

Abstract
Protein folding intermediates are often imperative to overall folding processes and consequent biological functions. However, the low population and transient nature of the intermediate states often hinder the biochemical and biophysical characterization. Previous studies have demonstrated that Bacillus subtilis ribonuclease P protein (P protein) is conformationally heterogeneous and folds with multiphasic kinetics, indicating the presence of an equilibrium and kinetic intermediate in its folding mechanism. In this study, NMR spectroscopy was used to study the ensemble corresponding to this intermediate (I). The results indicate that the N-terminal and C-terminal helical regions are mostly unfolded in I.1H-15N HSQC NMR spectra collected as a function of pH suggest that the protonation of His 22 may play a major role in the energetics of the equilibria between the unfolded, intermediate, and folded state ensembles of P protein. NMR paramagnetic relaxation enhancement experiments were also used to locate the small anion binding sites in both the intermediate and folded ensembles. The results for the folded protein are consistent with the previously modeled binding regions. These structural insights suggest a possible role for I in the RNase P holoenzyme assembly process.
Keywords: RNase P protein, NMR, Folding intermediate
An understanding of the processes by which a protein folds into its folded conformation requires knowledge of the various states that populate the folding pathways, the kinetic rate constants between these states, and their relative stabilities, as well as their structural properties. Despite the observation that many proteins fold through an intermediate state (1-3), the role of intermediates in productive folding is still the subject of fertile literature discussion (4). Intermediates between the unfolded and folded states may enhance the rate of folding by decreasing the conformational space through which the polypeptide chain has to search, or may reduce the rate of folding by sequestering the polypeptide chain in a stable, partially folded state (5, 6).
RNase P protein is the protein subunit in the RNase P holoenzyme. Our previous studies have established that P protein is an intrinsically disordered protein (IDP). Without the binding partners, P protein is predominantly unfolded but can be induced to fold in the presence of different small anions (e.g. sulfate). Many protein subunits in the ribonucleoprotein complexes exhibit conformational dynamics, which promote specific recognition of cognate RNA partners (7). The N-terminal RNA recognition domain (RRM) of human U1A binds to the 3′-UTR of U1A mRNA. In the absence of RNA, backbone and sidechain relaxation experiments reveal conformational heterogeneity on the μs-ms timescale in the regions that bind RNA (8, 9). Previous NMR studies showed that there are extra peaks in the 1H-15N HSQC spectrum of the sulfate-folded state of P protein, which is evidence of alternative conformations under these conditions (10). Backbone dynamics studies of sulfate-folded state of P protein under similar conditions also detected conformational heterogeneity (arising from motions on the μs to s timescale) (11). In addition, our recent kinetic and equilibrium studies of trimethylamine-N oxide (TMAO) induced folding of P protein established that there is an intermediate state in the folding process (kinetic studies of sulfate induced folding also detect an intermediate) (12 and unpublished data). Assembly of ribonucleoprotein complexes is usually accompanied by conformational changes in both protein and RNA components (13, 14). The folding intermediates of protein or RNA components can be crucial to the overall assembly process. The absence of key intermediate states could result in non-native and misfolded ribonucleoprotein complexes (15). Therefore, a primary motivation to structurally and energetically characterize this intermediate in the P protein system is to understand its role in the folding process and ultimately holoenzyme assembly.
For most small proteins, intermediates form within the dead time of a stopped-flow instrument, so it has not been feasible to determine their role in the folding process (16, 17). Recent development of ultra rapid mixing (18, 19) and temperature-jump (20) instruments enabled the direct detection of the initial folding phase and early folding events. The intermediate ensemble formed early in the folding of Im7 (2), as well as that of GB1 (21) have been shown to be on the folding pathway. In addition to the short lifetime of intermediate states, many of them are populated at very low levels, making their experimental characterization difficult in some systems. Most of the transient techniques that have been used to study protein folding are based on the use of a single probe or a single average property derived from many residues in the protein. It is thus difficult to obtain residue-specific information about the intermediate state. Therefore determination of the conformational properties of an intermediate state at high resolution is important for a full elucidation of the structural mechanism of folding. However, the transient nature and low population of the intermediate states relative to the unfolded and folded state are challenges for structural experiments.
A variety of NMR methods are available and designed to study the conformational properties of intermediate states. Relaxation dispersion NMR spectroscopy is extremely sensitive to processes that involve interconversion between a ground state (folded or unfolded state) and excited states (intermediate state) of molecules (22-24). The method also provides residue-specific information over the whole protein simultaneously. NMR analysis of partially folded proteins stabilized by changing solution conditions or mutagenesis have also provided insights about the nature of their conformational ensemble (25-27). Another approach to investigate protein folding at the residue level is to record a series of 1H-15N HSQC spectra at increasing denaturant concentrations under equilibrium conditions (28, 29). Lastly, NMR spectroscopy provides a powerful method for monitoring the exchange of a nucleus between different environments due to conformational transitions (30). If the transition rate (k) is in the fast-exchange regime (Δω << k, where Δω is the resonant frequency difference for the same nucleus in two magnetically inequivalent sites), a single sharp peak is observed. If the rate is in intermediate-exchange regime (Δω ≈ k), the peaks become very broad and can have similar intensity as baseline noise. When the rate is in slow-exchange (Δω >> k), two resonances are observed arising from the two distinct environments of the nucleus. In this study, all three scenarios were observed in folded and unfolded P protein NMR HSQC spectra, which enabled us to structurally characterize the intermediate state of P protein. Moreover, Δω values were useful for deducing structural characteristics of the intermediate. Overall, NMR offers several different ways to characterize the intermediate state kinetically, thermodynamically and structurally.
In this study, standard multidimensional NMR experiments were performed to determine the residue identities of the extra peaks in the 1H-15N HSQC spectrum of the sulfate-folded state of P protein. The assignment results showed that most of these residues are located in the vicinity of the N- and C-termini and their second resonances arise from the random coil conformation. The NMR data suggests that the central β-sheet and α-helix B are folded in the intermediate state and that the N-terminal α-helix A and C-terminal α-helix C are unfolded. Acid titration monitored by HSQC spectra indicated that, histidine protonation predominantly His 22, shifts the conformational equilibria of P protein at low pH.
Experimental Procedures
Chemicals and reagents
Ultra pure urea was purchased from Nacalai Tesque Inc. 15N ammonium chloride (99%), 13C glucose (U-13C6, 99%), and deuterated TMAO (d9, 98%) were purchased from Cambridge Isotope Laboratories Inc. Urea and TMAO concentration was measured by refractive index using the equation from (31) and (32), respectively.
Protein mutagenesis, expression and purification
Histidine variants of P protein were generated in a F107W background. These site-directed mutational plasmids were constructed using QuikChange® procedure (Stratagene) with the primer GGAGATATACCATGGCTAAGCTGAAAAAACGC and GCGTTTTTTCAGCTTAGCCATGGTATATCTCC for H3K, and CCAGAAAGTGTTTAAAAAGGGGACATCAGTTGC and GCAACTGATGTCCCCTTTTTAAACACTTTCTGG for H22K, and CGAAAAAAAGTCTGCAGAAGCTATGGAGAAAGTCTTC and GAAGACTTTCTCCATAGCTTCTGCAGACTTTTTTTCG for H105K. Single and triple histidine substitutions in F107W background were overexpressed to produce the P protein variants used for this study. The DNA sequence and the mass spectrum of each variant were determined to assure that it contained the desired substitution.
