

Abstract
Inflammatory (Ly6Chi CCR2+) monocytes provide defense against infections but also contribute to autoimmune diseases and atherosclerosis. Monocytes originate from bone marrow and their entry into the bloodstream requires stimulation of CCR2 chemokine receptor by monocyte chemotactic protein-1 (MCP1). How monocyte emigration from bone marrow is triggered by remote infections remains unclear. We demonstrated that low concentrations of Toll-like receptor (TLR) ligands in the bloodstream drive CCR2-dependent emigration of monocytes from bone marrow. Bone marrow mesenchymal stem cells (MSCs) and their progeny, including CXC chemokine ligand (CXCL)12-abundant reticular (CAR) cells, rapidly expressed MCP1 in response to circulating TLR ligands or bacterial infection and induced monocyte trafficking into the bloodstream. Targeted deletion of MCP1 from MSCs impaired monocyte emigration from bone marrow. Our findings suggest that bone marrow MSCs and CAR cells respond to circulating microbial molecules and regulate bloodstream monocyte frequencies by secreting MCP1 in proximity to bone marrow vascular sinuses.
Introduction
Recruitment of inflammatory cells to sites of infection is essential for innate immune defense against microbial pathogens (Medzhitov, 2007; Serbina et al., 2008). Induction of host chemokine secretion by microbes and establishment of gradients to guide inflammatory cells to sites of infection serves as a widely accepted paradigm for antimicrobial defense (Handel et al., 2005; Rot and von Andrian, 2004). Deficiencies in chemokine receptors or specific chemokines is associated with defective immune responses to infection and delayed clearance of viral, bacterial and protozoal pathogens (Dunay et al., 2008; Glass et al., 2005; Glass et al., 2006; Kurihara et al., 1997).
Although studies of chemokine-mediated recruitment of inflammatory cells have principally focused on trafficking across the endothelium from the bloodstream into infected tissues (Randolph et al., 1998), recent studies demonstrate that chemokines induced during early stages of microbial infection promote emigration of inflammatory cells from the bone marrow into the bloodstream. Neutrophils and inflammatory monocytes represent two bone marrow-derived cell populations that are essential for antimicrobial defense. Neutrophil emigration from bone marrow involves a finely modulated balance of retention and release mediated by the chemokine receptors CXCR2 and CXCR4 (Eash et al., 2010; Martin et al., 2003). In contrast to neutrophil recruitment, inflammatory monocyte recruitment during infection requires stimulation of the CCR2 chemokine receptor in order to trigger release of cells from the bone marrow into the bloodstream (Serbina and Pamer, 2006; Tsou et al., 2007). However, little is known about the regulation of monocyte emigration from bone marrow beyond a requirement for CCR2 signaling (Crane et al., 2009; Serbina and Pamer, 2006; Tsou et al., 2007).
It is unclear how infections in peripheral tissues or in central organs promote monocyte emigration from bone marrow. One possibility is that low-grade infection of the bone marrow directly stimulates monocyte emigration, a scenario for which there is little evidence and which seems counterintuitive (i.e. dispatching inflammatory cells away from a site of infection). Alternatively, cells at a site of focal infection might produce chemokines that enter the circulation and trigger responses in the bone marrow. This model, given the dilution of chemokines in plasma and their potentially rapid clearance by decoy chemokine-receptors (Jamieson et al., 2005; Rot, 2005), would require massive chemokine secretion at the site of infection in order to trigger responses in remote sites in the bone marrow. Although circulating chemokines can reach high serum concentrations during later stages of infection, in most scenarios inflammatory cells have already been recruited to primary sites of infection at earlier time points (Crane et al., 2009; Jia et al., 2008). A third possibility is that cells in the bone marrow detect low levels of circulating microbial molecules during infection and, in response, express chemokines that promote inflammatory cell emigration into the bloodstream. Evidence for this model is limited, and most studies of chemokine expression in bone marrow have focused on the retention and release of neutrophils and hematopoietic stem cells (Eash et al., 2010; Martin et al., 2003; Sugiyama et al., 2006).
