

Abstract
Activation of the maternal immune system in rodent models sets in motion a cascade of molecular pathways that ultimately result in autism- and schizophrenia-related behaviors in offspring. The finding that interleukin-6 (IL-6) is a crucial mediator of these effects led us to examine the mechanism by which this cytokine influences fetal development in vivo. Here we focus on the placenta as the site of direct interaction between mother and fetus and as a principal modulator of fetal development. We find that maternal immune activation (MIA) with a viral mimic, synthetic double-stranded RNA (poly(I:C)), increases IL-6 mRNA as well as maternally-derived IL-6 protein in the placenta. Placentas from MIA mothers exhibit increases in CD69+ decidual macrophages, granulocytes and uterine NK cells, indicating elevated early immune activation. Maternally-derived IL-6 mediates activation of the JAK/STAT3 pathway specifically in the spongiotrophoblast layer of the placenta, which results in expression of acute phase genes. Importantly, this parallels an IL-6-dependent disruption of the growth hormone-insulin-like growth factor (GH-IGF) axis that is characterized by decreased GH, IGFI and IGFBP3 levels. In addition, we observe an IL-6-dependent induction in pro-lactin-like protein-K (PLP-K) expression as well as MIA-related alterations in other placental endocrine factors. Together, these IL-6-mediated effects of MIA on the placenta represent an indirect mechanism by which MIA can alter fetal development.
Keywords: autism, schizophrenia, maternal immune activation, inflammation, growth hormone, insulin-like growth factor, JAK/STAT3, poly(I:C), maternal infection
Introduction
Both autism and schizophrenia are relatively common disorders with often tragic, lifelong consequences. Several susceptibility genes and environmental agents have been identified as risk factors, but few cases of autism or schizophrenia can be traced to a known cause. Maternal infection is regarded as a principal, non-genetic cause of schizophrenia, and is also associated with increased risk for autism in the offspring (Atladóttir et al., 2010; reviewed by Brown & Derkits, 2010; Patterson, 2009). In a mouse model of this risk factor, the offspring of mice injected with poly(I:C) dsRNA as a viral mimic develop behaviors and brain pathology consistent with those seen in human autism and schizophrenia (Ito et al., 2010; Lee et al., 2007; Li et al., 2009; Makinodan et al., 2008; Meyer et al., 2006, 2008b; Nyffeler et al., 2006; Ozawa et al., 2006; Piontkewitz et al., 2009; Shi et al., 2003, 2009; Smith et al., 2007; Winter et al., 2008; Zuckerman et al., 2003; Zuckerman & Weiner, 2005). These effects require a key mediator, the cytokine interleukin-6 (IL-6). Maternal injection of IL-6 alone is sufficient to cause the behavioral abnormalities seen in the offspring following maternal poly(I:C) injection or respiratory infection (Samuelsson et al., 2006; Smith et al., 2007). Conversely, co-injection of an antibody that neutralizes endogenous IL-6 along with poly(I:C) completely prevents the prepulse inhibition (PPI), latent inhibition (LI), exploratory and social deficits in the offspring caused by MIA (Smith et al., 2007). Furthermore, poly(I:C) injection of pregnant IL-6 knockout (KO) mice results in no such behavioral deficits. IL-6 is also required for induction of transcriptional changes as co-injection of anti-IL-6 and poly(I:C) normalizes 92% of the MIA-induced changes in gene expression in brains of adult offspring (Smith et al., 2007).
The importance of IL-6 in mediating the development of schizophrenic and autistic endophenotypes in rodents is further supported by post-mortem studies of human subjects. The significant increases in pro-inflammatory cytokines, including IL-6, in fetal brains shortly after MIA in rodents (Meyer et al., 2006, 2008a) are also observed in the brains of adult schizophrenic and autistic patients (Arion et al., 2007; Garbett et al., 2008; Li et al., 2009; Saetre et al., 2007;Vargas et al., 2005). IL-6 is also elevated in sera from living schizophrenic individuals (Maes et al., 2002; Potvin et al., 2008), and many pro-inflammatory cytokines are increased in the cerebrospinal fluid and sera from living autistic individuals (Ashwood et al., 2010; Chez, 2007; Pardo et al., 2006). Furthermore, an IL-6 receptor polymorphism is associated with schizophrenia (Sun et al., 2008), and increased IL-6 levels are associated with several other environmental risk factors for schizophrenia, including maternal stress, maternal malnutrition and obstetric complications.
Despite demonstration of the critical role of IL-6 in mouse MIA models and in human schizophrenia and autism, the mechanism underlying how IL-6 acts to disrupt early brain development is unknown. IL-6 can act directly on progenitor cells to regulate fetal neurogenesis and gliogenesis (reviewed by Deverman & Patterson, 2009). IL-6 can also act at the maternal-fetal interface to alter many parameters that influence fetal growth, including nutrient transfer, anoxia and vascular permeability (Desai et al., 2002; Jones et al., 2009; Kendall & Peebles, 2005). Moreover, IL-6 can disrupt the immunological balance of the placenta, altering Th1/Th2 homeostasis, activation of uterine immune cells, and maintenance of maternal tolerance (Arad et al., 2005; reviewed by Jonakait, 2007; Paul et al., 2003; Zimmerman et al., 2007).
We have studied the pathway of IL-6 action in the placenta to gain insight into the molecular processes that can lead to the postnatal manifestation of behavioral abnormalities. We confirm that MIA elevates IL-6 protein and mRNA expression in the placenta (Gilmore et al., 2005; Koga et al., 2009; Meyer et al., 2006, 2008a; Smith et al., 2007). We determine the relative contribution of maternally-derived versus fetally-derived IL-6 to the observed increases in placenta IL-6 levels. We identify activated decidual leukocytes as a likely source of MIA-induced placental IL-6, and we localize the site of maternal, IL-6-dependent STAT3 activation to fetal cells in the placental spongiotrophoblast layer. Finally, we report alterations in maternal-placental hormones and endocrine factors that may have important consequences for fetal development.
Methods
Generation of animals
Female C57BL/6J mice (Charles River; Wilmington, MA) were obtained from the Caltech breeding facility and housed under standard laboratory conditions. Mice were mated overnight and the presence of a vaginal plug on the following morning was noted as day E0.5.
IL-6 KO mice
IL-6 KO mice, strain B6.129S2-IL6tm1Kopf/J, were obtained from Jackson Laboratory (Bar Harbor, ME). IL-6 -/- females were mated with IL-6 +/- males, and IL-6 +/- females were mated with IL-6 -/- males. For assays involving placental samples, a small amount of tissue from the corresponding fetus was used for genotyping.
MIA
Pregnant C57BL/6J or IL-6 KO mice were injected on E12.5 with saline, poly(I:C), or recombinant IL-6 (rIL-6). For poly(I:C) injections, poly(I:C) potassium salt (Sigma Aldrich; St. Louis, MO) was freshly dissolved in saline and administered i.p. at 20 mg/kg based on the weight of the poly(I:C) itself, not including the total weight of the potassium salts. Control mice were injected with saline alone at 5 μl per gram body weight. For rIL-6 injections, 5 μg carrier-free, recombinant mouse IL-6 (eBioscience; San Diego, CA) was freshly dissolved in 150 μl saline and injected i.p.
Behavioral testing
After injection on E12.5, each pregnant mouse was single-housed. At 6 weeks of age, male and female offspring were behaviorally tested for PPI; at 8 weeks, for LI, and at 10 weeks, for open field exploration, according to methods described by Smith et al., 2007.
Briefly, for PPI testing, mice are acclimated to the testing chamber for 5 minutes, presented with six 120 db pulses of white noise (startle stimulus) and then subjected to 14 randomized blocks of either no startle, startle stimulus only, 5 db pre-pulse + startle or 15 db pre-pulse + startle. The startle response is recorded by a pliezo-electric sensor, and PPI is defined as (startle stimulus only − 5 or 15 db pre-pulse + startle)/startle stimulus only.
For LI testing, mice are presented with a pre-exposure of 40, 30 second tones followed by three pairings of the tone with a mild foot shock. Non-pre-exposed (NPE) are presented with the three pairings only. The following day, mice are placed in the same chambers for 8 minutes to record context freezing. On the third day, mice are placed in the same chambers and subjected to an 8 minute tone presentation, during which freezing is measured. LI is defined as the difference in freezing in response to the tone in pre-exposed mice compared to NPE mice.
For open field testing, mice are placed in 50 cm × 50 cm white plexiglass boxes for 10 minutes. An overhead video camera is used to record the session, and Ethovision software is used to analyze the distance traveled, and the number of entries and duration of time spent in the center arena (central 17 cm square).
Measurement of placental IL-6, growth hormone and IGFI
After injection on E12.5, wild type (WT) and IL-6 KO male and female mice were sacrificed at 3, 6 or 24 hours by an overdose of sodium pentobarbital (Nembutal). Placentas were dissected from the uterine horn and washed in PBS prior to snap-freezing in liquid nitrogen and storage at -80°C. To generate cell lysates, each placenta was placed in 1 ml of cell lysis buffer (50 mM Tris-HCl (pH 7.4) with 0.6 M NaCl, 0.2% Triton X-100, 1% BSA, and 1 EDTA-free protease inhibitor cocktail tablet/10 ml buffer) (Roche Applied Sciences; Indianapolis, IN). Each tissue was homogenized on ice using a syringe fitted with an 18G needle and then sonicated for 5 seconds at 10 mV. The homogenates were centrifuged at 4°C at 13,000 rpm for 20 minutes, and the supernatants aliquotted and frozen at -80°C until assayed. ELISAs for IL-6 (eBioscience), GH (Millipore; Billerica, MA), and IGFI (R&D Systems; Minneapolis, MN) were performed according to the manufacturers' instructions and analyzed against cell lysis buffer negative controls. Total protein was measured by BCA assay (Thermo Scientific; Rockford, IL) according to the manufacturer's instructions.
Measurement of placental gene expression by real-time PCR
Placentas were quickly dissected from the uterine horn and washed in PBS prior to preservation in 1 ml TRIzol solution (Invitrogen; Carlsbad, CA). Tissues were passed through an 18G needle and sonicated for two rounds of 3 seconds at 10 mV separated by incubation on ice. Homogenates were processed by chloroform extraction and washing with 70% ethanol according to standard procedures. RNA was further purified by applying the cleared lysate to an RNeasy mini column (Qiagen; Valencia, CA), and an on-column DNA digestion was performed according to the manufacturer's protocols. Samples were assayed using the 2100 Bioanalyzer (Agilent Technologies; Santa Clara, CA) and confirmed to contain high integrity RNA (RIN > 8). 5 μg RNA per sample was reverse-transcribed using the iScript cDNA synthesis kit (Biorad; Hercules, CA). Resultant cDNA was purified using a PCR purification kit (Qiagen) and eluted in 50 μl PCR-grade water (Roche). Gene expression was measured using SYBR Green master mix with Rox passive reference dye (Roche) on the ABI 7300 Real Time PCR system. Target gene expression was normalized against beta-actin transcript, and data are expressed as ratios of gene expression of poly(I:C) to saline samples. The primers used were adapted from the Primerbank database (Spandidos et al., 2010).