Wild type P protein and variants were overexpressed in E. coli (BL21 (DE3) pLysS) cells. For preparation of 15N singly labeled and 15N, 13C doubly labeled protein used in the NMR experiments, a single colony of positive transformants was used to inoculate 30mL LB media containing 50 μg/mL kanamycin and 17 μg/mL chloramphenicol and grown to an OD600 of 0.8-0.9. The culture was diluted (1:200) into the M9 minimum media culture (1L) containing the same antibiotics and enriched with 2g/L 13C glucose and/or 1g/L 15N ammonium chloride. The culture was incubated until the OD600 was 0.4 - 0.6, and protein overexpression induced by addition of IPTG to a final concentration of 0.4 mM. The culture was incubated with vigorous shaking (1 L in a 6L Erlenmeyer flask) for 10-12 hour, and the cells were harvested by centrifugation. Overexpressed P protein was purified as described (33) with the following modifications. The pooled fractions of P protein eluted from the second CM-Sepharose column were concentrated using a 3.5 kDa cutoff centriprep ultrafilter (Amicon) until the volume of the pooled fractions was reduced to 1~2 mL. The buffer in the sample was changed to 6 M Guanidine-HCl, 10mM Tris (pH 7.5) during this concentration step. The concentrated sample was then loaded on an S-100 column equilibrated with 6 M Guanidine-HCl, 10 mM Tris (pH 7.5) to remove any bound EDTA. Fractions containing P protein were pooled and concentrated again using a centriprep ultrafilter to desired concentration for NMR experiments. Protein purity was determined to be >99% by SDS-PAGE with coomassie blue detection. The molecular weight of each variant was confirmed by electrospray ionization mass spectrometry. The mass of unlabeled proteins were within ±0.5 Da of the expected mass and labeled proteins were 3-4 Da larger from the theoretical mass calculation, respectively. Protein was stored in 6 M Gdn-HCl at −80 °C for long term storage. The protein NMR sample was dialyzed extensively against water and then 20 mM sodium cacodylate (with proper pH value needed in the experiments). In all experiments, the protein concentration was calculated using the Edelhoch method (34) using an extinction coefficient of 11200 M−1 cm−1 at 276 nm.
NMR spectroscopy experiments
2D, 3D NMR experiments and sequential assignment
All three-dimensional NMR spectra were collected at 25 °C on a Varian INOVA 600 spectrometer with a triple resonance cryoprobe equipped with a z-field gradient coil. The two-dimensional spectra were collected on either a Varian INOVA 600 or a Varian INOVA 800 spectrometer with a triple resonance probe. The NMR sample used for assignment of the extra peaks consisted of 0.5 mM P protein in 5 mM sodium sulfate, 20 mM sodium cacodylate buffer at pH 5.0, 10% D2O and 0.05% sodium azide. The NMR sample used for the pH titrations of the unliganded protein consisted of 0.2 - 0.4 mM P protein in 20 mM sodium cacodylate buffer, 10% D2O and 0.05% sodium azide. The pH ranged from 5 to 7.5. Two-dimensional gradient-enhanced sensitivity-enhanced 1H-15N HSQC experiments were collected with a spectral width in the 1H dimension of 11990 Hz and 1024 complex points and a spectral width in the 15N dimension of 1994 Hz and 192 complex points. The resonance assignments of extra peaks were made using a suite of triple resonance experiments including (HCA)CO(CA)NH (35), HNCO (36), HN(CA)CO (37), CBCA(CO)NH (38), and H(CCO)NH (39, 40). In these three-dimensional NMR experiments, the spectral width in the 1H dimension was 7020 Hz with 1024 complex points and the spectral width of 15N dimension was 1944 Hz with 32,64 or 72 complex points. The spectral width in 13C dimension was 2000 Hz with 48,64 and 96 complex points for (HCA)CO(CA)NH, HN(CA)CO, and HNCO, respectively. The spectral width of 13C dimension was 12066 Hz with 64 complex points for the CBCA(CO)NH experiment. The NMR spectra were processed using NMRpipe (41), the processed spectra were viewed and examined by NMRDraw and NMRviewJ, and the intensities of the crosspeaks were quantified by NMRViewJ (42). Backbone sequential assignments were analyzed using CARA (CARA can be downloaded for free from www.nmr.ch.).
PRE Experiments
Two-dimensional gradient-enhanced sensitivity-enhanced 1H-15N HSQC were collected on a Varian INOVA 600 spectrometer with a triple resonance probe. The P protein was 200 μM with 20 mM sulfate in 20 mM cacodylate buffer, pH 6.5, 10% D2O, and 0.05% sodium azide. Potassium hexacyanochromate K3[Cr(CN)6] was titrated into the protein sample from 2.5 μM to 100 μM (with the concentration steps of 5, 10, 20, and 50 μM). The HSQC spectra were collected at each [Cr(CN)6]3− concentration with a spectral width in the 1H dimension of 7020Hz and 1024 complex points and a spectral width in the 15N dimension of 1994 Hz and 128 complex points.
Results
Sulfate and pH titration of folded P protein
Assignments for 95% of the backbone resonances of sulfate-folded (20mM sulfate) P protein have been reported previously (10). Although most of the peaks present in the 1H-15N HSQC spectrum under these conditions were assigned in the previous studies, a few residues were missing and some extra peaks could not be assigned. One interpretation of these observations is that one or more alternative conformations are in chemical exchange with folded state of P protein. In addition, the intensities of the extra peaks are dependent on ligand (sulfate) concentration and pH, suggesting that the population(s) of the alternative state(s) are influenced by ligand and pH. Sulfate has previously been shown to bind to P protein with an apparent affinity of 0.5 mM (43). In order to gain more insight into how ligand concentration and pH affect the equilibria between the alternative conformations and the folded state, a series of 1H-15N HSQC spectra were collected at various sodium sulfate concentrations and pH values. The sulfate concentration ranged from 0 to 100 mM, but focused on the range around 2 to 100 mM. As shown in the Figure 1A, the intensities of the extra peaks decreased as the ligand concentration increased, and the chemical shifts of some assigned peaks of folded state, many of which are in the N-terminal region, were strongly dependent on ligand concentration. These two observations indicate that both slow exchange and fast exchange processes contribute to the observed ligand-dependence.
Figure 1.
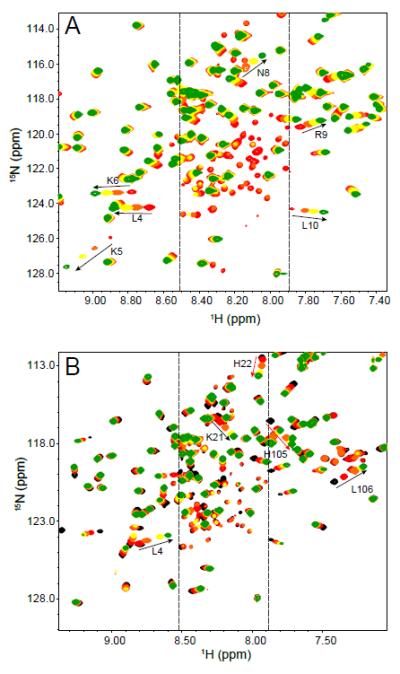
Sulfate titration and pH titration of the 1H-15N HSQC spectrum of 15N-labeled P protein, at pH 6.0 and 25°C. (A) The spectra of P protein in 5 mM sulfate (red), 10 mM (orange), 20 mM (yellow), and 100 mM (green) are overlaid in order of increasing concentration. The intensities of the extra peaks (between the two dashed lines) decrease with increasing sulfate concentrations as evidenced by the disappearance of the yellow and green peaks. The labeled peaks are assigned residues of folded P protein. The arrows show their change in frequency as the sulfate concentration increases from 5 mM to 100 mM. (B) The spectra of P protein in 20 mM sulfate at pH 5.3 (black), pH 5.5 (red), pH 6.0 (orange), pH 6.5 (yellow), and pH 7.0 (green) are overlaid in order of increasing pH. The labels for the two observable histidine residues are red. His3 is not observed in the spectrum. Residues next to histidine residues that exhibit significant chemical shift movement are labeled in black with arrows indicating their change in frequency with increasing pH. All extra peaks are located in the region between the two dashed lines and their intensities decrease with increasing pH.
