMed] [Google Scholar]
- Kelley, T. W., Prayson, R. A., Ruiz, A. I., Isada, C. M. & Gordon, S. M. (2003). The neuropathology of West Nile virus meningoencephalitis. A report of two cases and review of the literature. Am J Clin Pathol 119, 749–753. [DOI] [PubMed] [Google Scholar]
- Keogh, B., Sheahan, B. J., Atkins, G. J. & Mills, K. H. (2003). Inhibition of matrix metalloproteinases ameliorates blood−brain barrier disruption and neuropathological lesions caused by avirulent Semliki Forest virus infection. Vet Immunol Immunopathol 94, 185–190. [DOI] [PubMed] [Google Scholar]
- Klein, R. S., Lin, E., Zhang, B., Luster, A. D., Tollett, J., Samuel, M. A., Engle, M. & Diamond, M. S. (2005). Neuronal CXCL10 directs CD8+ T-cell recruitment and control of West Nile virus encephalitis. J Virol 79, 11457–11466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, M., Verma, S. & Nerurkar, V. R. (2010). Pro-inflammatory cytokines derived from West Nile virus (WNV)-infected SK-N-SH cells mediate neuroinflammatory markers and neuronal death. J Neuroinflammation 7, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyrkanides, S., Moore, A. H., Olschowka, J. A., Daeschner, J. C., Williams, J. P., Hansen, J. T. & Kerry O'Banion, M. (2002). Cyclooxygenase-2 modulates brain inflammation-related gene expression in central nervous system radiation injury. Brain Res Mol Brain Res 104, 159–169. [DOI] [PubMed] [Google Scholar]
- Lim, J. K., Glass, W. G., McDermott, D. H. & Murphy, P. M. (2006). CCR5: no longer a “good for nothing” gene – chemokine control of West Nile virus infection. Trends Immunol 27, 308–312. [DOI] [PubMed] [Google Scholar]
- Lu, L., Wei, L., Peng, G., Mu, Y., Wu, K., Kang, L., Yan, X., Zhu, Y. & Wu, J. (2008). NS3 protein of hepatitis C virus regulates cyclooxygenase-2 expression through multiple signaling pathways. Virology 371, 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark, K. S., Trickler, W. J. & Miller, D. W. (2001). Tumor necrosis factor-α induces cyclooxygenase-2 expression and prostaglandin release in brain microvessel endothelial cells. J Pharmacol Exp Ther 297, 1051–1058. [PubMed] [Google Scholar]
- McColl, B. W., Rothwell, N. J. & Allan, S. M. (2008). Systemic inflammation alters the kinetics of cerebrovascular tight junction disruption after experimental stroke in mice. J Neurosci 28, 9451–9462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina-Holgado, E., Arévalo-Martín, A., Ortiz, S., Vela, J. M. & Guaza, C. (2002). Theiler's virus infection induces the expression of cyclooxygenase-2 in murine astrocytes: inhibition by the anti-inflammatory cytokines interleukin-4 and interleukin-10. Neurosci Lett 324, 237–241. [DOI] [PubMed] [Google Scholar]
- Murono, S., Inoue, H., Tanabe, T., Joab, I., Yoshizaki, T., Furukawa, M. & Pagano, J. S. (2001). Induction of cyclooxygenase-2 by Epstein–Barr virus latent membrane protein 1 is involved in vascular endothelial growth factor production in nasopharyngeal carcinoma cells. Proc Natl Acad Sci U S A 98, 6905–6910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottino, P. & Bazan, H. E. (2001). Corneal stimulation of MMP-1, -9 and uPA by platelet-activating factor is mediated by cyclooxygenase-2 metabolites. Curr Eye Res 23, 77–85. [DOI] [PubMed] [Google Scholar]
- Paul, R., Winkler, F., Bayerlein, I., Popp, B., Pfister, H. W. & Koedel, U. (2005). Urokinase-type plasminogen activator receptor regulates leukocyte recruitment during experimental pneumococcal meningitis. J Infect Dis 191, 776–782. [DOI] [PubMed] [Google Scholar]
- Pompl, P. N., Ho, L., Bianchi, M., McManus, T., Qin, W. & Pasinetti, G. M. (2003). A therapeutic role for cyclooxygenase-2 inhibitors in a transgenic mouse model of amyotrophic lateral sclerosis. FASEB J 17, 725–727. [DOI] [PubMed] [Google Scholar]