We have generated CCR2 and monocyte chemotactic protein-1 (MCP1) reporter mice to investigate in vivo emigration of inflammatory monocytes from the bone marrow in response to low levels of circulating Toll-like receptor (TLR) ligands and following infection with the intracellular bacterial pathogen Listeria monocytogenes. Intravenous administration of nanomolar concentrations of lipopolysaccharide (LPS) induced emigration of monocytes from the bone marrow into the bloodstream. MCP1-reporter mice revealed that LPS administration or infection with L. monocytogenes induced rapid expression of MCP1 by cells that were tightly associated with endothelial cells lining bone marrow sinuses. MCP1-producing cells in the bone marrow expressed TLRs and, upon isolation and in vitro culture, could differentiate into osteoblasts, indicating that these cells included mesenchymal stem cells (MSCs). Targeted deletion of MCP1 in MSCs resulted in decreased monocyte emigration from the bone marrow upon LPS stimulation and also resulted in increased susceptibility to infection with the intracellular bacterial pathogen L. monocytogenes. Our findings suggest that MSCs and their progeny, including CXC chemokine ligand (CXCL)12-abundant reticular (CAR) cells, sense circulating microbial molecules and, by expressing MCP1, calibrate the circulating frequency of monocytes in the bloodstream.
Results
Systemic TLR ligands induce monocytosis
To determine whether circulating microbial molecules influence circulating monocyte levels, we administered highly purified LPS to mice and measured the frequency of circulating Ly6Chi monocytes (Fig. 1A and B). LPS administration induced a marked, TLR4-dependent, increase in monocyte frequency between 3 and 6 hours following inoculation (Fig. 1C). Stimulation of TLR2, TLR5 or TLR9 also increased circulating monocyte frequencies that depended on the expression of the corresponding TLR (Fig. S1). In contrast to monocytes, neutrophil frequencies continued to increase with increasing doses of LPS (data not shown). While low dose LPS markedly increased circulating monocyte frequencies, this effect was lost when the dosage of LPS was increased to 2 mg (Fig. 1D). This result may be explained by the finding that high doses of LPS induce sequestration of myeloid cells in different vascular beds (Andonegui et al., 2003; Andonegui et al., 2009), potentially obscuring increased emigration of monocytes from the bone marrow. Alternatively, it is possible that higher levels of circulating LPS promote retention of monocytes in the bone marrow. Titration of LPS doses and determination of monocyte frequencies at different times following LPS inoculation indicated that higher LPS doses principally result in retention of monocytes in the bone marrow with a minor contribution of monocyte sequestration in the lungs (Fig. S2). Administration of low dose LPS decreased the frequency of inflammatory monocytes in the bone marrow (Fig. 1E), suggesting that increased frequencies of circulating inflammatory monocytes do not result solely from de-margination of cells from the vascular endothelium or from a splenic reservoir (Swirski et al., 2009).
Fig. 1. Low amounts of LPS drive monocyte emigration from bone marrow.

(A) Increased circulating monocytes were identified as CD11b+Ly6Chi by flow cytometry. Flow cytometry plots are gated on CD45+ nucleated cells. (B) Wildtype (WT) and TLR4-deficient mice were injected intraperitoneally (i.p.) with 2ng LPS. Blood was obtained at the indicated time points following inoculation and analyzed by flow cytometry to determine CD11b+Ly6Chi monocyte percentages in CD45+ nucleated cells in the circulation. (C) WT mice were inoculated with PBS or 20pg to 2ug LPS and blood monocyte frequencies were determined 4 hr after LPS injection. (D) WT mice were inoculated with 2ng LPS. 4 hr later, blood, bone marrow and spleen monocyte frequencies were determined. Data are representative of 3–7 mice per group from at least three independent experiments, represented as mean +/− SEM.