Similar effects were observed when sample pH was varied. As shown in Figure 1B, the intensities of the unassigned extra peaks decreased and many native peaks shifted as the sample pH was increased from 5.0 to 7.5 with the sulfate concentration held constant at 20 mM. The pKa of the free histidine imidazole group of is 6.0 (44), which is within the range of pH values used for this experiment. For this reason, it is likely that the changes in folded resonance frequencies can be explained by the change of the protonation state of the three histidine sidechains in P protein. Figure 1B shows that the amide 15N/1H resonances assigned to His 22 and His 105 move as the pH changes, and the His 3 peak is missing in the spectrum, presumably due to intermediate chemical exchange (10). The 1H or 15N chemical shifts of the two histidine resonances plotted vs. pH can be fitted to the Henderson–Hasselbalch equation to estimate the pKa’s of His 22 and His 105 which are 6.08±0.16 and 5.65±0.17, respectively (supplementary material). Changes in chemical shift were also observed for residues in close proximity to all three histidine residues, which is expected because the ionization state of the histidine sidechain can also affect neighboring residues.
HSQC spectrum of folded P protein and unfolded P protein
In a previous study (10), the protein sample conditions of pH 6.0, 20 mM sodium sulfate reduced the intensity of the extra peaks, producing an HSQC spectrum dominated by folded P protein resonances. However, as described in the previous section, lowering the sulfate concentration to 5 mM and decreasing the sample pH from 6.0 to 5.0 increases the intensities of the unassigned extra peaks. As shown in Figure 2, there are approximately 50 unassigned peaks (colored in red) present in the spectrum, and most of them are located in the 8.0 to 8.5 ppm region of the 1H spectrum. The presence of extra peaks means that some residues in the folded P protein produce two (or multiple) crosspeaks in the HSQC spectrum. Therefore, the residues in P protein can be classified into three types. Residues that only have one peak are in fast-exchange (Δω << k) between the alternative and folded states. In the limit, Δω is equal to 0, implying that the magnetic environments of those residues in the alternative and folded states are similar or even the same. Residues that produce no detectable HSQC resonances presumably correspond to sites in intermediate-exchange (Δω ≅ k) between the different conformational states. The sensitivity of the INEPT coherence transfer steps in the HSQC pulse sequence to chemical exchange processes (45) and severe line broadening (46) both contribute to a severe loss of peak intensity in this regime. Residues assignable to two peaks correspond to sites in slow-exchange (Δω >> k) between the alternative and folded states. The large difference in frequency indicates that these residues are found in quite different magnetic environment in the alternative and folded states.
Figure 2.
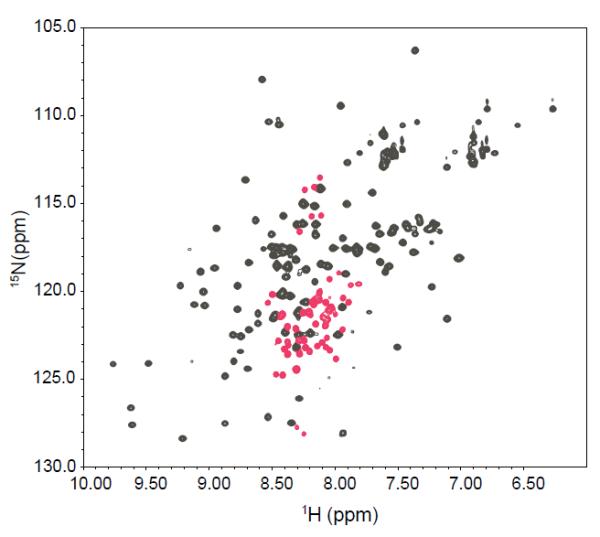
1H-15N HSQC spectrum of 15N-labeled P protein in 5 mM sulfate, pH 5.0 at 25°C. The peaks in grey are the folded state spectrum resonances assigned in a previous study (58). The peaks in red are extra peaks that appear under these buffer conditions. All of the extra peaks are located between 7.9 and 8.5 ppm in the 1H dimension. The labels give the assignments of the extra peaks.
Figure 3 shows the HSQC spectrum of P protein at the same pH values as those for Fig. 2, but in the absence of sulfate. Under these conditions, the protein is predominantly unfolded. This spectrum is characteristic of unfolded polypeptides, but there are two subsets of peaks (circled regions) whose positions are similar to assigned resonances in the HSQC spectrum of sulfate-folded P protein (10). As shown in the Fig. 3, the peaks in these circled regions overlapped well between the unfolded and folded conditions, which are strong evidences that these peaks correspond to the same residues in the protein. The presence of assigned peaks of folded P protein in the unfolded P protein spectrum can be explained by the presence of partially folded species that exchanges slowly (τex > 1 sec) with the fully unfolded protein. The presence of extra peaks and missing residues in the sulfate-folded state spectrum are evidence of the existence of one (or more) alternative conformation that exchanges slowly with the folded conformation. Taken together, both HSQC spectra are consistent with the presence of an alternative structural conformation that is in equilibrium between the unfolded and folded state. This interpretation is supported by our folding kinetic studies of P protein, which demonstrated that there is an intermediate state in the P protein folding process (47). Although our previous kinetic and equilibrium studies (12) of TMAO-folded protein did not involve ligand binding, similar studies of ligand induced folding also show the presence of an intermediate species in the folding process (unpublished data). Therefore, as a minimal complexity model that is consistent with all of the experimental data, we have assigned the alternative conformation found in the NMR spectra collected under both folding and unfolding conditions to the same partially folded intermediate state in the P protein folding mechanism. Assignment of the extra peaks in the HSQC spectrum (Figure 2) allowed identification of residues in the unfolded regions of the partially folded intermediate.
Figure 3.
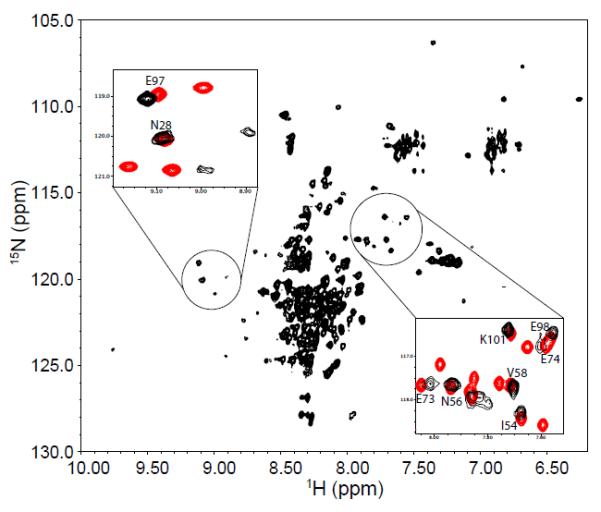
1H-15N HSQC spectrum of 15N-labeled P protein in 20 mM sodium cacodylate, pH 5.0, at 25°C. Sulfate is not present in the sample, so P protein is predominantly unfolded. The peaks in the spectrum are poorly dispersed compared to those of folded P protein spectrum in Figure 1. The peaks in the two circles form patterns similar to those found in the folded P protein spectrum. The intensities of these peaks are weaker than those of the remaining unassigned resonances from unfolded P protein. The two insets show the overlapped HSQC spectra of unfolded (black) and folded P protein (red) in the circled regions. Labeled peaks are previously assigned residues for folded P protein.
Assignment of extra peaks in the folded P protein HSQC spectrum
To assign the extra peaks, a protein sample in 5mM sodium sulfate, pH 5.0 was chosen to intensify the extra peaks. Standard multidimensional backbone assignment experiments with 13C/15N labeled protein were performed and a total of 50 extra peaks were observed in the HSQC spectrum. The clustering of the peaks in the 8 to 8.5 ppm 1H chemical shift range resulted in partial overlap of many of the extra peaks. This problem, combined with the relatively weak intensities of the extra peaks, made their assignments more challenging to determine than those of the folded resonances. Combination of data from HNCO (36), HN(CA)CO (48), and (HCA)CO(CA)NH (35) experiments, allowed the sequential assignments through the 13CO nuclei. The HNCACB (49) and CBCA(CO)NH (38) experiments were also included to detect 13Cα and 13Cβ resonance and the results further aid and validate the 13CO assignment. There were some short stretches consisting of three to five residues that could not be unambiguously assigned to definite fragments in the protein sequence. To resolve this ambiguity, an H(CCO)NH (39, 40) 3-dimensional experiment was performed to detect the sidechain proton spin system of the i-1 residue and subsequently each fragment was assigned to its correct sequence. This suite of experiments allowed for the assignment of 42 out of the observed 50 extra peaks. Five out of the remaining eight peaks exhibited no 13C chemical shift connectivity to each other or any of the peaks of folded state. The chemical shifts for the backbone atoms of the 42 assigned peaks and the 8 unassigned peaks can be found in Supporting Information.