- Radi, Z. A., Meyerholz, D. K. & Ackermann, M. R. (2010). Pulmonary cyclooxygenase-1 (COX-1) and COX-2 cellular expression and distribution after respiratory syncytial virus and parainfluenza virus infection. Viral Immunol 23, 43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salminen, A., Ojala, J., Suuronen, T., Kaarniranta, K. & Kauppinen, A. (2008). Amyloid-β oligomers set fire to inflammasomes and induce Alzheimer's pathology. J Cell Mol Med 12, 2255–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sejvar, J. J., Haddad, M. B., Tierney, B. C., Campbell, G. L., Marfin, A. A., Van Gerpen, J. A., Fleischauer, A., Leis, A. A., Stokic, D. S. & Petersen, L. R. (2003). Neurologic manifestations and outcome of West Nile virus infection. JAMA 290, 511–515. [DOI] [PubMed] [Google Scholar]
- Sellner, J., Simon, F., Meyding-Lamade, U. & Leib, S. L. (2006). Herpes-simplex virus encephalitis is characterized by an early MMP-9 increase and collagen type IV degradation. Brain Res 1125, 155–162. [DOI] [PubMed] [Google Scholar]
- Shrestha, B., Gottlieb, D. & Diamond, M. S. (2003). Infection and injury of neurons by West Nile encephalitis virus. J Virol 77, 13203–13213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, D. M. & Kuhn, D. M. (2005). Cyclooxygenase-2 is an obligatory factor in methamphetamine-induced neurotoxicity. J Pharmacol Exp Ther 313, 870–876. [DOI] [PubMed] [Google Scholar]
- Tzeng, S. F., Hsiao, H. Y. & Mak, O. T. (2005). Prostaglandins and cyclooxygenases in glial cells during brain inflammation. Curr Drug Targets Inflamm Allergy 4, 335–340. [DOI] [PubMed] [Google Scholar]
- van Marle, G., Antony, J., Ostermann, H., Dunham, C., Hunt, T., Halliday, W., Maingat, F., Urbanowski, M. D., Hobman, T. & other authors (2007). West Nile virus-induced neuroinflammation: glial infection and capsid protein-mediated neurovirulence. J Virol 81, 10933–10949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma, S., Molina, Y., Lo, Y. Y., Cropp, B., Nakano, C., Yanagihara, R. & Nerurkar, V. R. (2008). In vitro effects of selenium deficiency on West Nile virus replication and cytopathogenicity. Virol J 5, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma, S., Lo, Y., Chapagain, M., Lum, S., Kumar, M., Gurjav, U., Luo, H., Nakatsuka, A. & Nerurkar, V. R. (2009). West Nile virus infection modulates human brain microvascular endothelial cells tight junction proteins and cell adhesion molecules: transmigration across the in vitro blood−brain barrier. Virology 385, 425–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma, S., Kumar, M., Gurjav, U., Lum, S. & Nerurkar, V. R. (2010). Reversal of West Nile virus-induced blood−brain barrier disruption and tight junction proteins degradation by MMP inhibitor. Virology 397, 130–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, T., Town, T., Alexopoulou, L., Anderson, J. F., Fikrig, E. & Flavell, R. A. (2004). Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat Med 10, 1366–1373. [DOI] [PubMed] [Google Scholar]
- Wang, P., Dai, J., Bai, F., Kong, K. F., Wong, S. J., Montgomery, R. R., Madri, J. A. & Fikrig, E. (2008). Matrix metalloproteinase 9 facilitates West Nile virus entry into the brain. J Virol 82, 8978–8985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, H. & Chen, C. (2008). Cyclooxygenase-2 in synaptic signaling. Curr Pharm Des 14, 1443–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, Y., Lyons, C. E., Jr, Xiao, A., Templeton, D. J., Sang, Q. A., Brew, K. & Hussaini, I. M. (2008). Urokinase directly activates matrix metalloproteinases-9: a potential role in glioblastoma invasion. Biochem Biophys Res Commun 369, 1215–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.