Immunohistochemistry
Placentas were dissected and washed in PBS prior to fixation for 1.5 hours in 4% paraformaldehyde at 4°C and cryopreservation in 30% sucrose overnight at 4°C. After equilibration for 1 hr in Tissue-Tek OCT (Sakura Finetek; Torrance, CA), each placenta was hemi-sected laterally prior to freezing. Placentas were cut in 12 μm sagittal sections, with 8 medial sections spanning approximately 576 μm of tissue (every sixth section) taken from each placenta for each histological stain. Sections were stained for phospho-STAT3 and phospho-STAT1 (Cell Signaling; Danvers, MA) according to standard procedures. Briefly, antigen retrieval was conducted for 30 minutes in a 95°C water bath in 10 mM sodium citrate pH 6.0, for pSTAT3, and 1 mM EDTA pH 9.0, for pSTAT1. Slides were equilibrated to room temperature, washed and incubated for 10 minutes in 3% H2O2 in methanol. After washing, tissues were processed using Vectastain Elite ABC peroxide kits (Vector Labs; Burlingame, CA) according to the manufacturer's instructions, with overnight incubation of primary antibody at 4°C. Staining was developed using ImmPACT DAB substrate (Vector Labs) and mounted in Vectamount aqueous mounting medium (Vector Labs).
Flow cytometry of placental leukocytes
The decidua was dissected carefully from the labyrinth layer and washed in PBS on ice. 7-8 tissues from each litter were pooled in RPMI 1640 medium with 10% FBS and 1% penicillin/streptomycin and subjected to mechanical disruption by passage through a syringe fitted with a 16G needle followed by a syringe with an 18G needle. Tissue suspensions were then enzymatically disrupted with 0.1% collagenase for 30 minutes at 37°C, followed by 0.25% trypsin for 10 minutes at 37°C. Cell suspensions were washed with 3 volumes of RPMI and passed through 22G and 25G needles and subjected to 2 rounds of RBC lysis and filtration through 40 μm cell strainers. Cell counts were performed on a hemocytometer, and 1 million cells were aliquotted for each subsequent reaction. Single cell suspensions were treated with anti-mouse CD16/CD32 Fc block (eBioscience) for 10 minutes on ice prior to staining in CBSS with the following antibody conjugates: DBA-lectin-FITC (Sigma), CD69-PE (eBioscience), Ter119-PerCP-Cy5.5 (BioLegend; San Diego, CA), CD4-FITC (BioLegend), CD8-FITC (Biolegend), Gr-1-APC (BioLegend), B220-FITC (BioLegend) and CD11b-APC (BioLegend). Flow cytometry was performed using the FACSCalibur cytometer (BD Biosciences; San Jose, CA), and data were analyzed using FlowJo software (TreeStar Inc.; Ashland, OR) and presented as percent frequency of the parent population (non-erythroid (Ter119-) cells).
Laser capture microdissection (LMD)
Placentas were dissected, washed in PBS and immediately snap-frozen in liquid nitrogen. Tissues were then embedded in Tissue-Tek OCT and cut into 12 μm sections on PALM Membrane Slides (PALM Microlaser Technologies; Germany). Cryostat blades and workstations were cleaned with RNaseZap (Applied Biosystems; Austin, TX) to prevent RNA degradation. 6 medial sagittal sections were stained with hematoxylin QS (Vector Labs) and used for laser microdissection. Duration was limited to < 30 minutes per slide to prevent RNA degradation. For each placental section, the spongiotrophoblast layer was localized by morphology under the Axio Observer.Z1 confocal microscope (Zeiss; Thornwood, NY). Conservative regions of interest encompassing approximately 100 cell nuclei were microdissected (energy: 40-48; focus: 71) using the PALM Microbeam system and PALMRobo software 4.3 (Zeiss) and immediately expelled into an AdhesiveCap 500 microfuge tube (Zeiss). RNA isolation was performed immediately as described above, with the RNeasy Micro Plus kit (Qiagen). Genomic DNA was removed using gDNA eliminator columns (Qiagen). Total RNA was amplified and reverse transcribed using the Quantitect Whole Transcriptome Amplification kit (Qiagen). 100 ng cDNA was used for qPCR, according to the methods described above. No differences in relative ß-actin, IL-6Rα and gp130 expression were observed between saline and poly(I:C) samples, and treatment groups were therefore merged for greater statistical power.
Statistical analysis
Statistical analysis was performed with Prism software (Graphpad Software; La Jolla, CA). The statistical significance of differences between two treatment groups was assessed using the Student's t-test, and differences among multiple groups was assessed using one-way ANOVA followed by Bonferroni post-hoc tests. For PPI, 5 db pre-pulse and 15 db pre-pulse data from the same groups of mice were analyzed using two-way ANOVA.
Results
Maternal IL-6 exposure yields offspring with behavioral abnormalities similar to those seen in MIA offspring
In the mouse MIA model, it was previously shown that the cytokine IL-6 is necessary and that a single IL-6 injection is sufficient for the development of behavioral and transcriptional changes in the offspring (Smith et al., 2007). Here we confirm and extend those data. The offspring of pregnant mice injected with 5 μg of rIL-6 on E12.5 display decreased PPI compared to control offspring (Fig. 1A). Schizophrenic (Wynn et al., 2004) and autistic (Perry et al., 2006) individuals also exhibit abnormal PPI. PPI is a measure of sensorimotor gating, attention and distractibility. It refers to the inhibition of a startle response when the startling stimulus (120 db pulse) is preceded by a smaller, non-startling stimulus (5 or 15 db pre-pulse).
Fig. 1. Maternal IL-6 exposure yields offspring with behavioral abnormalities analogous to those seen in MIA offspring.

A. Compared to controls, offspring of mice treated with rIL-6 or poly(I:C) display a PPI deficit [F(2, 74)=4.40, *p < 0.05; n=10 saline, 10 poly(I:C), 19 rIL-6 offspring]. B. rIL-6 and poly(I:C) offspring display increased freezing in response to the conditioned acoustic cue during LI testing, as well as decreased LI compared to saline controls when measured against the non-pre-exposed (NPE) group [% freezing = 64.49 ± 6.05 (mean ± SEM)] [F(2, 41)=3.261, *p < 0.05; n=13 saline, 14 poly(I:C), 17 rIL-6 offspring]. NPE poly(I:C) and saline offspring display no significant difference in freezing response. C. Compared to controls, offspring of rIL-6 or poly(I:C)-injected mothers exhibit fewer entries into the center of the open field [F(2, 92)=8.596; p < 0.0005; n=36 saline, 36 poly(I:C), 23 rIL-6 offspring] and shorter duration spent in the center field [F(2, 92)=3.140; *p < 0.05]. rIL-6 offspring also present shorter total distance traveled in the open field [F(2, 92)=12.81; p < 0.0001].
LI is a measure of the ability to disregard irrelevant stimuli and refers to the inhibited acquisition of a conditioned stimulus when a subject has been exposed to the stimulus repeatedly prior to pairing with an unconditioned response. The disruption of LI seen in MIA and rIL-6 offspring mimics that which is characteristic of cognitive deficits in schizophrenia (Weiner, 2003). Adult offspring of rIL-6-injected mothers display elevated freezing compared to saline controls in response to the conditioned acoustic cue. This is indicative of decreased LI compared to the non-pre-exposed (NPE) group (Fig. 1B). A similar deficit is observed in poly(I:C) offspring compared to controls.
Offspring of rIL-6-injected mice exhibit decreased exploration of the open field (Fig. 1C). Compared to controls, rIL-6 offspring display fewer center entries, decreased center duration and decreased total distance traveled. Reluctance to enter the center area of an open arena is indicative of heightened anxiety under mildly stressful conditions. The decreased exploration exhibited by MIA and rIL-6 offspring is relevant to schizophrenia- and autism-related anxiety-like behavior.
Together, these data indicate that a single exposure to rIL-6 at mid-gestation causes offspring to develop autism- and schizophrenia-related behavioral abnormalities similar to those induced by maternal injection of poly(I:C). Previous studies involving the injection of poly(I:C) into IL-6 KO mice, or the co-injection of poly(I:C) with blocking antibody against IL-6, demonstrated that IL-6 is necessary for mediating the effect of MIA on the development of behavioral abnormalities in offspring (Smith et al., 2007). Therefore, in order to elucidate the mechanism by which the maternal immune response affects fetal brain development, we focus on the IL-6 signaling pathway.
The placenta is a site of IL-6 induction in response to MIA
To explore how an immune challenge that is administered to the mother can lead to changes in the development of offspring, we examined the placenta as the site of direct contact between the maternal and fetal systems. Shortly after poly(I:C) injection, pro-inflammatory cytokines including IL-6 are up-regulated in the maternal circulation (Gilmore et al., 2005; Koga et al., 2009; Meyer et al., 2006, 2008a; Smith et al., 2007). This signal is transmitted to the placenta, where levels of IL-6 and other pro-inflammatory cytokines are elevated. A striking increase in IL-6 protein is seen in the placenta by 3 hours post-poly(I:C) injection; poly(I:C) placentas exhibit an approximately 17-fold increase over saline injection controls (Fig. 2A). IL-6 levels remain elevated for over 24 hours after MIA, at levels over 4 times greater than in placentas from saline-injected mothers. Moreover, maternal poly(I:C) injection leads to a 16-fold up-regulation of placental IL-6 mRNA expression by 3 hours post-poly(I:C) injection (Fig. 2B). Tumor necrosis factor α (TNFα) and IL-1β expression are up-regulated in poly(I:C) placentas as well, approximately 3-fold over the level in the saline control. A trend for increases is also observed for placental expression of IL-17 and IL-10 in MIA placentas. However, there is no significant difference in expression of leukemia inhibitory factor (LIF), and interferon γ (IFNγ) is not detected in poly(I:C) and saline placentas.
Fig. 2. MIA upregulates maternally-derived IL-6 in the placenta.

A. Compared to controls, placentas from poly(I:C)-injected mothers exhibit increased IL-6 protein at 3, 6 and 24 hours post-injection [n= 9 placentas per treatment per time point (pooled from 3 independent litters); * p < 0.05, ** p < 0.001]. B. At 3 hours post-injection, compared to saline controls, placentas from poly(I:C)-injected mothers also display increased mRNA expression of pro-inflammatory cytokines, including a markedly elevated level of IL-6 [n=9 placentas per treatment group (pooled from 3 independent litters); ***p < 0.0001]. C. Eliminating the contribution of fetally-derived IL-6 has no effect on total placental IL-6 levels, whereas eliminating the contribution of maternally-derived IL-6 completely diminishes total placental IL-6 to levels below those seen in saline controls at both 3 hours and 6 hours post-injection. This indicates that basal and MIA-induced placental IL-6 is maternally-derived. [F(3,80)=24.89; ***p < 0.0001; n= 6-15 placentas per treatment group per genotype pair (pooled from 3-6 litters)].