Most of the extra peaks were assigned to residues located near the N- and C-termini in the protein sequence. There is one long N-terminal fragment from Leu 4 to Val 19, which is half way through helix A in the folded structure (Figure 4). Four short fragments Ala 91-Thr 95, Glu 98- Gln 104, Leu 106-Arg 109, and Leu 112-Ser 116 are located in the C-terminal region, which includes the loop between β sheet 4 and helix C and as well as most of the helix C. A short fragment from Lys 52 to Ile 54 is located outside of these two structural regions. As shown in the Figure 1, many extra peaks assigned to the same residue type are clustered near each other in the HSQC spectrum, which suggests that these residues are located in a highly solvated magnetic environment in the intermediate.
Figure 4.
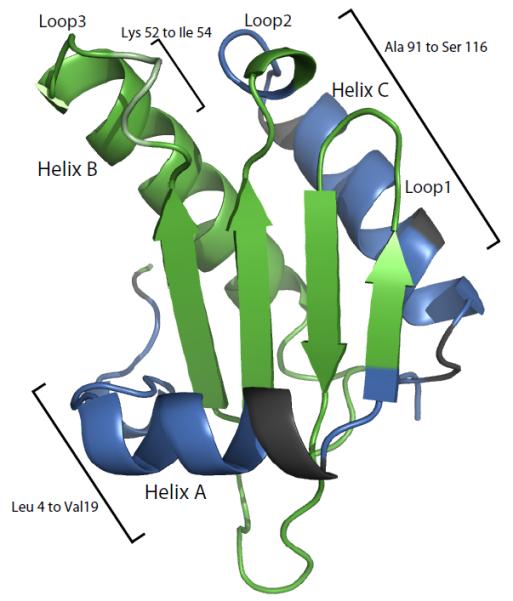
Mapping of the residues assigned to extra HSQC resonances on the crystal structure of folded P protein. The residues colored in blue are the extra peaks that are assigned in the study. The residues in dark grey are the residues that might have to the extra peaks but could not be unambiguously assigned. The remaining residues in green have a single resonance in the spectrum.
Chemical shift indices (CSI) for 13Cα and 13Cβ assignments can be used to deduce secondary structure from established empirical relationships (50, 51). 13Cα resonances experience a downfield shift relative to random coil shifts in α-helical conformations and the 13Cα chemical shift is typically upfield of random coil shifts when the residue is in a β-strand conformation. The chemical shift index values as a function of protein sequence for 13Cα and 13Cβ atoms of the extra peaks are shown in Figure 5. The N-terminal region of folded P protein is a flexible structure followed by helix A to residue S25. Most of the 13Cα CSI values of the stretch of 18 assigned extra peaks in this region are smaller than 1 ppm and there is no significant deviation from random coil shifts, indicating that these residues have a random-coil-like structure in the intermediate state. Extra peaks assigned to residues found in the C-terminal helix C region of the folded state have 13Cα CSI values indicative of random coil, supporting the conclusion that this region of the intermediate state is also unfolded. The 13Cβ CSI values are consistent with these conclusions. For the extra peaks assigned to these two regions, the differences in chemical shifts from the folded state assignments are greater than 3 ppm. The three residues in the middle of the sequence in Figure 5 are colored light green to indicate that their CSI values are not consistent between 13Cα and 13Cβ and some of them are larger than 2 ppm. Low chemical shift dispersion of the extra peak 1H/15N HSQC resonances and their low 13Cα and 13Cβ CSI values are consistent with the conclusion that these residues are unfolded in the partially folded intermediate. In addition, the TALOS+ program (52) uses available chemical shifts (Hα, HN, H, Cα, Cβ and CO) to calculate a random coil index (RCI) and predict the secondary structure. The secondary structure prediction from TALOS+ is consistent with CSI values. The order parameter (S2) prediction from the program shows these regions are dynamic (S2 < 0.5), which agrees with that these residues are in the unfolded state.
Figure 5.

Chemical shift analysis of the extra peaks in the folded P protein HSQC spectrum. Residues 52 to 54 are shown in light green because their CSI values are larger than 2 ppm.
Effect of histidine replacement in P protein
As shown in the Figure 3, the presence of assigned peaks of folded state in the unfolded P protein spectrum can be explained by the presence of the intermediate state, which exchanges slowly with the unfolded state. In order to investigate how the histidine residues in the protein perturb the equilibrium between U and I states, different single His to Lys variants (H3K, H22K, and H105K) and a triple His to Lys variant (HtriK) were constructed to study the effects. These variants were made in the background of the F107W variant to allow both NMR and fluorescence stopped-flow experiments. Since the F107W substitution has little effect on the overall structure and stability of P protein, the 1H-15N HSQC spectrum of unfolded F107W is similar to that of WT, including the peaks of folded state observed in the unfolded WT spectrum (circled area in Figure 6). A series of pH titration experiments from 5.0 to 7.0 monitored by HSQC spectra were performed to investigate the pH effect on the equilibrium between the U and I states. As shown in the Figure 7A, when the pH is increased from 5.0 to 7.0, the intensities of the folded-like peaks due to I also increase. This means that the relative population of I increases as the pH increases. These results suggest that the ionization of histidine residues in the P protein affect the equilibrium transition between U and I, because the histidine side chain is the only likely titratable group in this pH range. The HtriK variant was constructed to mimic the charge state of the F107W variant at pH 5.0, since the histidines are more than 80% protonated (His 22 > 93% and His 105 > 82%) and carry a positive charge at that pH. As shown in Figure 7B, the HtriK substitutions almost completely abolish the intensities of the peaks of folded state in the HSQC spectrum, which is now independent of pH. This is the expected result if the extra charge on the variant depopulates I. Because P protein is a highly basic protein containing 19 Lys and 11 Arg residues, introducing 3 more Lys residues into the protein increases electrostatic repulsion in the fully or partially folded forms, favoring the unfolded state of the protein.
Figure 6.
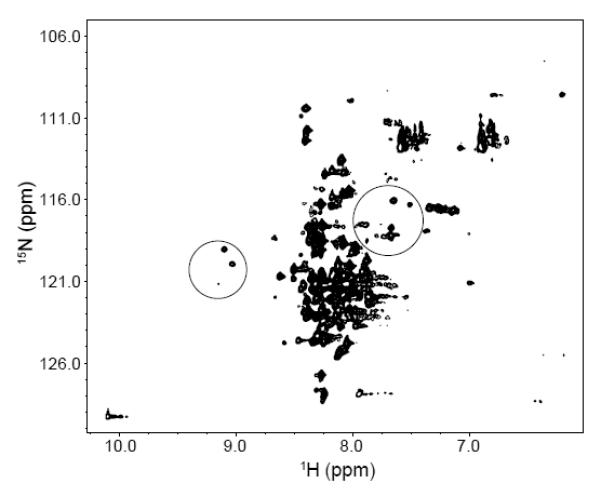
1H-15N HSQC spectrum of 15N-labeled F107W variant of P protein in 20 mM sodium cacodylate, pH 6.0, at 25°C. The peaks in the spectrum are as poorly dispersed as those of unfolded WT P protein shown in Figure 3. The peaks in the two circles are assigned folded state P protein residues also present in the unfolded P protein spectrum. The intensities of these peaks are weaker than those of the remaining unassigned peaks of unfolded P protein.
Figure 7.

HSQC spectra of various histidine variants of P protein under unfolding conditions at three pH values. The pH titration experiments were done at pH 5.0 (yellow), 6.0 (red) and 7.0 (black) for each variant. The name of the variant is in the bottom right corner of each plot. The spectra are overlaid and the peaks in the circles area are assigned peaks of folded state of F107W. As shown in the plots, the intensities of the peaks in the circled area increase as the pH increases.