The increase in circulating inflammatory monocytes was dependent on CCR2 and partially dependent on MCP1 (Fig. 2A). Previous studies have demonstrated that MCP1 and MCP3 both contribute to CCR2-mediated monocyte emigration from the bone marrow (Jia et al., 2008; Tsou et al., 2007). Although MCP1 expression can be induced by TLR stimulation (Tsuboi et al., 2002), inflammatory cytokines such as tumor necrosis factor (TNF) and type I interferons induce and amplify MCP1 expression during infection (Jia et al., 2009). Low-dose LPS, however, induced MyD88-dependent monocyte emigration from the bone marrow that is independent of TNF and type I interferon expression (Fig. 2B).
Fig. 2. LPS-induced emigration is CCR2, MCP1 and MyD88 dependent but TNF and type I interferon independent.
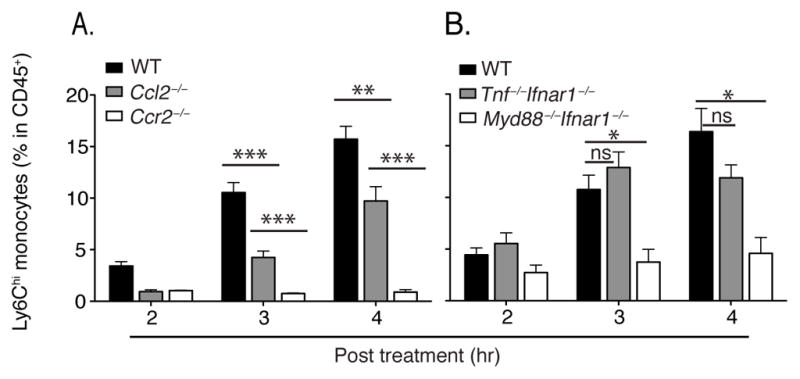
(A, B) WT, Ccl2−/−, Ccr2−/−, Tnf−/−Ifnar1−/− and Myd88−/−Ifnar1−/− mice were inoculated with 20ng LPS. Blood monocyte frequencies were determined at indicated time points. Data are represented as mean +/− SEM.
CCR2+ monocytes traffic to vascular sinuses following TLR stimulation
Since the localization of monocytes in the bone marrow has not been defined, we used a CCR2 reporter mouse strain to visualize CCR2+ inflammatory monocytes (which represents the Ly6Chi population), on the basis of the Green Fluorescent Protein (GFP) expression (Hohl et al., 2009). At baseline, CCR2+ monocytes were widely and evenly dispersed throughout the bone marrow of the mouse femur, generally not directly associated with vascular endothelial (VE)-cadherin positive endothelial cells and only rarely within the lumen of blood vessels or marrow sinuses (Fig. 3A). Administration of LPS to mice, however, changed the localization of CCR2+ monocytes within 2 hours, triggering association with VE-cadherin positive cells at first and leading to increased numbers of CCR2+ monocytes in the lumen of blood vessels (Fig. 3A). To determine whether CCR2 signaling is required for this trafficking, we crossed the CCR2 reporter to the Ccr2−/− background and investigated the response to LPS administration in the bone marrow (Fig. 3B). Trafficking of CCR2+ monocytes into the circulation depended on CCR2 signaling, and CCR2-deficient monocytes did not enter the lumen of blood vessels following LPS administration (Fig. 3C & D).
Fig. 3. Monocyte emigration is CCR2-dependent and is mediated by MCP1 produced by non-hematopoietic cells.

(A and B) Fixed-frozen bone marrow sections from naïve and LPS-treated (2ng/mouse) CCR2 reporter mice (A) and CCR2 reporter-Ccr2−/− mice (B) were stained with Goat anti-mouse VE-cadherin and counterstained with Hoechst (63X). (C) Quantification of exiting monocyte percentage from CCR2 reporter and CCR2 reporter-Ccr2−/− mice following LPS treatment. Exiting monocyte percentages were calculated by dividing the total number of total GFP+ cells by the number of GFP+ cells within one cell distance of VE-cadherin+ endothelium. (E) Ccr2.GFP>WT and Ccr2.GFP>Ccl2−/− bone marrow chimeric mice were treated with 2ng/mouse LPS for 4hr. Fixed-frozen femurs were stained with anti-VE-cadherin. (F) Circulating monocyte frequencies in blood were determined in bone marrow chimeric mice. (G) The percentage of monocytes associated with VE-cadherin expessing endothelials, defined as exiting monocytes, in bone marrow was calculated. Data are representative of three independent experiments. Error bars indicate SEM. Scale bars indicate 90μm.