Increased levels of placental IL-6 are maternally-derived
Elevated IL-6 protein in MIA placentas can come from the mother (e.g. bloodstream, decidual cells, uterine immune cells) and/or the fetus (e.g. bloodstream, trophoblasts, endothelial cells). To explore the relative contribution of the maternal and fetal pools of IL-6, we crossed IL-6 +/- females with IL-6 -/- males, and injected pregnant females with either poly(I:C) or saline on E12.5. This creates a system to assay the role of IL-6 production from the fetal compartment: pregnant mothers are able to produce IL-6, whereas approximately one-half of the offspring (and the cells that comprise the spongiotrophoblast and labyrinth) will be null for the IL-6 allele, and thus incapable of generating IL-6 transcript. The remaining heterozygous conceptuses serve as internal controls. We also mated IL-6 -/-females with IL-6 +/- males to test the necessity of maternal/decidual IL-6 production (and sufficiency of fetal IL-6 production) in mediating the effects of MIA on embryonic development. It is important to note that in this latter paradigm, the decidua (the superficial layer of the placenta) and cells that fill the maternal blood spaces within the spongiotrophoblast and labyrinth, will still be of maternal genotype.
Injection of poly(I:C) into pregnant IL-6 +/- females induces the expected MIA response that is characterized by elevated placental IL-6 levels by 3 hours post-injection (Fig. 2C). Note that, compared to WT, IL-6 heterozygous mice display a weaker and more transient MIA response at 3 hours post-injection; placental IL-6 levels are increased ∼5-fold over controls, while a ∼17-fold induction is seen in WT mice. Also, the placental IL-6 induction lasts between 6 and 24 hours in heterozygous mice, but lasts over 24 hours in WT mice. This indicates that maternal IL-6 gene dosage affects the IL-6 response to MIA in the placenta.
Fetal or trophoblast expression of IL-6 contributes little to the pool of IL-6 protein, as there is no significant difference between IL-6 levels in IL-6 -/- placentas from +/-mothers compared to IL-6 +/- placentas from +/- mothers (Fig. 2C). Negligible levels of IL-6 are detected in IL-6 +/- and -/- placentas derived from saline or poly(I:C)-injected IL-6 -/- mothers (Fig. S1). Thus, the IL-6 protein response to MIA in the placenta is maternally-derived.
MIA activates uterine NK cells, granulocytes and macrophages that may contribute maternal IL-6 in the placenta
The finding that the placenta exhibits elevated levels of both IL-6 protein and mRNA in response to MIA suggests that placental IL-6 is supplied by not only by the maternal bloodstream, but also by the maternal cells within the placenta. We examined decidual immune cells as candidate sources of maternal IL-6 production. The decidua is the lining of the placenta that contains maternal vasculature and leukocytes, including a distinctive lymphocyte population of uterine NK (uNK) cells. By flow cytometry we assessed immune subtypes in the placenta for expression of CD69, a surface glycoprotein acquired during early lymphoid activation and inducibly expressed by T cells, B cells, NK cells, monocytes, neutrophils and eosinophils.
Compared to saline controls, decidual cell suspensions generated from poly(I:C) injected mothers exhibit elevated CD69 expression in DBA lectin+ uNK cells, CD11b+ macrophages and Gr-1+ granulocytes (Fig. 3A,B). Similarly activated cells are observed in decidual cell suspensions from IL-6 -/- mothers injected with poly(I:C), indicating that initial activation of placental immune cells occurs independently of IL-6 action. The increase in CD69+ innate immune cells coincides with an overall increase in placental CD69 expression (Fig. 3C). There is no significant difference in the percentages of these immune subtypes in response to MIA (Fig. S2). Furthermore, there is no apparent difference in localization of uNK or macrophage cells, as demonstrated by immunofluorescence staining with DBA lectin and F4/80 (data not shown). Because CD4+, CD8+ and B220+ cells represent minor lymphocyte populations in the E12.5 placenta, low levels of these cells are detected in saline and poly(I:C) decidual cell suspensions (Fig. S2). The significant increase in activated maternal leukocytes and lymphocytes in MIA placentas suggests that decidual uNK cells, macrophages and granulocytes are a likely source of maternally-derived IL-6 in response to MIA.
Fig. 3. MIA activates decidual leukocytes to express CD69.
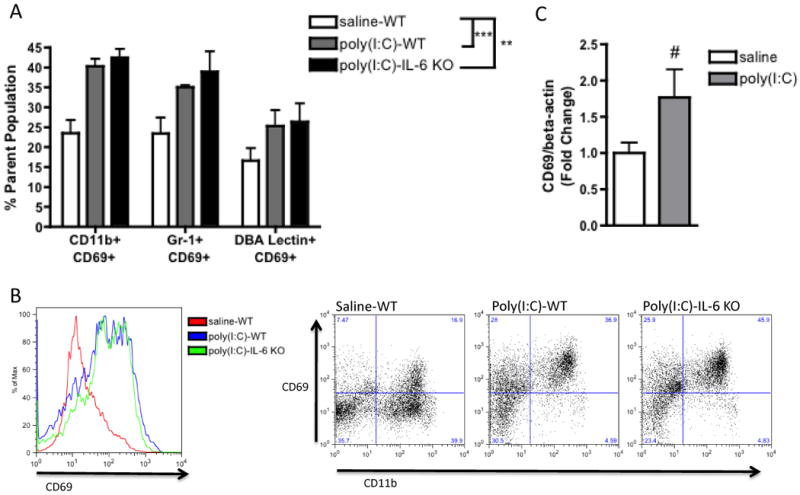
A. Compared to controls, placentas from poly(I:C)-injected mothers harbor increased numbers of CD69-expressing CD11b+, Gr-1+ and DBA lectin+ cells [F(1,12)=24.76, ***p=0.0003; n=5 litters per treatment group (7-8 deciduas pooled per litter)]. These are similarly increased in poly(I:C)-injected IL-6-/- mothers, suggesting that this effect occurs independently of IL-6 action [F(1,12)=21.87, **p=0.0005]. B. Representative flow cytometry spectra show increased total CD69+ cells in poly(I:C) decidual suspensions (left) and increased CD69+ CD11b+ cells from a non-erythroid parent population (right) C. Increased activation of decidual leukocytes corresponds with a trending increase in total CD69+ gene expression (# p = 0.08; n=9 placentas per treatment group (pooled from 3 independent litters)
Maternal IL-6 activates the JAK/STAT pathway in the fetal compartment of the placenta
In order to localize the site(s) of IL-6 action within the placenta, we searched for downstream markers of IL-6 signaling. IL-6 and other IL-6 family cytokines, such as LIF, IL-11 and ciliary neurotrophic factor, signal by binding cytokine-specific membrane receptors to trigger further signal transduction: intracellular JAK proteins dimerize and auto-phosphorylate to activate downstream kinase activity, which ultimately leads to the phosphorylation of STAT transcription factors and global changes in gene expression, including increased transcription of the acute phase genes SOCS3, Pim1, TIMP1 and NOS2. In particular, IL-6 is known to signal through STAT3 and STAT1. We performed histological staining for phosphorylated (p)STAT3 and pSTAT1 in WT mice injected with poly(I:C) or saline, and also assayed gene expression of downstream markers of IL-6 activity. To evaluate whether changes were initiated specifically by IL-6, we assessed STAT activation and downstream gene expression in IL-6 +/- or -/- placentas from poly(I:C) or saline-injected IL-6 +/- or -/- mothers.
Unlike saline controls, STAT3 is phosphorylated in the poly(I:C) spongiotrophoblast layer (Fig. 4) at 3 and 6 hours post-injection. This region of the placenta is comprised of a layer of fetal trophoblast cells that provides both structural support and hormones to maternal and fetal tissues. Widespread pSTAT3 staining is seen in IL-6 +/- and -/- spongiotrophoblasts from IL-6 +/- mothers injected with poly(I:C). This staining is, however, absent in IL-6 +/- and -/- spongiotrophoblasts from IL-6 -/-mothers, further confirming a specific effect of maternally-derived IL-6 on STAT3 activation in the placenta. There is no significant difference in the number of pSTAT3-positive cells in IL-6 +/- versus -/- spongiotrophoblasts from +/- mothers, indicating a negligible effect of fetally-derived IL-6 on STAT3 activation (Fig. S3-A). No pSTAT1 staining is detected in the spongiotrophoblast layer of poly(I:C) or saline placentas (Fig. S3-B). Together, these data indicate that maternal IL-6 plays a specific role in mediating gene expression changes through STAT3 activation in the spongiotrophoblast layer of MIA placentas.
Fig. 4. Maternally-derived IL-6 activates the JAK/STAT3 pathway in the fetal placental compartment in response to MIA.
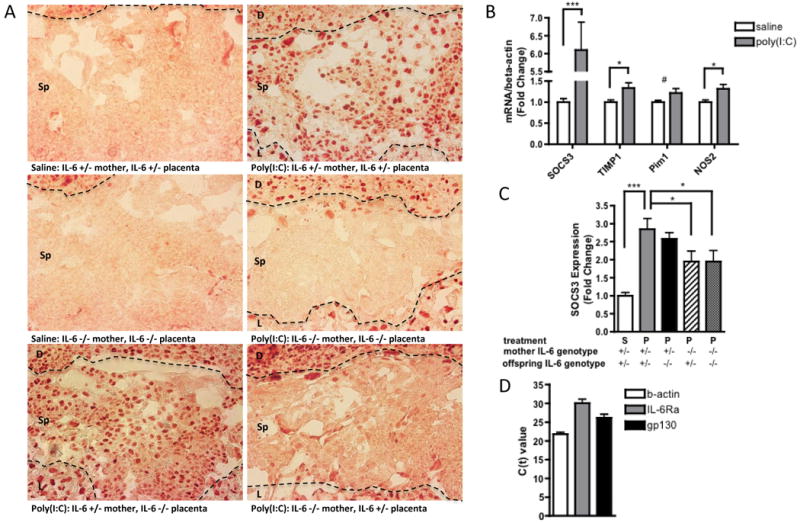
A. Representative sections of the spongiotrophoblast layer of the placenta (with decidua along the top edge and labyrinth along the bottom edge of each image) show positive pSTAT3 staining when placentas are isolated from poly(I:C)-injected mothers. Eliminating the contribution of fetally-derived IL-6 has no effect on pSTAT3 staining, while eliminating the contribution of maternally-derived IL-6 completely abrogates pSTAT3 staining in the spongiotrophoblast. [n=5-7 placentas per treatment group per genotype pair (pooled from 3-5 independent litters)]. Dotted lines = boundary between decidua and spongiotrophoblast layers (upper) and boundary between spongiotrophoblast and labyrinth (lower); D=decidua, Sp=spongiotrophoblast, L=labyrinth; B. MIA-induced JAK/STAT3 activation is accompanied by increased expression of the downstream acute phase genes, SOCS3, TIMP1, Pim1 and NOS2, in poly(I:C) placentas [n=9 placentas per treatment group (pooled from 3 independent litters); ***p < 0.0001, *p < 0.05, # p = 0.07]. C. Eliminating the contribution of maternally-derived IL-6 reduces placental SOCS3 induction [n=9 placentas per treatment group per genotype pair (pooled from 3 independent litters); ***p < 0.0001, *p < 0.05]. D. Murine spongiotrophoblast cells express IL-6Rα and gp130 mRNA, indicating that they may respond directly to placental IL-6 activation. [n=3 saline, 3 poly(I:C} placentas; merged].