Subsequent pH titration experiments were performed on each single H→K variant to determine which histidine residue(s) account for the observed effects. As shown in Figures 7C to 7E, the HSQC spectrum of the H22K variant is nearly the same as the HtriK variant suggesting that protonation of this histidine, located at the end of the helix A, is the source of the pH-dependence of the U  I equilibrium. The spectrum of the H105K shows a pH dependence similar to that of F107W, indicating that the protonation of His 105 has little effect on the stability of I. The pH dependence of peak intensities in the H3K variant spectrum shows that His 3 does have an effect on the population of I, but the effect is smaller than that of H22.
I equilibrium. The spectrum of the H105K shows a pH dependence similar to that of F107W, indicating that the protonation of His 105 has little effect on the stability of I. The pH dependence of peak intensities in the H3K variant spectrum shows that His 3 does have an effect on the population of I, but the effect is smaller than that of H22.
Based on the pH titration experiments performed with these different variants, we conclude that as the pH is raised from 5 to 7, the equilibrium between U and I is shifted toward I primarily by the deprotonation of His 22, with some contribution from His 3 and little or none from H105.
Identification of ligand binding sites by PRE
Our previous studies showed that the stoichiometry of anion binding to P protein is two and proposed the locations of the two high affinity binding sites responsible for the folding coupled binding transition of P protein (43). The electron density of one sulfate was clearly observed by x-ray crystallography near the N-terminus with sulfate coordinated by His3, Arg9 and Arg68 (53). We later proposed that the second site is located near Arg88 and Lys89, although Stams, et al. had assigned the observed electron density at that site to a water molecule instead of a sulfate ion. In order to determine the anion binding sites more definitively, we used paramagnetic relaxation enhancement (PRE) NMR experiments. PRE is caused by magnetic dipole interactions between a nucleus and the unpaired electrons (54), and results in an increase in the relaxation rate of the nuclear magnetization. Its magnitude is proportional to <r−6> (where r is the distance between the nucleus of interest and the paramagnetic center) (55). As a result of the large magnetic moment of an unpaired electron, the effect of the PRE can be detected at a fairly long range (15-24 Å). Previous studies used the paramagnetic hexacyanochromate(III) ion ([Cr(CN)6]3−) to probe the active site of RNase A(56) and the anion binding site of phosphoglycerate kinase (57). We have used the same anion for the PRE studies of the P protein anion binding site.
The apparent affinity of P protein for [Cr(CN)6]3− was determined by CD detected 6 titration with this ligand yielding an apparent Kd of approximately 20 μM (data not shown), consistent with high affinity binding expected for a trivalent anion. Subsequently, [Cr(CN)6]3− titration experiments monitored by 1H-15N HSQC spectrum were performed to identify the P protein residues in close proximity to the anion binding sites. The initial NMR sample contained 200 μM of pre-folded P protein in 20mM sulfate, pH 6.5. [Cr(CN)6]3− was added to the sample at concentrations ranging from 2.5 μM to 100 μM. Because [Cr(CN)6]3− is more tightly bound than sulfate, 2.5 μM of [Cr(CN)6]3− was sufficient to displace some of the bound sulfate. It is also reasonable to assume that the on rates of the paramagnetic ligand is diffusion limited, at least 106 M−1 s−1. Under these conditions, there would be at least 20 [Cr(CN)6]3− binding events within the T1 of the 1H or 15N spins. Thus, even if the population of the sites with [Cr(CN)6]3− is small (<1%), this minor species can still strongly affect the apparent relaxation rate and cause line broadening of the 1H-15N HSQC resonances from the backbone amides of nearby residues. Figure 8 shows HSQC spectra from the [Cr(CN)6]3− titration experiments. With the addition of the [Cr(CN)6]3− ion, the amide peak intensities of the residues that are in vicinity of the binding sites start to decrease or completely broaden out. These residues are labeled in Figure 8. The degree of the intensity decrease of a residue as a function of [Cr(CN)6]3− concentration provides information about the distance between the amide and the paramagnetic center. The lower the [Cr(CN)6]3− concentration needed to broaden the peak, the closer the residue is to the binding site. As seen in Figure 9, mapping the affected residues to the structure of the P protein identified two anion binding sites as expected. In general, the PRE NMR results agree with the previously proposed anion binding sites, especially the one located in the N- terminal region. The other site is also close to the previously proposed site, but Lys89 and Lys52 amide resonances were more strongly affected by the paramagnetic anion than the Arg88 amide. This suggests that the second anion binding site might be close to the loop 2 and loop 3 regions indicated in Figure 9, rather than loop1 and loop2 region as previously proposed.
Figure 8.

1H-15N HSQC spectrum of folded P protein in various concentrations of K3[Cr(CN)6]. The protein concentration was 200 μM with 20 mM sulfate in 20 mM cacodylate buffer pH 6.5. The [Cr(CN)6]3− concentration ranged from 2.5 μM to 50 μM. The spectra with [Cr(CN)6]3− concentrations higher than 50 μM are not shown in the plot. Higher concentrations of [Cr(CN)6]3− broaden the resonances of many residues, which may result from nonspecific binding of [Cr(CN)6]3− thereby masking the effects due binding at the two high affinity sites.
Figure 9.

PRE results mapped on the P protein crystal structure. The results of the NMR PRE experiments show two regions of P protein involved in the anion binding. The color of each residue indicates the [Cr(CN)6]3− concentration at which the associated resonance intensity is affected: red is 2.5 μM, orange is 5 to 10 μM, and yellow is 25 to 50 μM. Residues colored gray correspond to resonances already missing in the 0M [Cr(CN)6]3− HSQC spectrum, prolines or unresolved peaks. Residues colored green are unaffected by [Cr(CN)6]3− between 2.5 μM and 100 μM concentration range.
Discussion
Structural description of the intermediate state
Figure 4 shows the residues assigned to the extra peaks present in the folded P protein 1H-15N HSQC spectrum mapped on the crystal structure of P protein. The extra peak residues are colored in blue on the structure and most are located in the N-terminal (residues 3 to 19) and C-terminal regions (residues 91 to 116). Three of the extra peaks were assigned to Lys 52 to Ile 54, which are located in the middle of loop 3. The low chemical shift dispersion of the extra 1H-15N HSQC resonances and their low 13Cα and 13Cβ CSI values indicate that the associated residues are part of random-coil-like structure in I. It should be noted that each of the assigned residues in these three regions has two separate crosspeaks in the HSQC spectrum. One peak corresponds to the folded state resonance assigned in previous studies (10, 58), and the other corresponds to a resonance whose magnetic environment appears to be very similar to that of an unfolded protein. No extra peaks were assigned to the residues colored in green in Figure 4, suggesting that these residues retain a folded-like magnetic environment in both the intermediate and folded states. The residues colored dark grey in Figure 4 (Phe 20, Lys 21, His 22, Tyr 96, Glu97, His 105, Ser 110, and Ser 111) are near the N and C termini, but cannot be unambiguously assigned to any of the extra peaks. Residues unfolded in I would not have an extra peak if the difference between their folded and unfolded frequencies are small, i.e. if their folded state frequencies happen to be near the random coil values. This might be the case for residues 20, 21, 22, 105, 110, and 111, since their folded state 1H chemical shifts are between 8 and 8.5 ppm, near the random coil values. This explanation, however, is not valid for Tyr 96 and Glu 97, because their 1H chemical shifts are down field at 9.7 and 9.5 ppm, respectively. An alternative explanation could be that since only 42 out of 50 extra peaks have been assigned, the remaining unassigned eight peaks might correspond to these residues. The intensities of these unassigned peaks are weakest among the extra peaks in the HSQC spectrum and they correspond to even weaker or missing crosspeaks in the three-dimensional heteronuclear triple-resonance experiments. Therefore, it was difficult to obtain sequential assignments for these eight peaks. An ideal solution would be assignment of the spectrum of completely unfolded P protein allowing the identification of the remaining unassigned peaks. The last possibility is that these 8 peaks belong to residues outside the N and C-terminal regions and could be some of the residues colored dark green in Figure 4. Based on the results of the NMR experiments, the likely structure of the intermediate includes a folded-like central β-sheet and α-helix B but an unfolded and N-terminal region through helix A and C-terminal helix C.