Monocyte emigration requires MCP1 production by non-hematopoietic cells
To begin to determine the cellular source of MCP1 that drives the repositioning of CCR2+ monocytes in the bone marrow following LPS administration, we generated chimeric mice in which bone marrow from CCR2 reporter mice was transferred into either wild type C57BL/6 recipient mice or mice with a deletion of the Ccl2 gene, which encodes MCP1 (Gu et al., 1998). LPS treatment of the bone marrow chimeric mice revealed that MCP1 production by host-derived, non-hematopoietic cells drove emigration of CCR2+ monocytes from the bone marrow and that monocytes were retained in the bone marrow of MCP1-deficient recipient mice (Fig. 3E). MCP1 deficiency in recipient mice resulted in diminished frequencies of circulating inflammatory monocytes (Fig. 3F) and diminished association of CCR2+ monocytes with bone marrow endothelial cells following LPS administration (Fig. 3G). These results indicate that radiation-insensitive and presumably non-hematopoietic cells produce MCP1 in response to LPS administration and thus induce monocyte emigration from the bone marrow into the circulation.
TLR stimulation induces MCP1 production in proximity to vascular sinuses
Many cell populations, including macrophages, fibroblasts, endothelial cells, vascular smooth muscle cells and renal tubular epithelial cells are capable of producing MCP1 upon stimulation with microbial molecules or inflammatory cytokines (Gu et al., 1998; Ping et al., 1996; Poon et al., 1996; Tsuboi et al., 2002). We hypothesized that non-hematopoietic cells in the bone marrow respond to LPS administration and orchestrate monocyte emigration by producing MCP1. Because tissue staining with existing MCP1-specific antibodies was ineffective, we generated a transgenic reporter mouse strain in which MCP1 was expressed with a linked GFP that is proteolytically cleaved to mark MCP1 producing cells (Fig. 4A). Transgenic mice were normal in appearance and the transgene, when expressed on the Ccl2−/− background, corrected MCP1 deficiency (Fig. S3A–C). Primary hepatocyte cultures generated from MCP1-reporter (M1R) mice became fluorescent upon stimulation with LPS (Fig. S3D) and the fluorescence intensity of bone marrow derived macrophages stimulated with a range of LPS concentrations correlated with the amount of secreted MCP1 (Fig. S3E). Because MCP1 production by non-hematopoietic cells drives monocyte emigration from the bone marrow (Fig. 3E & G), we generated bone marrow chimeric mice in which M1R mice were the recipients of bone marrow from normal C57BL/6 mice. We administered LPS to these mice and, 4 hours later, obtained muscle, brain, liver and bone for histological analyses. While we were unable to detect GFP expression in muscle, brain and liver, we detected marked fluorescence in the bone marrow (Fig. 4B). Much, but not all, GFP expression was closely associated with CD31 staining cells, suggesting that cells associated with blood vessels in the bone marrow, but not other tested tissues, might be a source of MCP1 following LPS administration. Kinetic analysis demonstrated GFP expression in bone marrow as early as 2 hours and increasing at 4 hours after LPS administration (Fig. 4C).
Fig. 4. MCP1-producing cells in bone marrow co-localize with vascular sinuses.

(A) WT>M1R bone marrow chimeric mice were inoculated with 20ng LPS and 4 hr later muscle, brain, liver and bone marrow were isolated, fixed and frozen and stained for CD31 expression and examined by confocal microscopy for GFP expression. Scale bars indicate 90μm. (B) Fixed-frozen bone marrow samples at the indicated time points following 20ng LPS inoculation were stained for CD31 expression and examined by confocal microscopy. Scale bars indicate 47μm. Data are representative of at least three independent experiments.