MIA-induced activation of the STAT3 pathway coincides with a strong increase in SOCS3 expression in placentas from poly(I:C)-injected mothers (Fig. 4B). Expression of Pim1, TIMP1 and NOS2 is also increased in MIA placentas. Increased SOCS3 expression is similarly observed in IL-6 +/- placentas from poly(I:C)-injected IL-6 +/-mothers (Fig. 4C). Note that the up-regulation of SOCS3 expression in IL-6 heterozygous MIA placentas is significantly attenuated compared to that in IL-6 WT placentas. This effect is similarly observed with MIA-induced IL-6 expression, as described above, and provides further evidence that maternal IL-6 gene dosage regulates the intensity and duration of the placental pro-inflammatory response to poly(I:C). Moreover, eliminating the contribution of maternal IL-6 significantly decreases the level of SOCS3 expression in MIA placentas, while eliminating the contribution of fetal/placental IL-6 has no significant effect. These data indicate that MIA leads to increases in maternally-derived IL-6 and subsequent downstream action in the placenta.
To examine whether maternally-derived IL-6 may act directly on spongiotrophoblasts to activate the JAK/STAT3 pathway, we used laser capture microdissection of the spongiotrophoblast layer and qRT-PCR to test for expression of IL-6 receptor components. On its target cells, IL-6 binds to its membrane-bound receptor, IL-6Rα, which associates with the signaling subunit of the receptor, gp130, to initiate intracellular signal transduction. Spongiotrophoblast cells from placentas of both saline and poly(I:C)-injected mothers indeed express IL-6Rα as well as gp130 (Fig. 4D) and are therefore likely to be capable of responding to placental IL-6. This offers the potential for direct IL-6 signaling in spongiotrophoblast cells and subsequent activation of STAT3.
In addition to IL-6, other factors also mediate STAT3 and STAT1 activation in the placenta after MIA. Both pSTAT3 and pSTAT1 staining are localized to parietal trophoblast giant cells (pTGCs) and mononuclear sinusoidal trophoblast giant cells (sTGCs) that reside in the junctional zone and labyrinth (Fig. S3-B,C). This staining is present with the same localization and abundance in placentas from WT and IL-6 KO mice injected with poly(I:C), suggesting that MIA-induced pro-inflammatory factors other than IL-6 mediate this effect. Furthermore, the staining is not blocked by co-injection of anti-IL-6 antibody and poly(I:C) in pregnant mothers (data not shown). Thus, MIA promotes widespread STAT3 and STAT1 activation in the fetal compartments of the placenta.
IL-6 specific signaling in response to MIA alters placental expression of factors that can influence fetal development
The finding that IL-6 mediates STAT3 activation specifically in the spongiotrophoblasts inhabiting the fetal compartment of MIA placentas suggests that IL-6 may affect offspring development by altering expression of critical placental hormones and growth factors. In order to explore the downstream effects of MIA-induced STAT3 activation in the placenta, we conducted real time qRT-PCR for relevant placental factors using WT placentas from poly(I:C)- or saline-injected mothers at 3 hours post-injection, a time at which pro-inflammatory cytokines are highly up-regulated. To assess the functional relevance of IL-6-mediated signaling in the placenta, we conducted similar real time qRT-PCR assays utilizing placentas from pregnant IL-6 KO mice injected with poly(I:C).
Maternal poly(I:C) injection leads to disruption of the GH-IGF axis in the placenta. Levels of GH protein are significantly reduced in MIA placentas (Fig. 5A-D), and there is also a corresponding decrease in IGFI expression, suggesting that reduced levels of GH may lead to decreased stimulation of IGFI production in the placenta (Forbes & Westwood, 2008). Furthermore, compared to saline-injected controls, placentas from poly(I:C)-injected mothers exhibit decreased IGFBP3 expression. IGFBP3 is a primary carrier binding protein responsible for increasing the half-life of IGFI, and its reduction is often associated with lower IGFI levels (Donahue & Beamer, 1993; De Benedetti, 2001; Liao et al., 2008). Reductions in both IGFI and IGFBP3 are absent in placentas from IL-6 -/- mice injected with poly(I:C). In fact, placentas from MIA IL-6 KO mice exhibit increased expression of IGFI and IGFBP3, suggesting that basal levels of IL-6 are also critical for regulation of IGFI and IGFBP3 expression. A similar effect is seen with levels of placental IGFI protein, where WT MIA placentas display a decreased concentration of IGFI compared to saline controls, while IL-6 KO MIA placentas display higher levels of IGFI. This provides further evidence that both basal IL-6 and MIA-induced IL-6 in the placenta play a role in suppressing IGFI and IGFBP3 production.
Fig. 5. MIA-induced IL-6 action reduces placental IGFI and IGFBP3.
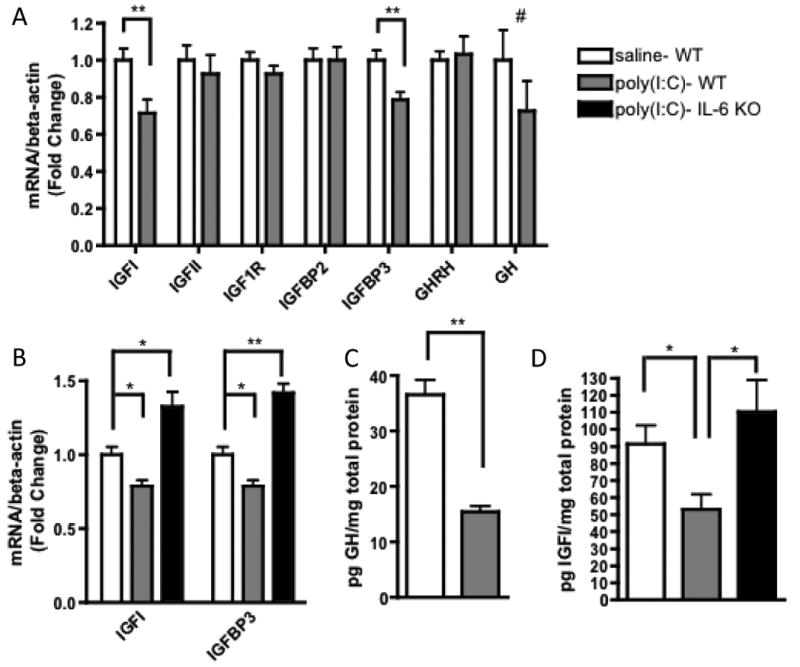
A. Compared to saline controls, placentas from poly(I:C)-injected mothers have significantly reduced expression of IGFI and IGFBP3 mRNA and a trending decrease in GH expression [n=9 placentas per treatment group (pooled from 3 independent litters); **p < 0.001, # p = 0.24]. B. The deficits in IGFI and IGFBP3 expression observed in MIA placentas require IL-6 production [IGFI: F(2,23)=15.95; *p < 0.05; IGFBP3: F(2,23)=37.77; **p < 0.001; n=8-9 placentas per treatment group (pooled from 3 independent litters]. C. MIA placentas exhibit significantly decreased GH protein [n=7 placentas per treatment group (pooled from 3 independent litters); **p < 0.001]. D. MIA placentas exhibit significantly decreased IGFI protein, and this deficiency is dependent on IL-6 production [F(2, 17)=5.908; *p < 0.05; n=5-7 placentas per treatment group (pooled from 3 independent litters)].
There are no differences between placentas from saline- or poly(I:C)-injected mothers in the levels of GHRH, IGFII, IGF1R and IGFBP2, an inhibitory IGFI binding protein (Fig. 5A). Thus, MIA selectively reduces levels of placental GH and IGFI, two important hormones responsible for promoting embryonic development.
MIA also alters the expression of placental lactogen (PL) and pro-lactin-like proteins (PLPs) in the placenta. The PL and PLPs comprise families of prominent placenta-specific factors that regulate pregnancy, placental physiology and fetal development (Soares, 2004). Maternal poly(I:C) injection up-regulates placental PLP-K expression at 3 hours post-injection and this effect is abrogated in placentas from poly(I:C)-injected IL-6 -/- mice (Fig. 6A, B). PLP-K is a placenta-specific protein that is widely expressed in the sTGCs of the labyrinth and in the spongiotrophoblast layer (Wiemers et al., 2003), an expression pattern that correlates precisely with IL-6-induced STAT3 activation in the placenta. Taken together, these data suggest that the activation of STAT3 driven by maternal IL-6 in response to MIA is responsible for the observed elevation in PLP-K expression. In addition, there is a trend towards increased expression of placental growth factor (PGF), a key a protein in angiogenesis and vasculogenesis in the placenta; placental lactogen 2 (PLII), a lactogen regulated by the inhibitory control of GH; as well as PLP-F (Fig. 6A), a pro-lactin-like protein known to regulate hematopoiesis (Wiemers et al., 2003). No significant difference is seen in expression of PLI or PLP-E between saline and poly(I:C) placentas. Thus, MIA-induced IL-6 regulates expression of placenta-specific hormones that influence the maternal response to pregnancy and the regulation of fetal growth.
Fig. 6. MIA-induced IL-6 action increases PLP-K and related growth factor gene expression.
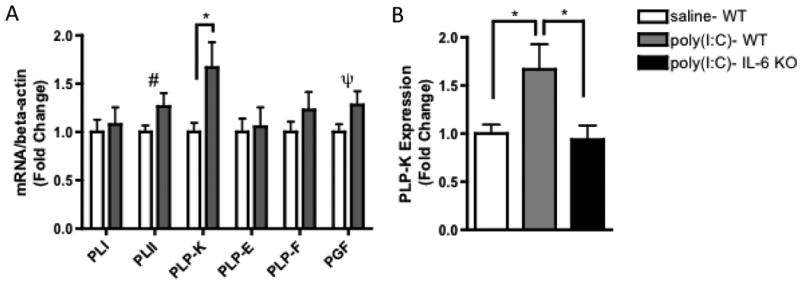
A. Placentas from poly(I:C)-injected mothers have significantly elevated expression of PLP-K and trending increases in PGF and PLII [n=9 per treatment group (pooled from 3 independent litters); *p < 0.05, # p = 0.10, ψ p = 0.11]. B. The upregulation of PLP-K expression observed in MIA placentas require IL-6 action [F(2,23)=5.033; *p < 0.05].
Discussion
The finding that IL-6 is critical for mediating the effects of MIA on the development of schizophrenia- and autism-related endophenotypes in the mouse model offers the opportunity to trace the molecular and cellular pathways by which maternal infection increases risk for these neurodevelopmental disorders. We show that a single injection of rIL-6 into pregnant mice yields offspring with PPI, LI and open field deficits that are similar to those seen in MIA offspring. Although there is no significant difference between poly(I:C) and rIL-6 offspring in PPI, LI or open field performance, rIL-6 offspring trend toward more severe behavioral deficits in all three tests. This may be attributed to the dosage of IL-6 administered (5 μg) compared to levels reported on the order of 10-20 ng IL-6 per mL of maternal serum at 3 hours post-poly(I:C) injection (Meyer et al., 2006). In addition, poly(I:C), compared to rIL-6 injection, leads to TLR3 activation and mounting of a complex immune response involving production of pro-inflammatory and anti-inflammatory cytokines. IL-10, for example, is upregulated in placentas shortly after poly(I:C) injection (Fig. 2B), and is traditionally known to regulate the effects of proinflammatory cytokines such as IL-6 and IFNγ. Overall, the fact that maternal rIL-6 injection yields offspring with behavioral deficits that are equal, if not more severe, than those seen in MIA offspring demonstrates the importance of IL-6 in mediating neural development and adult behavioral outcome.