Reconsideration of previous dynamic and amide-hydrogen exchange data
Previous backbone dynamics studies of P protein demonstrated the conformational heterogeneity of P protein under sulfate-folded conditions. A significant Rex, the conformational exchange contribution of R2, was found for some residues in N- and C-terminal regions. These are the same regions that are unfolded in the intermediate state. On this basis, we propose that previously observed, the Rex values might due to the folding/unfolding of the helix A and helix C to form I under conditions that mostly populate the folded protein. Some residues in the RNR motif of P protein exhibit significant Rex values, but are not unfolded in the intermediate conformation we propose. These could correspond to the 8 unassigned extra peaks in the HSQC spectrum.
Our previous studies have shown that there are two anion binding sites in P protein (43). In principle, each state (U, I and N) can bind either one or two ligands (L) (e.g. IL IL2, NL, and NL2). Our previous hydrogen exchange (HX) studies showed that the ligand concentration could highly perturb the exchange pathway (58). In the presence of 20mM sulfate, the primary routes of HX are principally through N and NL (singly liganded folded state). In 200mM sulfate, the primary routes of HX change and are through NL and NL2 states. However in this previous exchange scheme, the intermediate state was not included in the analysis. If the intermediate state is taken into account, it is very likely that the HX from I, or IL (singly liganded I) is more significant than the HX from N and NL. This mechanism may be particularly dominant at low ligand concentration, since the population of IL is significant at 20mM sulfate (the population of I+IL+IL2 is ~5% of the total protein). Under the experimental conditions used to assign the extra peaks (5mM sulfate), the total population of I is ~10% of the total protein (unpublished). Therefore, the intermediate state likely plays a major role in the HX pathway of P protein. Because our previous studies did not recognize the existence of an intermediate, the mechanisms of exchange presented were probably incomplete.
Unstructured N and C termini of I may fold independently to N
As shown in Figure 4, α-helix A and α-helix C are spatially separated from each other. It is possible that the two α-helices fold independently, which would lead to three intermediate states: both helices unfolded, helix A unfolded, and helix B unfolded. There are some hints in the data that this might be the case. As shown in Figure 1A, as additional sulfate is added to P protein all the extra peaks disappear at roughly the same sulfate concentration. However, a more detailed comparison of the extra peaks from the N- and C-termini indicates that the C-terminal residues completely disappear between 10 and 20 mM, and N-terminal residues completely disappear above 20 mM. In addition, the HSQC peaks in the folded state spectrum that appear to correspond to the peaks in the unfolded state spectrum were previously assigned to residues E97, Glu 98, and Lys 101 (circled regions in Fig. 3). These residues are close to the C-terminal helix are observed as extra peaks in the sulfate folded spectrum. Thus, the population of intermediate species with helix A unfolded and helix C folded appears to exceed that of the other two intermediate species under some conditions. The kinetic mechanism developed in our previous kinetic studies (47) of P protein is the simplest mechanistic model consistent with the experimental data. If the folding rate constants of N-terminal and C-terminal helices are similar to each other, the kinetic experiments would be unable to distinguish multiple intermediate conformations. For this reason, we will still treat I as a single thermodynamic state with incompletely characterized conformational heterogeneity.
Effect of histidine ionization on conformational equilibria
The results of the pH titration the various H→K variants indicate that ionization of both His 3 and His 22 perturbs the relative populations of U and I between pH 5.0 and 7.0 and His 22 has a major effect on this equilibrium. Because only three different pH values were examined for each variant, the NMR experiments provide only qualitative information about the effect of histidine ionization on stability of U and I. As shown in Figures 3 and 7, the effect of increasing pH (i.e., deprotonating the histidines) is to shift the I  N conformational equilibrium toward N in the presence of sulfate, and the U
N conformational equilibrium toward N in the presence of sulfate, and the U  I equilibrium toward I in the absence of sulfate. These effects likely arise from unfavorable electrostatic interactions in more compact forms of the protein. At pH 5.0 in the absence of sulfate, the repulsive forces due to additional net positive charge from the protonated histidine residues presumably raises the free energy of I relative to the less compact U, so P protein favors the U state. When the pH is increased, the neutralization of the histidines relieves some repulsive forces and lowers the free energy of I relative to U. The same principle can be used to explain the effect of pH on the I to N equilibrium in the presence of sulfate.
I equilibrium toward I in the absence of sulfate. These effects likely arise from unfavorable electrostatic interactions in more compact forms of the protein. At pH 5.0 in the absence of sulfate, the repulsive forces due to additional net positive charge from the protonated histidine residues presumably raises the free energy of I relative to the less compact U, so P protein favors the U state. When the pH is increased, the neutralization of the histidines relieves some repulsive forces and lowers the free energy of I relative to U. The same principle can be used to explain the effect of pH on the I to N equilibrium in the presence of sulfate.
In order to obtain more quantitative estimates of the effect of pH on U, I and N free energies in F107W and the histidine variants, a more finely stepped pH titration followed by NMR is necessary. The relative populations of U and I could be estimated as a function of pH using the relative peak intensities in the HSQC spectrum for each variant. In addition, the chemical shifts of the 13C2-1H spin pair in selectively labeled His sidechains would provide a sensitive probe for the pKa of each. Thermodynamic coupling between His proton binding and protein conformational change would allow quantitative estimation of the energetic contributions of electrostatic interactions in conformational equilibria.
Ligand interactions and biological relevance of the intermediate state
The results of the PRE NMR experiments, summarized in Figure 9, showed good agreement with the previously modeled binding sites in the P protein (43). Leu 4, Lys 6 and Arg 9 in the N-terminal region are strongly affected by the paramagnetic anions, indicating that they are in close proximity to the sulfate binding site (formed by the His 3, Arg 9, and Arg 68 sidechains) observed in the crystal structure (53). The cross peak of His 3 is missing in the 1H-15N HSQC spectrum and the cross peak of Arg 68 is overlapped with other two strong peaks, Ser 116 and Ser 117. For these reasons, the PRE of H3 and R68 could not be determined and they are accordingly colored gray in Figure 9. The second binding region probed by paramagnetic anion is also close to the previously proposed region (43), based on excess electron density observed in water 108 in the crystal structure (53). Interestingly, Lys 89 (colored in red) is affected most strongly by the paramagnetic anion, followed by Lys 52 (colored in orange). Arg 88 (colored in yellow) is actually affected than either Lys 89 and Lys 52. These results point out some discrepancies between the crystal structure shown in Figure 9 and the PRE NMR data. In the crystal structure, the amide group of Lys 53 is closer to that of Lys 89 than to the Lys 52 amide. However, the peak intensity of Lys 52 is broadened out at lower concentrations of paramagnetic anion compared to Lys 53 indicating that amide group of Lys 52 is closer to Lys 89. The loop region between residue 51 to 60 has the highest B-factor values in the crystal structure, suggesting that the loop has significant flexibility (53). The loop orientation observed in the crystal structure may be one of several conformations of this region present in solution. Based on the PRE NMR experiments, we conclude that the Lys 89 and Lys 52 likely play an important role in forming the second anion binding site of P protein.
Based on our proposed structure of the intermediate state, only one high affinity anion binding site would be present in the forms of the intermediate lacking the N-terminal helix. This conclusion is supported by our ligand induced folding kinetics data (in preparation), which suggests that one anion ligand binds with much higher affinity to the partially folded intermediate than the other. Interestingly, the majority of helix B and the central β-sheet are formed in the intermediate state. Helix B contains the conserved RNR motif in the P protein and has many crucial contacts with P RNA (59). The central β-sheet also has many interactions with pre-tRNA leading sequence during the catalysis (60, 61). Given the preservation of the structural elements key to RNA interactions in the intermediate state, these regions may represent the functional core of the protein and may be key to the holoenzyme assembly process. Moreover, the population of I under physiological condition which is surrounded by different anions will also be important to the process. Future mechanistic studies of holoenzyme assembly will be aimed at delineating the role of the partially folded intermediate ensemble in this process.