Bone marrow mesenchymal stem cells respond to TLR-ligands by producing MCP1
To determine which cell population expresses MCP1 in the bone marrow following LPS administration, we stained bone marrow sections for CD31, a marker for endothelial cells. Fig. 5A demonstrates that GFP expression occurred in both CD31+ and CD31-negative cells, but that GFP-expressing, CD31-negative cells were tightly apposed to endothelial cells. Flow cytometric characterization of GFP-expressing cells obtained from collagenase-dissociated bone marrow demonstrated that nearly 90% of MCP1 producing cells were CD31-negative and VE-cadherin-negative (Fig. 5B).
Fig. 5. Marrow endothelial cells and tightly associated MSCs produce MCP1 and promote monocyte emigration.

(A) M1R mice were inoculated with 20ng LPS and 4 hr later bone marrow was sectioned and stained. GFP+ MCP1 producing cells after LPS stimulation overlap (upper) or are tightly associated (lower) with CD31+ endothelial cells. Scale bars indicate 12μm. (B) Bone marrow from stimulated mice was harvested, dissociated and CD45−GFP+ cells (left panel) were stained for expression of CD31 and VE-cadherin (right panel). (C) CD45−GFP+ and CD45−GFP− cells were flow cytometrically-sorted from M1R mice following LPS inoculation and cultured for CFU-F. (D) CD45−GFP+ cells were further differentiated in vitro to osteoblasts. (E) Pdgfrβ, Eng and Tlr4 expression in sorted cells were measured by Q-PCR. (F) Expression of GFP and PDGFRβ by CD45−Ter119−CD31−Sca1− population in the bone marrow. (G) CD45−Ter119−GFP+ cells and CD45−Ter119− CD31+Sca1+ endothelial cells were sorted and CXCL12 mRNA expression was quantified by Q-PCR. (H) CD45−CD31−GFP+ and CD45−CD31+GFP+ cells were sorted and MCP1 mRNA expression was quantified by Q-PCR. Data are represented as mean +/− SEM.
Recent studies have suggested that MSCs might be positioned adjacent to venous sinuses in the bone marrow and could give rise to a wide range of cell populations, including osteoblasts, adipocytes, chondrocytes and fibroblasts (Crisan et al., 2008; Mendez-Ferrer et al., 2010; Sacchetti et al., 2007). MSCs also produce immunomodulatory cytokines but are not known to contribute to antimicrobial defense or to monocyte trafficking within the bone marrow (BM). CXCL12-abundant reticular (CAR) cells have been shown to regulate the localization of hematopoietic stem and B lymphoid progenitor cells in the bone marrow (Sugiyama et al., 2006) and also have progenitor activity, giving rise to multiple mesenchymal cell types (Omatsu et al., 2010). To determine whether CD31−GFP+ cells contained mesenchymal progenitor activity, CD45−GFP+ and CD45−GFP− cells were sorted from the BM of M1R mice 4 hours after LPS administration. All the fibroblastic colony-forming units (CFU-F) and clonogenic capacity of the BM under these culture conditions resided in the CD45−GFP+ fraction (Fig. 5C) and was restricted to the CD31− subset (data not shown). Plating of CD45−GFP− cells at equal or higher density did not generate CFU-F. CFU-F derived from CD45−GFP+ cells could produce osteoblasts following culture and differentiation for three weeks (Fig. 5D). Compared to the CD45−GFP− population, CD45−GFP+ cells were also enriched in the expression platelet-derived growth factor (PDGF) receptor b and endoglin, two markers of bone marrow MSCs (Fig. 5E). In addition, the expression of TLR-2, -3, -4, -8 and -9 was higher in CD45−GFP+ than in CD45−GFP− cells (Fig. 5E and Fig. S4). Since MCP1 expressing cells following LPS administration did not express CD45, Ter119 (a lineage marker for erythroid cells), Sca1 (stem cell antigen-1) or CD31 and express PDGFRb (Fig. 5F), they could also be classified as CAR cells. Indeed, MCP1 expressing cells also expressed high amounts of CXCL12 (Fig. 5G). These results suggest that BM perivascular MSCs and/or CAR cells can detect circulating TLR ligands and produce MCP1, thus directing monocyte emigration from the bone marrow into the bloodstream. While some endothelial cells express MCP1 in response to LPS administration, the majority of cells expressing MCP1 were VE-cadherin and CD31 negative. To further measure MCP1 production by bone marrow endothelial cells and MSCs, we sorted these two populations and determined MCP1 mRNA amounts. Fig. 5H demonstrates that MCP1 expression was induced in both populations after LPS stimulation, and that MSCs expressed slightly higher MCP1 mRNA amounts than endothelial cells on a per cell basis.