We find that maternally-derived IL-6 accounts for the increased pool of IL-6 in placentas from poly(I:C)-injected mothers. Potential sources of maternally-derived IL-6 in the placenta include circulating IL-6 in the maternal bloodstream and IL-6 that is secreted by maternal cells that reside in the placenta, such as decidual immune cells, stromal cells and endothelial cells. It is clear that maternal poly(I:C) injection increases the level of IL-6 and other pro-inflammatory cytokines in the maternal circulation (Gilmore et al., 2005; Koga et al., 2009; Meyer et al., 2006; Smith et al., 2007). However, our finding that IL-6 mRNA expression is dramatically increased in MIA placentas suggests that resident placental cells of maternal origin may also be a source of increased placental IL-6. Furthermore, we show that MIA increases the number of CD69-expressing decidual macrophages, granulocytes and uNK cells in poly(I:C) placentas. CD69 is a marker of an early activation response, and its expression is associated with production of pro-inflammatory cytokines including IL-6 (Saito, 2000). Thus, following MIA, activated decidual leukocytes may contribute to the induction of IL-6 in the placenta (Lockwood et al., 2008). Some studies report that maternal stromal cells and endothelial cells in the placenta can also generate cytokines in response to other types of immune activation (Montes et al., 1995; Semer et al., 1991). Interestingly, in the CBA × DBA/2 model of miscarriage, early maternal poly(I:C) injection leads to increased expression of CD69, IFNγ and TNFα by uNK cells (Zhang et al., 2007). Altogether, these findings demonstrate that the MIA response is relayed to the maternal cells in the placenta, leading to increased levels of maternally-derived IL-6 directly at the materno-fetal interface.
Interestingly, compared to saline-injected controls, IL-6 protein is increased in amniotic fluid (AF) from poly(I:C)-injected mothers (Mandel et al., 2010). While the importance of the fetal placental component was stressed as the source of AF IL-6, it appears that much of the AF IL-6 is dependent on maternal IL-6 production. Eliminating the contribution of maternal IL-6 in poly(I:C)-injected mice decreases the concentration of AF IL-6 by over 14-fold compared to that observed in WT poly(I:C)-injected mice, and brings the final AF IL-6 concentration to a level lower than that observed in saline controls (Mandel et al., 2010). This indicates that maternal IL-6 production is critical for MIA-induced increases in AF IL-6. It will be important to compare AF from IL-6 +/- offspring of IL-6 -/- mothers with AF from IL-6 +/- offspring of IL-6 +/- mothers as a control.
We further demonstrate that maternally-derived IL-6 is responsible for activation of the JAK/STAT3 pathway specifically in the spongiotrophoblast layer, a fetal compartment of the placenta. It is likely that this STAT3 activation occurs by a direct effect of maternally-derived IL-6 on spongiotrophoblast cells, since we find that they express IL-6Rα and gp130. Moreover, paracrine signaling is a well-established mechanism of materno-fetal communication between distinct placental layers (Bischof et al., 2000; Hess et al., 2006; Lacey et al., 2002; Petraglia et al., 1996). The spatially-localized activation of STAT3 at the junctional zone of the placenta suggests that MIA induces maternally-derived IL-6 in the decidual layer (Fig. 7), which then signals in a paracrine manner to fetal spongiotrophoblast cells. It is important to note that, while a direct effect of IL-6 on placental cells is likely, we have not excluded the possibility that IL-6 may act as an upstream, indirect mediator of MIA effects on the placenta, and that IL-6 may act by so-called trans-signaling, or binding soluble IL-6Rα before complexing with membrane-bound gp130. Nonetheless, the maternal IL-6-dependent activation of the STAT3 pathway in spongiotrophoblast cells demonstrates a direct transfer of the MIA response from maternal to fetal cells.
Fig. 7. Model of MIA-induced IL-6 effects on the placenta.
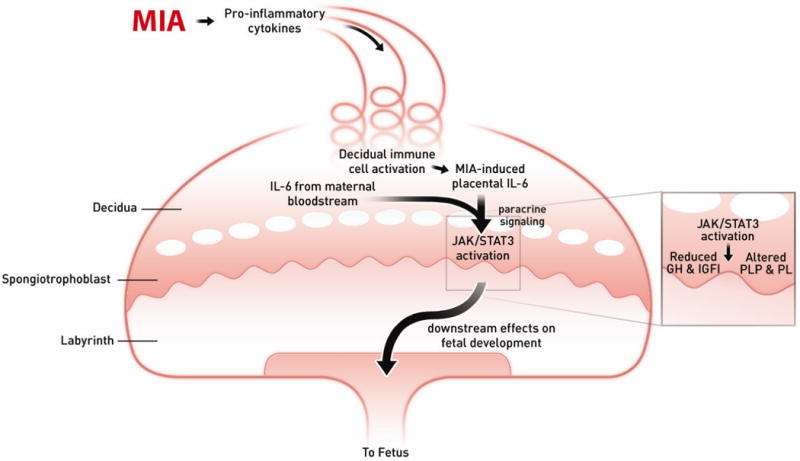
Maternal injection of poly(I:C) leads to the activation of the maternal immune system via a TLR3-mediated anti-viral response. Pro-inflammatory factors, including IL-6, are secreted by activated TLR3+ cells into the maternal bloodstream. As maternal blood circulates continuously through the placenta, IL-6 and soluble pro-inflammatory factors increase in the spiral arteries that descend through the decidua and spongiotrophoblast, as well as in the maternal blood spaces of the labyrinth, and circulate back up into the maternal compartment. Resident immune cells in the decidua are activated by maternal cytokines and other signaling factors to express CD69, and they propagate the inflammatory response by further cytokine release. IL-6 derived from decidual cells acts in a paracrine manner on target cells in the spongiotrophoblast layer. Ligation with the cognate receptor IL-6Ra and gp130 leads to signal transduction resulting in JAK/STAT3 activation and downstream changes in gene expression. Increases in acute phase proteins, such as SOCS3, down-regulate placental GH production and signaling. This leads to reduced IGFBP3 and IGFI. Global changes in STAT3 activation in the spongiotrophoblast layer alter the production of placenta-specific PLP and pro-lactin proteins. These changes in endocrine factors lead to acute placental pathophysiology and subsequent effects on fetal development.
Spongiotrophoblast cells arise just prior to mid-gestation and are a prominent source of endocrine factors throughout the latter half of gestation (Soares, 2004). We find an IL-6-dependent elevation in placental PLP-K expression after MIA, along with mild increases in several other pro-lactogen and pro-lactin-like-protein family members. As a recently discovered PLP, PLP-K is characterized as a placenta-specific protein produced primarily by spongiotrophoblast cells and also by sTGCs (Wiemers et al., 2003). The correlation of maternal-IL-6-dependent STAT3 activation in the spongiotrophoblast with maternal-IL-6-dependent up-regulation of PLP-K suggests that modulation of PLP-K expression lies downstream of STAT3 activity. While its function is unclear, PLP-K possesses structural similarity to proliferin (PLF), a protein that binds mannose-6/IGFII complexes to regulate vasculogenesis and trophoblast proliferation. Thus, we demonstrate that MIA-induced maternal IL-6 alters expression of PLP-K and potentially other genes that encode endocrine or paracrine modulators of maternal and fetal cells.
Furthermore, we find an IL-6-dependent dysregulation of the GH-IGF axis in MIA placentas, characterized by decreased levels of GH and IGFI mRNA, with corresponding decreases in placental IGFI and IGFBP3 protein. The actions of GH are achieved through the stimulation of IGFI production in target tissues. In addition, GH regulates the activity of IGFI by altering the production of either facilitatory or inhibitory binding proteins, including the IGFI stabilizing protein, IGFBP3. This suggests that the decreased GH levels seen in MIA placentas leads to the observed downstream suppression of IGFBP3 and IGFI production. It is believed that IGFs in the maternal circulation do not enter the placenta, and therefore IGFs in the placenta are derived from the placental compartment itself (Kanai-Azuma et al., 1993).
We demonstrate that the changes in IGFI and IGFBP3 expression are mediated by IL-6. However, it is unclear whether decreases in placental GH and subsequent effects on IGF production are downstream of IL-6-specific STAT3 activation. IL-6 does modulate IGFI and IGFBPs in several tissues, including placenta and cord blood (De Benedetti et al., 2001; Street et al., 2009). Pro-inflammatory cytokines, including IL-6, decrease circulating and tissue concentrations of GH and IGFI (Lang et al., 2005). We observe that IL-6-mediated STAT3 activation is associated with the expected IL-6-mediated increase in SOCS3 expression, along with other acute phase genes. Factors like SOCS play an important role in the down-regulation of GH and GH signaling (Herrington et al., 2000; Lang et al., 2005). Importantly, it is reported that IL-6 inhibits hepatic GH signaling through up-regulation of SOCS3 (Denson et al., 2003). As such, it is possible that, in MIA placentas, maternal IL-6-induced STAT3 activation and downstream sequelae lead to suppression of placental GH levels, disruption of IGFI production and further consequences on maternal physiology, placental function and fetal development.
Altered placental physiology and release of deleterious mediators to the fetus are important risk factors for the pathogenesis of neurodevelopmental disorders. Placental IGFI in particular regulates trophoblast function (Forbes & Westwood, 2008), nutrient partitioning and placental efficiency (Fowden et al., 2009). Moreover, altered IGFI levels are associated with intrauterine growth restriction (IUGR) and abnormal development (Crossey et al., 2002; Laviola et al., 2008). Animal models of IUGR and intrauterine infection, where the immune insult is confined to the uteroplacental compartment, highlight the key role of placental inflammation in perinatal brain damage, involving altered cortical astrocyte development (Bassan et al., 2010), white-matter damage (reviewed by Dammann & Leviton, 1997), microglial activation, cell death (Hutton et al., 2008) and reduced effectiveness of the fetal blood-brain-barrier (Yan et al., 2004). In addition, adult pathophysiology is subject to feto-placental “programming”, wherein molecular changes that occur prenatally reflect permanent changes that persist throughout postnatal life (Bilbo & Schwarz, 2009; Barker et al., 2010a, 2010b; Frost & Moore, 2010; Merlot et al., 2008; Seckl & Holmes, 2007; Zhang et al., 2005). Interestingly, placental responses to maternal insults can potentiate sexually dimorphic effects on fetal development (Clifton, 2005; Mueller & Bale, 2008).
Obstetric complications are linked to schizophrenia risk (reviewed by Preti et al., 2000) and to the treatment responses of schizophrenic individuals (Alivir et al., 1999). Notably, a greater occurrence of placental trophoblast inclusions was observed in placental tissue from children who develop autism spectrum disorder (ASD) compared to non-ASD controls (Anderson et al., 2007). Chorioamnionitis and other obstetric complications are significantly associated with socialization and communication deficitis in autistic infants (Limperopoulos, 2008). The characterization of placental pathophysiology and obstetric outcome in ASD and schizophrenic individuals will be useful for the identification of molecular mechanisms that underlie these disorders and for potential biomarkers for early risk diagnosis.