Supplementary Material
Acknowledgements
We thank Dr. Ronald Venters for technical assistance with the NMR experiments. We also thank all the members of the Oas laboratory for insightful discussion and advice.
† Supported by National Institute of Health Grants 5RO1GM061367 and 5RO1GM081666 (to T.G.O.).
Abbreviations
- HSQC
heteronuclear single quantum coherence
- PRE
paramagnetic relaxation enhancement
Footnotes
Supporting information available Table S1 lists the chemical shifts of backbone NH, N, CO, Cα, and Cβ of the extra peaks in the folded P protein spectra. Figure S1 shows the titration curves of His 22 and His 105 between pH 5 to pH 8 monitored by nitrogen chemical shifts from 15N HSQC spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Teilum K, Maki K, Kragelund BB, Poulsen FM, Roder H. Early kinetic intermediate in the folding of acyl-CoA binding protein detected by fluorescence labeling and ultrarapid mixing. Proc Natl Acad Sci USA. 2002;99:9807–9812. doi: 10.1073/pnas.152321499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Capaldi AP, Shastry MC, Kleanthous C, Roder H, Radford SE. Ultrarapid mixing experiments reveal that Im7 folds via an on-pathway intermediate. Nat Struct Biol. 2001;8:68–72. doi: 10.1038/83074. [DOI] [PubMed] [Google Scholar]
- 3.Sánchez I, Kiefhaber T. Evidence for sequential barriers and obligatory intermediates in apparent two-state protein folding. J Mol Biol. 2003;325:367–376. doi: 10.1016/s0022-2836(02)01230-5. [DOI] [PubMed] [Google Scholar]
- 4.Clark AC. Protein folding: are we there yet? Arch Biochem Biophys. 2008;469:1–3. doi: 10.1016/j.abb.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krantz BA, Mayne L, Rumbley J, Englander SW, Sosnick TR. Fast and slow intermediate accumulation and the initial barrier mechanism in protein folding. J Mol Biol. 2002;324:359–371. doi: 10.1016/s0022-2836(02)01029-x. [DOI] [PubMed] [Google Scholar]
- 6.Fersht AR. Optimization of rates of protein folding: the nucleation-condensation mechanism and its implications. Proc Natl Acad Sci USA. 1995;92:10869–10873. doi: 10.1073/pnas.92.24.10869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deka P, Rajan PK, Perez-Canadillas JM, Varani G. Protein and RNA dynamics play key roles in determining the specific recognition of GU-rich polyadenylation regulatory elements by human Cstf-64 protein. J Mol Biol. 2005;347:719–733. doi: 10.1016/j.jmb.2005.01.046. [DOI] [PubMed] [Google Scholar]
- 8.Shajani Z, Varani G. 13C NMR relaxation studies of RNA base and ribose nuclei reveal a complex pattern of motions in the RNA binding site for human U1A protein. J Mol Biol. 2005;349:699–715. doi: 10.1016/j.jmb.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 9.Mittermaier A, Varani L, Muhandiram DR, Kay LE, Varani G. Changes in side-chain and backbone dynamics identify determinants of specificity in RNA recognition by human U1A protein. J Mol Biol. 1999;294:967–979. doi: 10.1006/jmbi.1999.3311. [DOI] [PubMed] [Google Scholar]
- 10.Henkels CH. Biophysical studies of Bacillus subtilis ribonuclease P protein. Duke University; 2005. p. xi. 303 leaves. 2005. [Google Scholar]
- 11.Henkels CH, Chang Y-C, Chamberlin SI, Oas TG. Dynamics of backbone conformational heterogeneity in Bacillus subtilis ribonuclease P protein. Biochemistry. 2007;46:15062–15075. doi: 10.1021/bi701425n. [DOI] [PubMed] [Google Scholar]
- 12.Chang Y-C, Oas TG. Osmolyte-Induced Folding of an Intrinsically Disordered Protein: Folding Mechanism in the Absence of Ligand. Biochemistry. 2010 doi: 10.1021/bi100222h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Williamson J. Induced fit in RNA-protein recognition. Nat Struct Biol. 2000;7:834–837. doi: 10.1038/79575. [DOI] [PubMed] [Google Scholar]
- 14.Weeks K. Protein-facilitated RNA folding. Curr Opin Struct Biol. 1997;7:336–342. doi: 10.1016/s0959-440x(97)80048-6. [DOI] [PubMed] [Google Scholar]
- 15.Maity TS, Weeks KM. A threefold RNA-protein interface in the signal recognition particle gates native complex assembly. Journal of Molecular Biology. 2007;369:512–524. doi: 10.1016/j.jmb.2007.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roder H, Colón W. Kinetic role of early intermediates in protein folding. Current Opinion in Structural Biology. 1997;7:15–28. doi: 10.1016/s0959-440x(97)80004-8. [DOI] [PubMed] [Google Scholar]
- 17.Baldwin RL. On-pathway versus off-pathway folding intermediates. Folding & design. 1996;1:R1–8. doi: 10.1016/S1359-0278(96)00003-X. [DOI] [PubMed] [Google Scholar]
- 18.Maki K, Cheng H, Dolgikh DA, Roder H. Folding kinetics of staphylococcal nuclease studied by tryptophan engineering and rapid mixing methods. J Mol Biol. 2007;368:244–255. doi: 10.1016/j.jmb.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shastry MC, Luck SD, Roder H. A continuous-flow capillary mixing method to monitor reactions on the microsecond time scale. Biophys J. 1998;74:2714–2721. doi: 10.1016/S0006-3495(98)77977-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mayor U, Johnson CM, Daggett V, Fersht AR. Protein folding and unfolding in microseconds to nanoseconds by experiment and simulation. Proc Natl Acad Sci USA. 2000;97:13518–13522. doi: 10.1073/pnas.250473497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Park SH, Shastry MC, Roder H. Folding dynamics of the B1 domain of protein G explored by ultrarapid mixing. Nat Struct Biol. 1999;6:943–947. doi: 10.1038/13311. [DOI] [PubMed] [Google Scholar]
- 22.Korzhnev DM, Neudecker P, Mittermaier A, Orekhov VY, Kay LE. Multiple-site exchange in proteins studied with a suite of six NMR relaxation dispersion experiments: an application to the folding of a Fyn SH3 domain mutant. J Am Chem Soc. 2005;127:15602–15611. doi: 10.1021/ja054550e. [DOI] [PubMed] [Google Scholar]
- 23.Korzhnev DM, Salvatella X, Vendruscolo M, Di Nardo AA, Davidson AR, Dobson C, Kay LE. Low-populated folding intermediates of Fyn SH3 characterized by relaxation dispersion NMR. Nature. 2004;430:586–590. doi: 10.1038/nature02655. [DOI] [PubMed] [Google Scholar]
- 24.Palmer AG. NMR characterization of the dynamics of biomacromolecules. Chem Rev. 2004;104:3623–3640. doi: 10.1021/cr030413t. [DOI] [PubMed] [Google Scholar]
- 25.Religa TL, Markson JS, Mayor U, Freund SM, Fersht AR. Solution structure of a protein denatured state and folding intermediate. Nature. 2005;437:1053–1056. doi: 10.1038/nature04054. [DOI] [PubMed] [Google Scholar]
- 26.Nishimura C, Dyson HJ, Wright PE. Identification of native and non-native structure in kinetic folding intermediates of apomyoglobin. J Mol Biol. 2006;355:139–156. doi: 10.1016/j.jmb.2005.10.047. [DOI] [PubMed] [Google Scholar]
- 27.Feng H, Zhou Z, Bai Y. A protein folding pathway with multiple folding intermediates at atomic resolution. Proc Natl Acad Sci USA. 2005;102:5026–5031. doi: 10.1073/pnas.0501372102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van Mierlo CP, van den Oever JM, Steensma E. Apoflavodoxin (un)folding followed at the residue level by NMR. Protein Sci. 2000;9:145–157. doi: 10.1110/ps.9.1.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schulman BA, Kim PS, Dobson CM, Redfield C. A residue-specific NMR view of the non-cooperative unfolding of a molten globule. Nat Struct Biol. 1997;4:630–634. doi: 10.1038/nsb0897-630. [DOI] [PubMed] [Google Scholar]