To determine whether bone marrow MSCs and their progeny induce monocyte emigration by secreting MCP1 in response to LPS stimulation, we used a genetic approach to conditionally delete the Ccl2 gene from MSCs. Recent studies have demonstrated that bone marrow MSCs express Nestin and that the mesenchymal activity and clonogenicity of CD45− cells reside within the Nestin+ population (Mendez-Ferrer et al., 2010). We generated a conditional Ccl2 targeted mouse strain by inserting loxP sites that flank exons 2 and 3 of the Ccl2 gene and also tagged MCP1 with the HA epitope and a linked but cleaved Red Fluorescent Protein (RFP) gene to act as a reporter (Fig. S5A). Conditional deletion of Ccl2 in MSCs and their progeny was accomplished by crossing Ccl2-RFPflox/flox mice with mice expressing Cre recombinase under the control of Nes promoter. Of note, MCP1-expressing cells were found to express higher amounts of Nestin transcript than endothelial cells (Fig. S5B). Histological analyses of bone marrow sections after LPS administration revealed that MCP1 expression, as visualized by RFP induction, was markedly diminished in Nes-Cre expressing mice (Fig. 6A and Fig. S5C). Expression of MCP1 transcripts in bone marrow endothelial cells of LPS-treated mice was unaffected by Nes-Cre expression in Ccl2-RFPflox/flox mice (Fig. S5D). In contrast, in Tek-Cre expressing mice, in which the Ccl2 gene would be deleted in endothelial and myeloid cells, MCP1 expression was only modestly diminished (Fig. 6A right panel, and Fig. S5C). The percentage of circulating Ly6Chi monocytes was significantly decreased after LPS stimulation in Nes-Cre.Ccl2-RFPflox/flox mice (Fig. 6B and C), suggesting that MCP1 production by Nestin expressing cells drives monocyte emigration from the bone marrow. In contrast, circulating Ly6G+ neutrophils were not affected (Fig. 6D). The lack of MCP1 in endothelial cells, as determined in Tek-Cre.Ccl2-RFPflox/flox mice, significantly decreased monocyte emigration, though to a lesser extent (Fig. 6B). LPS induced monocyte emigration from the bone marrow was not affected in Ccl2-RFPflox/flox expressing Lyz2-Cre or Alb-Cre, which removes MCP1 from myeloid lineage and hepatocytes, respectively (Fig. S6). These findings suggest that MSCs and CAR cells function as the predominant sensors of circulating LPS amounts and, by producing MCP1 in the bone marrow, determine the frequency of circulating inflammatory monocytes.
Fig. 6. MCP1 produced by Nestin-expressing MSCs is required for optimal monocyte emigration after LPS stimulation.

Ccl2-RFPflox/flox, Ccl2-RFPflox/flox expressing Nes-Cre or Tek-Cre, and Ccl2−/− were inoculated with LPS. 4 hr post stimulation, (A) bone marrow was sectioned and stained. Scale bars indicate 40μm. (B) blood was obtained analyzed by flow cytometry to determine CD11b+Ly6Chi monocyte frequencies in the circulation. The emigration kinetics of CD11b+Ly6Chi monocytes and CD11b+Ly6G+ neutrophils are shown in (C) and (D). Error bars indicate SEM.