In addition to the observed effects of IL-6 on placental physiology and its downstream effects on fetal brain development and postnatal growth, direct effects of IL-6 on the fetal brain are also likely. Maternal IL-6 can potentially cross the placenta and enter the fetus after MIA (Dahlgren et al., 2006). Furthermore, IL-6 mRNA and protein are elevated and STAT3 is phosphorylated in the fetal brain itself following MIA (Gilmore et al., 2005; Hsiao & Patterson, 2009; Meyer et al., 2006), raising the obvious possibility that IL-6 acts directly on the developing brain to influence astrogliosis, neurogenesis, microglial activation and/or synaptic pruning (Conroy et al., 2004; reviewed in Deverman & Patterson, 2010; Gilmore et al., 2004). However, recall that the identification of IL-6 as a critical mediator of MIA is based on maternal co-injection of poly(I:C) and anti-IL-6 blocking antibody, in addition to experiments inducing MIA in IL-6 KO animals. As such, in considering which pool(s) of IL-6 (e.g. maternal, placental, fetal brain, fetal periphery) is the “critical mediator”, it will be important to understand the potential interaction between maternal IL-6 and fetal brain IL-6 expression. While we believe that the endocrine changes triggered by maternal-IL-6 signaling in the placenta reported here are important for fetal growth, it will be crucial to assess the potential impact of these placental changes on offspring behavior and neuropathology. We are currently exploring the effects of MIA in targeted IL-6Rα KOs in order to tie tissue- and cell-specific IL-6 activity to the manifestation of schizophrenia- and autism-related endophenotypes.
Supplementary Material
Fig. S1. Fetally-derived IL-6 contributes negligible amounts of IL-6 protein to the placenta after MIA. Placentas were collected to assess levels of placental IL-6 by ELISA. A low-level absorbance signal is observed in IL-6 -/- placentas from IL-6 -/-mothers, indicating a degree of non-specific protein binding during the experimental procedure. All placentas from saline- or poly(I:C)-injected IL-6 -/- mothers exhibit similarly negligible amounts of absorbance signal (n=3-6 placentas per treatment group per genotype pair (pooled from 2-3 litters).
Fig. S2. MIA has no affect on percentages of decidual immune cell subtypes. Compared to controls, placentas from poly(I:C)-injected mothers have no significant difference in the percentages of macrophages, granulocytes, uNK cells, CD4+ T cells, CD8+ T cells or B cells (n=5 litters per treatment group (7-8 deciduas pooled per litter).
Fig. S3. MIA induces phosphorylation of STAT1 and STAT3 in sTGCs and pTGCs in an IL-6-independent manner. A. IL-6 +/- and IL-6 -/- placentas from poly(I:C)-injected IL-6 +/- mothers display no significant difference in the number of pSTAT3-positive spongiotrophoblast cells. Thus, fetally-derived IL-6 contributes little to MIA-induced STAT3 activation. B. Representative images taken at 20× magnification depict saline placenta (left) and MIA placenta (right). Compared to saline controls, MIA placentas (right) stain positively for pSTAT1 in sTGCs and pTGCs (arrows). Dotted lines = boundary between decidua and spongiotrophoblast layers (upper) and boundary between spongiotrophoblast and labyrinth (lower); D=decidua, Sp=spongiotrophoblast, L=labyrinth; C. Representative images taken at 40× magnification depict pSTAT3 staining in saline labyrinth (left) and MIA labyrinth (right). Compared to saline controls, MIA placentas stain positively for pSTAT3 in sTGCs. [n=5-7 placentas per treatment group per genotype pair (pooled from 3-5 independent litters)].
Acknowledgments
The authors acknowledge the kind assistance of B. Deverman and A. Bonnin with reviewing the manuscript; A. Colon, L. Sandoval and R. Sauza with caring for and maintaining the animals; and N. Tetreault and D. Andersen with providing the LMD and qPCR equipment. This research was supported by an Autism Speaks Dennis Weatherstone Pre-Doctoral Fellowship and by a Caltech training grant from the National Institutes of Health (NIH/NRSA 5 T32 GM07737).
Footnotes
Conflict of Interest Statement: All authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alvir JM, Woerner MG, Gunduz H, Degreef G, Lieberman JA. Obstetric complications predict treatment response in first-episode schizophrenia. Psychol Med. 1999;29:621–627. doi: 10.1017/s0033291799008363. [DOI] [PubMed] [Google Scholar]
- Anderson GM, Jacobs-Stannard A, Chawarska K, Volkmar FR, Kliman HJ. Placental trophoblast inclusions in autism spectrum disorder. Biol Psychiatry. 2007;61:487–491. doi: 10.1016/j.biopsych.2006.03.068. [DOI] [PubMed] [Google Scholar]
- Arad M, Atzil S, Shakhar K, Adoni A, Ben-Eliyahu S. Poly I-C induces early embryo loss in f344 rats: a potential role for NK cells. Am J Reprod Immunol. 2005;54:49–53. doi: 10.1111/j.1600-0897.2005.00286.x. [DOI] [PubMed] [Google Scholar]
- Arion D, Unger T, Lewis DA, Levitt P, Mirnics K. Molecular evidence for increased expression of genes related to immune and chaperone function in the prefrontal cortex in schizophrenia. Biol Psychiatry. 2007;62:711–721. doi: 10.1016/j.biopsych.2006.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashwood P, Krakowiak P, Hertz-Picciotto I, Hansen R, Pessah I, Van de Water J. Elevated plasma cytokines in autism spectrum disorders provide evidence of immune dysfunction and are associated with impaired behavioral outcome. Brain, Behav, Imm. 2010 doi: 10.1016/j.bbi.2010.08.003. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atladóttir HO, Thorsen P, Ostergaard L, Schendel DE, Lemcke S, Abdallah M, Parner ET. Maternal Infection Requiring Hospitalization During Pregnancy and Autism Spectrum Disorders. J Autism Dev Disord. 2010 doi: 10.1007/s10803-010-1006-y. [DOI] [PubMed] [Google Scholar]
- Barker DJP, Thornburg KL, Osmond C, Kajantie E, Eriksson JG. The surface area of the placenta and hypertension in the offspring in later life. Int J Dev Biol. 2010a;54:525–530. doi: 10.1387/ijdb.082760db. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker DJP, Thornburg KL, Osmond C, Kajantie E, Eriksson JG. The prenatal origins of lung cancer. II. The Placenta. Am J Hum Biol. 2010b;2:512–516. doi: 10.1002/ajhb.21041. [DOI] [PubMed] [Google Scholar]
- Bassan H, Kildren D, Bassan M, Rotstein M, Kariv N, Giladi E, Davidson A, Gozes I, Harel S. The effects of vascular intrauterine growth retardation on cortical astrocytes. J Matern Fetal Neonatal Med. 2010;23:595–600. doi: 10.1080/14767050903197068. [DOI] [PubMed] [Google Scholar]
- Bilbo SD, Schwarz JM. Early-life programming of later-life brain and behavior: critical role for the immune system. Front Behav Neurosci. 2009;3:1–14. doi: 10.3389/neuro.08.014.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischof P, Meisser A, Campana A. Paracrine and autocrine regulators of trophoblast invasion – a review. Placenta. 2000;21(suppl A):S55–S60. doi: 10.1053/plac.2000.0521. [DOI] [PubMed] [Google Scholar]
- Boksa P. Effects of prenatal infection on brain development and behavior: a review of findings from animal models. Brain Behav Immun. 2010;24:881–897. doi: 10.1016/j.bbi.2010.03.005. [DOI] [PubMed] [Google Scholar]
- Bauer S, Kerr BJ, Patterson PH. The neuropoietic cytokine family in development, plasticity, disease and injury. Nat Rev Neurosci. 2007;8:221–232. doi: 10.1038/nrn2054. [DOI] [PubMed] [Google Scholar]
- Brown AS, Derkits EJ. Prenatal infection and schizophrenia: a review of epidemiological and translational studies. Am J Psychiatry. 2010;167:261–280. doi: 10.1176/appi.ajp.2009.09030361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chez MG, Dowling T, Patel PB, Khanna P, Kominsky M. Elevation of tumor necrosis factor-alpha in cerebrospinal fluid of autistic children. Pediatr Neurol. 2007;36:361–365. doi: 10.1016/j.pediatrneurol.2007.01.012. [DOI] [PubMed] [Google Scholar]
- Clifton VL. Sexually dimorphic effects of maternal asthma during pregnancy on placental glucocorticoid metabolism and fetal growth. Cell Tissue Res. 2005;322:63–71. doi: 10.1007/s00441-005-1117-5. [DOI] [PubMed] [Google Scholar]
- Conroy S, Nguyen V, Quina L, Blakely-Gonzales P, Ur C, Netzeband J, Prieto A. Interleukin-6 produces neuronal loss in developing cerebellar granule neuron cultures. J Neuroimmunol. 2004;155:43–54. doi: 10.1016/j.jneuroim.2004.06.014. [DOI] [PubMed] [Google Scholar]
- Crossey PA, Pillai CC, Miell JP. Altered placental development and intrauterine growth restriction in IGF binding protein-1 transgenic mice. J Clin Invest. 2002;110:411–418. doi: 10.1172/JCI10077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlgren K, Samuelsson AM, Jansson T, Halmong A. Interleukin-6 in the maternal cirulation reaches the rat fetus in mid-gestation. Pediatr Res. 2006;60:147–151. doi: 10.1203/01.pdr.0000230026.74139.18. [DOI] [PubMed] [Google Scholar]
- Dammann O, Leviton A. Maternal intrauterine infection, cytokines, and brain damage in the preterm newborn. Pediatr Res. 1997;42:1–8. doi: 10.1203/00006450-199707000-00001. [DOI] [PubMed] [Google Scholar]
- De Benedetti F, Meazza C, Oliveri M, Pignatti P, Vivarelli M, Alonzi T. Effect of IL-6 on IGF Binding Protein-3: a study in IL-6 transgenic mice and in patients with systemic juvenile idiopathic arthritis. Endocrinology. 2001;142:4818–4826. doi: 10.1210/endo.142.11.8511. [DOI] [PubMed] [Google Scholar]
- Densen LA, Held MA, Menon RK, Frank SJ, Parlow AF, Arnold DL. Interleukin-6 inhibits hepatic growth hormone signaling via upregulation of Cis and Socs-3. Am J Physiol Gastrointest Liver Physiol. 2003;284:G646–G654. doi: 10.1152/ajpgi.00178.2002. [DOI] [PubMed] [Google Scholar]
- Desai TR, Leeper NJ, Hynes KL, Gewertz BL. Interleukin-6 casues endothelial barrier dysrunction via the protein kinase C pathway. J Surg Res. 2002;104:118–123. doi: 10.1006/jsre.2002.6415. [DOI] [PubMed] [Google Scholar]