- 30.Cavanagh J. Protein NMR spectroscopy : principles and practice. Academic Press; San Diego: 1996. [Google Scholar]
- 31.Warren JR, Gordon JA. On the Refractive Indices of Aqueous Solutions of Urea. J Phys Chem. 1966;70:297–300. [Google Scholar]
- 32.Wang A, Bolen DW. A naturally occurring protective system in urea-rich cells: mechanism of osmolyte protection of proteins against urea denaturation. Biochemistry. 1997;36:9101–9108. doi: 10.1021/bi970247h. [DOI] [PubMed] [Google Scholar]
- 33.Niranjanakumari S, Kurz J, Fierke C. Expression, purification and characterization of the recombinant ribonuclease P protein component from Bacillus subtilis. Nucleic Acids Res. 1998;26:3090–3096. doi: 10.1093/nar/26.13.3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Edelhoch H. Spectroscopic determination of tryptophan and tyrosine in proteins. Biochemistry. 1967;6:1948–1954. doi: 10.1021/bi00859a010. [DOI] [PubMed] [Google Scholar]
- 35.Löhr F, Rüterjans H. A new triple-resonance experiment for the sequential assignment of backbone resonances in proteins. [DOI] [PubMed]
- 36.Ikura M, Kay LE, Bax A. A novel approach for sequential assignment of 1H, 13C, and 15N spectra of proteins: heteronuclear triple-resonance three-dimensional NMR spectroscopy. Application to calmodulin. Biochemistry. 1990;29:4659–4667. doi: 10.1021/bi00471a022. [DOI] [PubMed] [Google Scholar]
- 37.Yamazaki T, Lee W, Arrowsmith CH, Muhandiram DR, Kay LE. A Suite of Triple Resonance NMR Experiments for the Backbone Assignment of 15N, 13C, 2H Labeled Proteins with High Sensitivity. Journal of the American Chemical Society. 1994;116:11655–11666. [Google Scholar]
- 38.Grzesiek S, Bax A. Correlating backbone amide and side chain resonances in larger proteins by multiple relayed triple resonance NMR. Journal of the American Chemical Society. 1992;114:6291–6293. [Google Scholar]
- 39.Grzesiek S, Anglister J, Bax A. Correlation of Backbone Amide and Aliphatic Side-Chain Resonances in 13C/15N-Enriched Proteins by Isotropic Mixing of 13C Magnetization. Journal of Magnetic Resonance, Series B. 1993;101:114–119. [Google Scholar]
- 40.Lyons BA, Montelione GT. An HCCNH Triple-Resonance Experiment Using Carbon-13 Isotropic Mixing for Correlating Backbone Amide and Side-Chain Aliphatic Resonances in Isotopically Enriched Proteins. Journal of Magnetic Resonance, Series B. 1993;101:206–209. [Google Scholar]
- 41.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. Journal of Biomolecular NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 42.Johnson BA, Blevins RA. NMR View: A computer program for the visualization and analysis of NMR data. Journal of Biomolecular NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 43.Henkels CH, Kurz JC, Fierke CA, Oas TG. Linked folding and anion binding of the Bacillus subtilis ribonuclease P protein. Biochemistry. 2001;40:2777–2789. doi: 10.1021/bi002078y. [DOI] [PubMed] [Google Scholar]
- 44.Berg JM, Tymoczko JL, Stryer L. Biochemistry. 5th ed W.H. Freeman; New York: 2002. [Google Scholar]
- 45.Kim S, Bracken C, Baum J. Characterization of millisecond time-scale dynamics in the molten globule state of alpha-lactalbumin by NMR. Journal of Molecular Biology. 1999;294:551–560. doi: 10.1006/jmbi.1999.3250. [DOI] [PubMed] [Google Scholar]
- 46.McConnell HM. Reaction Rates by Nuclear Magnetic Resonance. The Journal of Chemical Physics. 1958;28:430–431. [Google Scholar]
- 47.Chang Y-C, Oas TG. Osmolyte-Induced Folding of an Intrinsically Disordered Protein: Folding Mechanism in the Absence of Ligand. Biochemistry. 49:5086–5096. doi: 10.1021/bi100222h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yamazaki T, Lee W, Arrowsmith C, Muhandiram D, Kay L. A Suite of Triple Resonance NMR Experiments for the Backbone Assignment of 15N, 13C, 2H Labeled Proteins with High Sensitivity. J Am Chem Soc. 1994;116:11655–11666. [Google Scholar]
- 49.Wittekind M, Mueller L. HNCACB, a High-Sensitivity 3D NMR Experiment to Correlate Amide-Proton and Nitrogen Resonances with the Alpha- and Beta-Carbon Resonances in Proteins. Journal of Magnetic Resonance, Series B. 1993;101:201–205. [Google Scholar]
- 50.Wishart DS, Sykes BD. The 13C chemical-shift index: a simple method for the identification of protein secondary structure using 13C chemical-shift data. J Biomol NMR. 1994;4:171–180. doi: 10.1007/BF00175245. [DOI] [PubMed] [Google Scholar]
- 51.Spera S, Bax A. Empirical correlation between protein backbone conformation and C.alpha. and C.beta. 13C nuclear magnetic resonance chemical shifts. J. Am. Chem. Soc. 1991;113:5490–5492. [Google Scholar]
- 52.Shen Y, Delaglio F, Cornilescu G, Bax A. TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J Biomol NMR. 2009;44:213–223. doi: 10.1007/s10858-009-9333-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stams T, Niranjanakumari S, Fierke C, Christianson D. Ribonuclease P protein structure: evolutionary origins in the translational apparatus. Science. 1998;280:752–755. doi: 10.1126/science.280.5364.752. [DOI] [PubMed] [Google Scholar]
- 54.Solomon I. Relaxation Processes in a System of Two Spins. Physical Review. 1955;99:559. [Google Scholar]
- 55.Bloembergen N, Morgan LO. Proton Relaxation Times in Paramagnetic Solutions. Effects of Electron Spin Relaxation. The Journal of Chemical Physics. 1961;34:842–850. [Google Scholar]
- 56.Inagaki F, Watanabe K, Miyazawa T. Hexacyanochromate ion as a paramagnetic anion probe for active sites of enzymes. Application to ribonuclease A. J Biochem. 1979;86:591–594. doi: 10.1093/oxfordjournals.jbchem.a132561. [DOI] [PubMed] [Google Scholar]
- 57.Fairbrother WJ, Graham HC, Williams RJ. An NMR study of anion binding to yeast phosphoglycerate kinase. Eur J Biochem. 1990;190:161–169. doi: 10.1111/j.1432-1033.1990.tb15560.x. [DOI] [PubMed] [Google Scholar]
- 58.Henkels CH, Oas TG. Ligation-state hydrogen exchange: coupled binding and folding equilibria in ribonuclease P protein. J Am Chem Soc. 2006;128:7772–7781. doi: 10.1021/ja057279+. [DOI] [PubMed] [Google Scholar]
- 59.Niranjanakumari S, Day-Storms JJ, Ahmed M, Hsieh J, Zahler NH, Venters RA, Fierke CA. Probing the architecture of the B. subtilis RNase P holoenzyme active site by cross-linking and affinity cleavage. RNA. 2007;13:521–535. doi: 10.1261/rna.308707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Crary S, Niranjanakumari S, Fierke C. The protein component of Bacillus subtilis ribonuclease P increases catalytic efficiency by enhancing interactions with the 5′ leader sequence of pre-tRNAAsp. Biochemistry. 1998;37:9409–9416. doi: 10.1021/bi980613c. [DOI] [PubMed] [Google Scholar]
- 61.Niranjanakumari S, Stams T, Crary S, Christianson D, Fierke C. Protein component of the ribozyme ribonuclease P alters substrate recognition by directly contacting precursor tRNA. Proc Natl Acad Sci U S A. 1998;95:15212–15217. doi: 10.1073/pnas.95.26.15212. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.