Infection with Listeria monocytogenes induces MCP1 expression by bone marrow mesenchymal stem cells
Previous studies have demonstrated that infection with L. monocytogenes, an intracellular bacterial pathogen, induces CCR2-dependent emigration of monocytes from the bone marrow (Serbina and Pamer, 2006). To determine whether non-hematopoietic cells produce MCP1 and promote monocyte trafficking during bacterial infection, we used wild type and Ccl2−/− mice to generate bone marrow chimeric mice. Following L. monocytogenes infection, the majority of MCP1 was produced by non-hematopoietic cells and MCP1 production by non-hematopoietic cells was required for monocyte emigration from bone marrow (Fig. 7A). We infected CCR2 reporter mice with L. monocytogenes and characterized monocyte localization in the bone marrow. Within 4 hr of infection, we detected mobilization of monocytes and by 12 hr, most CCR2+ monocytes were associated with cells lining bone marrow venous sinuses (Fig. 7B and Fig. S7). Along similar lines, MCP1-GFP expression in bone marrow was detected 12 hr and 24 hr post infection (Fig. 7C) and, as following LPS administration, was tightly associated with CD31+ endothelial cells. The delay relative to LPS administration likely reflected the requirement for threshold levels of microbial molecules to be introduced into the circulation by expanding bacterial populations. Infection of Nes-Cre.Ccl2-RFPflox/flox mice with L. monocytogenes resulted in diminished emigration of Ly6Chi monocytes from the bone marrow into the bloodstream (Fig. 7D & E). Impaired monocyte recruitment resulted in diminished clearance of bacteria form the spleen (Fig. 7F). Deletion of MCP1 from endothelial cells and hematopoietic cells by Tek-Cre also diminished the emigration of Ly6Chi monocytes, albeit to a lesser extent (Fig. 7G & H). Infected Tek-Cre.Ccl2-RFPflox/flox mice had higher bacterial counts in the spleen, though the difference did not reached a statistically significant level (Fig. 7I). These findings indicate MSCs and CAR cells, and to a lesser extent endothelial cells, are functionally relevant sources of MCP1 during bacterial infection.
Fig. 7. MCP1 production by bone marrow MSCs enhances resistance to Listeria monocytogenes infection.

(A) Bone marrow chimeric mice were infected with 3000 L. monocytogenes. 24 hr after infection, serum was harvested for MCP1 ELISA (upper panel) and bone marrow cells were harvested to quantify Ly6Chi monocytes (lower panel). (B) CCR2 reporter (upper panel) and CCR2 reporter-Ccr2−/− mice (lower panel) were infected with L. monocytogenes. The percentages of exiting monocytes were quantified at different time points following infection. (C) WT-M1R bone marrow chimeric mice were infected with L. monocytogenes. Fixed-frozen sections were prepared at different time points and stained for CD31 expression, and examined by confocal microscopy. Data are representative of at least three independent experiments. Ccl2-RFPflox/flox, Ccl2-RFPflox/flox expressing Nes-Cre or Tek-Cre, and Ccl2−/− mice were infected with L. monocytogenes. CD11b+Ly6Chi monocyte frequencies in the circulation (D, G) and the bone marrow (E, H) were determined by flow cytometry at d1 post infection and the number of viable L. monocytogenes the spleens was quantified at d3 post infection (F, I). Data are represented as mean +/− SEM.
Discussion
Inflammatory monocytes have been implicated in defense against viral, bacterial, fungal and protozoal infections (Serbina et al., 2008). Most of these infections do not directly involve the bone marrow, raising the question of how monocytes are instructed to emigrate from the bone marrow into the circulation. Our studies demonstrated that very low levels of circulating TLR ligands promote the release of bone marrow monocytes into the bloodstream, thus providing these pluripotent cells with an opportunity to circulate to sites of infection or inflammation. Ideally, inflammatory monocytes should reach their target early during infection, so they can optimize pathogen clearance by differentiating into microbicidal cells and marshaling additional inflammatory cell populations. While inflammatory monocytes generally enhance defense against infection, in some circumstances, such as during infection by highly virulent strains of influenza virus, robust recruitment of inflammatory monocytes is associated with increased mortality (Aldridge et al., 2009).