- Deverman BE, Patterson PH. Cytokines and CNS development. Neuron. 2009;64:61–78. doi: 10.1016/j.neuron.2009.09.002. [DOI] [PubMed] [Google Scholar]
- Donahue LR, Beamer WG. Growth hormone deficiency in “little” mice results in aberrant body composition, reduced insulin-like growth factor-I and insulinlike growth factor binding protein 3, IGFBP3), but does not affect IGFBP-2, -1 or -4. J Endocrinology. 1993;136:91–104. doi: 10.1677/joe.0.1360091. [DOI] [PubMed] [Google Scholar]
- Forbes K, Westwood M. The IGF axis and placental function. Horm Res. 2008;69:129–137. doi: 10.1159/000112585. [DOI] [PubMed] [Google Scholar]
- Fowden AL, Sferruzzi-Perri AN, Coan PM, Constancia M, Burton GJ. Placental efficiency and adaptation: endocrine regulation. Am J Physiol. 2009;587:3459–3472. doi: 10.1113/jphysiol.2009.173013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost JM, Moore GE. The importance of imprinting in the human placenta. PLoS Genet. 2010;6:e1001015. doi: 10.1371/journal.pgen.1001015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbett K, Ebert PJ, Mitchell A, Lintas C, Manzi B, Mirnics K, Persico AM. Immune transcriptome alterations in the temporal cortex of subjects with autism. Neurobiol Dis. 2008;30:303–311. doi: 10.1016/j.nbd.2008.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmore J, Fredrik J, Vadlamudi S, Lauder J. Prenatal infection and risk for schizophrenia: IL-1beta, IL-6, and TNFalpha inhibit cortical neuron dendrite development. Neuropsychopharmacology. 2004;29:1221–1229. doi: 10.1038/sj.npp.1300446. [DOI] [PubMed] [Google Scholar]
- Gilmore JH, Jarskog LF, Vadlamudi S. Maternal poly(I:C) exposure during pregnancy regulates TNFalpha, BDNF, and NGF expression in neonatal brain and the maternal-fetal unit of the rat. J Neuroimmunology. 2005;159:106–112. doi: 10.1016/j.jneuroim.2004.10.008. [DOI] [PubMed] [Google Scholar]
- He F, Ge W, Martinowich K, Becker-Catania S, Coskun V, Zhu W, Wu H, Castro D, Guillemot F, Fan G, de Vellis J, Sun YE. A positive autoregulatory loop of Jak-STAT signaling controls the onset of astrogliogenesis. Nat Neurosci. 2005;8:616–625. doi: 10.1038/nn1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrington J, Smit LS, Schwartz J, Carter-Su C. The role of STAT proteins in growth hormone signaling. Oncogene. 2000;19:2585–2597. doi: 10.1038/sj.onc.1203526. [DOI] [PubMed] [Google Scholar]
- Hess AP, Hamilton AE, Talbi S, Dosiou M, Nyegaard N, Nayak O, Genbecev-Krtolica O, Mavrogianis P, Ferrer K, Fruessel J, Fazleabas AT, Fisher SJ, Giudice LC. Decidual stromal cell response to paracrine signals from the trophoblast: amplifcation of immune and angiogenic modulators. Biol Reprod. 2006;76:102–117. doi: 10.1095/biolreprod.106.054791. [DOI] [PubMed] [Google Scholar]
- Hsiao E, Patterson PH. Maternal immune activation evokes IL-6-dependent downstream signaling in the placenta and fetal brain Program No 436.19. Chicago, IL: Society for Neuroscience; 2009. Online. [Google Scholar]
- Hutton LC, Castillo-Melendez M, Smythe GA, Walker DW. Microglial activation, macrophage infiltration, and evidence of cell death in the fetal brain after ueteroplacental administration of lipopolysaccharide in sheep in later gestation. Am J Obstet Gynecol. 2008;117:e1–11. doi: 10.1016/j.ajog.2007.06.035. [DOI] [PubMed] [Google Scholar]
- Ito HT, Smith SE, Hsiao E, Patterson PH. Maternal immune activation alters nonspatial information processing in the hippocampus of the adult offspring. Brain Behav Immun. 2010;24:930–941. doi: 10.1016/j.bbi.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonakait G. The effects of maternal inflammation on neuronal development: 684 possible mechanisms. Int J Dev Neurosci. 2007;25:415–425. doi: 10.1016/j.ijdevneu.2007.08.017. [DOI] [PubMed] [Google Scholar]
- Jones HN, Jansson T, Powell TL. IL-6 stimulates system A amino acid transporter activity in trophoblast cells through STAT3 and increased expression of SNAT2. Am J Physiol Cell Physiol. 2009;297:C1228–35. doi: 10.1152/ajpcell.00195.2009. [DOI] [PubMed] [Google Scholar]
- Kanai-Azuma M, Kanai Y, Kurohmaru M, Sakai S, Hayashi Y. Insulin-like growth factor-I stimulates proliferation and migration of mouse ectoplacental cone cells, while IGF-II transforms them into trophoblastic giant cells in vitro. Biol Reprod. 1993;48:252–261. doi: 10.1095/biolreprod48.2.252. [DOI] [PubMed] [Google Scholar]
- Kendall G, Peebles D. Acute fetal hypoxia: the modulating effect of infection. Early Hum Dev. 2005;81:27–34. doi: 10.1016/j.earlhumdev.2004.10.012. [DOI] [PubMed] [Google Scholar]
- Koga K, Cardenas I, Aldo P, Abrahams VK, Peng B, Fill S, Romero R, Mor G. Activation of TLR3 in the trophoblast is associated with preterm delivery. Am J Reprod Immunol. 2009;61:192–212. doi: 10.1111/j.1600-0897.2008.00682.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacey H, Haigh T, Westwood M, Aplin JD. Mesenchymally-derived insulin-like growth factor 1provides a paracrine stimulus for trophoblast migration. BMC Dev Biol. 2002;2:5. doi: 10.1186/1471-213X-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang CH, Hong-Brown L, Frost RA. Cytokine inhibition of JAK-STAT signaling: a new mechanism of growth hormone resistance. Pediatr Nephrol. 2005;20:306–312. doi: 10.1007/s00467-004-1607-9. [DOI] [PubMed] [Google Scholar]
- Laviola L, Natalicchio A, Perrini S, Giorgino F. Abnormalities of IGF-I signaling in the pathogenesis of diseases of the bone, brain, and fetoplacental unit in humans. Am J Physiol Endocrinol Metab. 2008;295:E991–E999. doi: 10.1152/ajpendo.90452.2008. [DOI] [PubMed] [Google Scholar]
- Lee KH, Smith SEP, Kim S, Patterson PH, Thompson RF. Maternal immune activation impairs extinction of the conditioned eyeblink response in the adult offspring. Soc Neurosci Abs 209.4 2007 [Google Scholar]
- Li Q, Cheung C, Wei R, Hui ES, Feldon J, Meyer U, Chung S, Chua SE, Sham PC, Wu EX, McAlonan GM. Prenatal immune challenge is an environmental risk factor for brain and behavior change relevant to schizophrenia: evidence from MRI in a mouse model. PLoS One. 2009;24:e6354. doi: 10.1371/journal.pone.0006354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao L, Chen X, Shu W, Parlow AF, Xu J. Steroid receptor coactivator 3 maintains circulating IGF-I by controlling IGF-binding protein 3 expression. Mol Cell Biol. 2008;28:2460–2569. doi: 10.1128/MCB.01163-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limperopoulos C, Bassan H, Sullivan NR, Soul JS, Robertson RL, Jr, Moore M, Ringer SA, Volpe JJ, du Plessis AJ. Positive screening for autism in ex-preterm infants: prevalence and risk factors. Pediatrics. 2008;121:758–765. doi: 10.1542/peds.2007-2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockwood CJ, Yen C, Basar M, Kayisli UA, Martel M, Buhimschi I, Buhimschi C, Huang SJ, Krikun G, Schatz F. Pre-eclampsia-related inflammatory cytokines regulate interleukin-6 expression in human decidual cells. Am J Path. 2008;172:1571–1579. doi: 10.2353/ajpath.2008.070629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maes M, Bocchio Chiavetto L, Bignotti S, Battisa Tura GJ, Pioli R, Boin F, Kenis G, Bosmans E, de Jongh R, Altamura CA. Increased serum interleukin-8 and interleukin-10 in schizophrenic patients resistant to treatment with neuroleptics and the stimulatory effects of clozapine on serum leukemia inhibitory factor receptor. Schizophr Res. 2002;54:281–91. doi: 10.1016/s0920-9964(00)00094-3. [DOI] [PubMed] [Google Scholar]
- Makinodan M, Tatsumi K, Manabe T, Yamauchi T, Makinodan E, Matsuyoshi H, Shimoda S, Noriyama Y, Kishimoto T, Wanaka A. Maternal immune activation in mice delays myelination and axonal development in the hippocampus of the offspring. J Neurosci. 2008;86:2190–2200. doi: 10.1002/jnr.21673. [DOI] [PubMed] [Google Scholar]
- Mandel M, Marzouk AC, Donnelly R, Ponzio NM. Maternal immune stimulation during pregnancy affects adaptive immunity in offspring to promote development of TH17 cells. Brain Behav Immun. 2010 doi: 10.1016/j.bbi.2010.09.011. in press. [DOI] [PubMed] [Google Scholar]
- Merlot E, Couret D, Otten W. Prenatal stress, fetal imprinting and 722 immunity. Brain Behav Immun. 2008;22:42–51. doi: 10.1016/j.bbi.2007.05.007. [DOI] [PubMed] [Google Scholar]
- Meyer U, Nyffeler M, Engler A, Urwyler A, Schedlowski M, Knuesel I, Yee BK, Feldon J. The time of prenatal immune challenge determines the specificity of inflammation-mediated brain and behavioral pathology. J Neurosci. 2006;26:4752–62. doi: 10.1523/JNEUROSCI.0099-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer U, Murray PJ, Urwyler A, Yee BK, Schedlowski M, Feldon J. Adult behavioral and pharmacological dysfunctions following disrupting of the fetal brain balance between pro-inflammatory and IL-10-mediated anti-inflammatory signaling. Mol Psych. 2008a;13:208–221. doi: 10.1038/sj.mp.4002042. [DOI] [PubMed] [Google Scholar]
- Meyer U, Nyffeler M, Yee BK, Kneusel I, Feldon J. Adult brain and behavioral pathological markers of prenatal immune challenge during early/middle and late fetal development in mice. Brain Behav Immun. 2008b;22:469–486. doi: 10.1016/j.bbi.2007.09.012. [DOI] [PubMed] [Google Scholar]
- Montes MJ, Tortosa CG, Borja C, Abadia AC, Gonzalez-Gomez F, Ruiz C, Olivares EG. Constitutive secretion of interleukin-6 by human decidual stromal cells in culture. Am J Reprod Immunol. 1995;34:188–194. doi: 10.1111/j.1600-0897.1995.tb00937.x. [DOI] [PubMed] [Google Scholar]
- Mueller BR, Bale TL. Sex-specific programming of offspring emotionality after stress early in pregnancy. J Neurosci. 2008;28:9055–9065. doi: 10.1523/JNEUROSCI.1424-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyffeler M, Meyer U, Yee BK, Feldon K, Kneusel I. Maternal immune activation during pregnancy increases limbic GABAA receptor immunoreactivity in the adult offspring: implications for schizophrenia. Neuroscience. 2006;143:51–62. doi: 10.1016/j.neuroscience.2006.07.029. [DOI] [PubMed] [Google Scholar]
- Ozawa K, Hashimoto K, Kishimoto T, Shimizu E, Ishikura H, Iyo M. Immune activation during pregnancy in mice leads to dopaminergic hyperfunction and cognitive impairment in the offspring: a neurodevelopmental animal model of schizophrenia. Biol Psychiatry. 2006;59:546–554. doi: 10.1016/j.biopsych.2005.07.031. [DOI] [PubMed] [Google Scholar]
- Pardo CA, Vargas DL, Zimmerman AW. Immunity, neuroglia and neuroinflammation in autism. Int Rev Psychiatry. 2006;17:485–495. doi: 10.1080/02646830500381930. [DOI] [PubMed] [Google Scholar]
- Patterson PH. Immune involvement in schizophrenia and autism: etiology, pathology and animal models. Behav Brain Res. 2009;204:313–321. doi: 10.1016/j.bbr.2008.12.016. [DOI] [PubMed] [Google Scholar]
- Paul R, Koedel U, Winkler F, Kieseier BC, Fontana A, Kopf M, Hartung HP, Pfister HW. Lack of IL-6 augments inflammatory response but decreases vascular permeability in bacterial meningitis. Brain. 2003;126:1873–1882. doi: 10.1093/brain/awg171. [DOI] [PubMed] [Google Scholar]
- Perry W, Minassian A, Lopez B, Maron L, Lincoln A. Sensorimotor Gating Deficits in Adults with Autism. Biol Psychiatry. 2007;61:482–486. doi: 10.1016/j.biopsych.2005.09.025. [DOI] [PubMed] [Google Scholar]
- Petraglia F, Florio P, Nappi C, Genazzi AR. Peptide signaling in human placenta and membranes: autocrine, paracrine and endocrine mechanisms. Endocr Rev. 1996;17:156–186. doi: 10.1210/edrv-17-2-156. [DOI] [PubMed] [Google Scholar]
- Piontkewitz Y, Assaf Y, Weiner I. Clozapine administration in adolescence prevents postpubertal emergence of brain structural pathology in an animal model of schizophrenia. Biol Psychiatry. 2009;66:1038–46. doi: 10.1016/j.biopsych.2009.07.005. [DOI] [PubMed] [Google Scholar]
- Potvin S, Stip E, Sepehry AA, Gendron A, Bah R, Kouassi E. Inflammatory cytokine alterations in schizophrenia: a systematic quantitative review. Biol Psychiatry. 2008;63:802–808. doi: 10.1016/j.biopsych.2007.09.024. [DOI] [PubMed] [Google Scholar]
- Preti A, Cardascia L, Zen T, Marchetti M, Favaretto G, Miotto P. Risk for obstetric complications and schizophrenia. Psychiatry Res. 2000;96:127–139. doi: 10.1016/s0165-1781(00)00185-2. [DOI] [PubMed] [Google Scholar]
- Saetre P, Emilsson L, Axelsson E, Kreuger J, Lindholm E, Jazin E. Inflammation-related genes up-regulated in schizophrenia brains. BMC Psychiatry. 2007;7:46. doi: 10.1186/1471-244X-7-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito S. Cytokine network at the feto-maternal interface. J Reprod Immunol. 2000;47:87–103. doi: 10.1016/s0165-0378(00)00060-7. [DOI] [PubMed] [Google Scholar]
- Samuelsson AM, Jennische E, Hansson HA, Holmang A. Prenatal exposure to interleukin-6 results in inflammatory neurodegeneration in hippocampus with NMDA/GABA(A) dysregulation and impaired spatial learning. Am J Physiol. 2006;290:1345–1356. doi: 10.1152/ajpregu.00268.2005. [DOI] [PubMed] [Google Scholar]
- Seckl JR, Holmes MC. Mechanisms of disease: glucocorticoids, their placental metabolism and fetal “programming” of adult pathophysiology. Nat Clin Pract Endocrinol Metab. 2007;3:479–488. doi: 10.1038/ncpendmet0515. [DOI] [PubMed] [Google Scholar]
- Semer D, Reisler K, MacDonald PC, Casey ML. Responsiveness of human endometrial stromal cells to cytokines. Ann NY Acad Sci. 1991;622:99–110. doi: 10.1111/j.1749-6632.1991.tb37854.x. [DOI] [PubMed] [Google Scholar]
- Shi L, Fatemi SH, Sidwell RW, Patterson PH. Maternal influenza infection causes marked behavioral and pharmacological changes in the offspring. J Neurosci. 2003;23:297–302. doi: 10.1523/JNEUROSCI.23-01-00297.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Smith SE, Malkova N, Tse D, Su Y, Patterson PH. Activation of the maternal immune system alters cerebellar development in the offspring. Brain Behav Immun. 2009;23:116–123. doi: 10.1016/j.bbi.2008.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SE, Li J, Garbett K, Mirnics K, Patterson PH. Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci. 2007;27:10695–10702. doi: 10.1523/JNEUROSCI.2178-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares MJ. The prolactin and growth hormone families: pregnancy-specific hormones/cytokines at the maternal-fetal interface. Reprod Biol Endocrinol. 2004;2:51. doi: 10.1186/1477-7827-2-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spandidos A, Wang X, Wang H, Seed B. Primerbank: a resource of human and mouse PCR primer pairs for gene expression detection and quantification. Nucleic Acids Res. 2010;38:D793–799. doi: 10.1093/nar/gkp1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Street M, Enzo G, Volta C, Faleshini E, Bernasconi S. Placental determinants of fetal growth: identification of key factors in the insulin-like growth factor and cytokine systems using artificial neural networks. BMC Pediatrics. 2009;8 doi: 10.1186/1471-2431-8-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Wang F, Wei J, Cao LY, Qi LY, Xiu MH, Chen S, Li XH, Kosten TA, Kosten TR, Zhang XY. Association between interleukin-6 receptor polymorphism and patients with schizophrenia. Schizophr Res. 2008;102:346–347. doi: 10.1016/j.schres.2008.04.018. [DOI] [PubMed] [Google Scholar]
- Vargas DL, Nascimbene C, Krishnan C, Zimmerman AW, Pardo CA. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann Neurol. 2005;57:67–81. doi: 10.1002/ana.20315. [DOI] [PubMed] [Google Scholar]
- Weiner I. The “two-headed” latent inhibition model of schizophrenia: modeling positive and negative symptoms and their treatment. Psychopharmacology. 2003;169:257–297. doi: 10.1007/s00213-002-1313-x. [DOI] [PubMed] [Google Scholar]
- Wiemers DO, Shao L, Ain R, Dai G, Soares MJ. The mouse prolactin gene family locus. Endocrinology. 2003;144:313–325. doi: 10.1210/en.2002-220724. [DOI] [PubMed] [Google Scholar]
- Winter C, Reutiman TJ, Folsom TD, Sohr R, Wolf RJ, Juckel G, Fatemi SH. Dopamine and serotonin levels following prenatal viral infection in mouse-implications for psychiatric disorders such as schizophrenia and autism. Eur Neuropsychopharmacol. 2008;18:712–716. doi: 10.1016/j.euroneuro.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn JK, Dawson ME, Schell AM, McGee M, Salveson D, Green MF. Prepulse facilitation and prepulse inhibition in schizophrenia patients and their unaffected siblings. Biol Psychiatry. 2004;55:518–523. doi: 10.1016/j.biopsych.2003.10.018. [DOI] [PubMed] [Google Scholar]
- Zhang J, Sun R, Wei H, Wu D, Tian Z. Toll-like receptor 3 agonist enhances IFNγ and TNFα production by murine uterine NK cells. Int Immunopharmacol. 2007;7:588–596. doi: 10.1016/j.intimp.2006.12.014. [DOI] [PubMed] [Google Scholar]
- Zhang X, Sliwowska JH, Weinberg J. Prenatal alcohol exposure and fetal programming: effects on neuroendocrine and immune function. Exp Biol Med. 2005;230:376–388. doi: 10.1177/15353702-0323006-05. [DOI] [PubMed] [Google Scholar]
- Zimmerman AW, Connors SL, Matteson KJ, Lee LC, Singer HS, Castaneda JA, Pearce DA. Maternal antibrain antibodies in autism. Brain Behav Immun. 2007;21:351–357. doi: 10.1016/j.bbi.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Zuckerman L, Rehavi M, Nachman R, Weiner I. Immune activation during pregnancy in rats leads to a postpubertal emergence of disrupted latent inhibition, dopaminergic hyperfunction, and altered limbic morphology in the offspring: a novel neurodevelopmental model of schizophrenia. Neuropsychopharmacology. 2003;28:1778–89. doi: 10.1038/sj.npp.1300248. [DOI] [PubMed] [Google Scholar]
- Zuckerman L, Weiner I. Maternal immune activation leads to behavioral and pharmacological changes in the adult offspring. J Psychiatr Res. 2005;39:311–323. doi: 10.1016/j.jpsychires.2004.08.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Fetally-derived IL-6 contributes negligible amounts of IL-6 protein to the placenta after MIA. Placentas were collected to assess levels of placental IL-6 by ELISA. A low-level absorbance signal is observed in IL-6 -/- placentas from IL-6 -/-mothers, indicating a degree of non-specific protein binding during the experimental procedure. All placentas from saline- or poly(I:C)-injected IL-6 -/- mothers exhibit similarly negligible amounts of absorbance signal (n=3-6 placentas per treatment group per genotype pair (pooled from 2-3 litters).
Fig. S2. MIA has no affect on percentages of decidual immune cell subtypes. Compared to controls, placentas from poly(I:C)-injected mothers have no significant difference in the percentages of macrophages, granulocytes, uNK cells, CD4+ T cells, CD8+ T cells or B cells (n=5 litters per treatment group (7-8 deciduas pooled per litter).
Fig. S3. MIA induces phosphorylation of STAT1 and STAT3 in sTGCs and pTGCs in an IL-6-independent manner. A. IL-6 +/- and IL-6 -/- placentas from poly(I:C)-injected IL-6 +/- mothers display no significant difference in the number of pSTAT3-positive spongiotrophoblast cells. Thus, fetally-derived IL-6 contributes little to MIA-induced STAT3 activation. B. Representative images taken at 20× magnification depict saline placenta (left) and MIA placenta (right). Compared to saline controls, MIA placentas (right) stain positively for pSTAT1 in sTGCs and pTGCs (arrows). Dotted lines = boundary between decidua and spongiotrophoblast layers (upper) and boundary between spongiotrophoblast and labyrinth (lower); D=decidua, Sp=spongiotrophoblast, L=labyrinth; C. Representative images taken at 40× magnification depict pSTAT3 staining in saline labyrinth (left) and MIA labyrinth (right). Compared to saline controls, MIA placentas stain positively for pSTAT3 in sTGCs. [n=5-7 placentas per treatment group per genotype pair (pooled from 3-5 independent litters)].