Inflammatory monocytes have also been implicated in the pathogenesis of autoimmunity (Huang et al., 2001), atherosclerosis (Boring et al., 1998; Gu et al., 1998) and other clinical conditions (Swirski et al., 2009). In these circumstances, the density of circulating inflammatory monocytes may contribute to the initiation and/or progression of pathology. Given our finding that low doses of intravenously administered TLR ligands markedly increased circulating numbers of inflammatory monocytes, it is possible that fluctuations in the amounts of microbial molecules absorbed from mucosal surfaces contribute to the progression of inflammatory diseases. In this scenario, increases in epithelial permeability to microbial molecules could result in increased expression of MCP1 by bone marrow MSCs and CAR cells and the release of monocytes into the circulation. The intestinal microbial flora introduces peptidoglycan-derived molecules into the circulation and stimulates Nod1 receptors in bone marrow neutrophils, demonstrating the plausibility of a model of innate immune activation by intestinally absorbed microbial molecules (Clarke et al., 2010). In other studies, marked variation in the amount of circulating LPS in humans has been associated with the lipid composition of the diet (Erridge et al., 2007; Ghoshal et al., 2009). Circulating LPS has also been implicated in the progression of HIV infection (Brenchley et al., 2006).
We found that in vivo MCP1 expression under baseline conditions (i.e. in the absence of LPS administration and in uninfected mice) was very low in the blood and in the bone marrow, as determined by MCP1 ELISA or by fluorescence microscopy of the M1R mouse strain, and that LPS administration or L. monocytogenes infection induced de novo production of MCP1. This result contrasted with a recent study that uses transgenic mice to report MCP1 expression in the bone marrow before and after LPS administration (Jung et al., 2009). One important difference between our studies is the dosage of LPS. While Jung et al. administered roughly 200 mg of LPS, we found that monocyte emigration from the bone marrow was induced by much lower doses of LPS and, surprisingly, not induced by LPS doses exceeding 2 mg. Thus, we believe that our experiments are characterizing vastly different events, with Jung et al.’s system approximating the pathophysiology of septic shock, while our system more closely resembled early stage and far less severe infections. Furthermore, monocyte responses to very low amounts of circulating LPS may model events that occur during low-level fluctuations in circulating microbial molecules following their absorption from the gut. A second difference between our studies is the disparity in baseline expression of MCP1 in mice. Here we speculate that colonization of mice with different flora, and perhaps the presence or absence of intestinal microbes such as Helicobacter hepaticus or Segmented Filamentous Bacterium may have enhanced MCP1 expression in the bone marrow. Determining whether the composition of the intestinal microbial flora influences MCP1 expression and monocyte trafficking from the bone marrow into the blood stream requires further investigation.
Recruitment of monocytes from the bone marrow during systemic infection with complex organisms such as Listeria monocytogenes or Toxoplasma gondii is likely mediated by multiple redundant pathways that include TLR signaling but also TLR-independent induction of inflammatory cytokines and chemokines. Thus, while we demonstrated TLR-stimulation by LPS can induce MCP1 production by bone marrow MSCs and CAR cells, leading to the emigration of Ly6Chigh monocytes into the bloodstream, previous studies from our laboratory have also demonstrated that in the setting of L. monocytogenes infection, monocyte emigration from the bone during early time points following bacterial inoculation is MyD88 independent (Serbina et al., 2009; Serbina et al., 2003). Monocyte emigration from the bone marrow is markedly diminished, however, in the combined absence of both MyD88 and the type I interferon receptor (Jia et al., 2009), suggesting that, in the setting of bacterial infection, bone marrow MSCs can express MCP1 in response to inflammatory cytokines, such as type I interferon, in the absence of TLR-mediated signals.
Our studies demonstrated mesenchymal stem cells and CAR cells in the bone marrow sense circulating TLR ligands and, by producing MCP1, modulate the frequency of bloodstream Ly6Chi CCR2+ monocytes. This mechanism calibrates the host’s innate immune tone, on one hand enhancing antimicrobial defenses, while, on the other hand, potentially exacerbating non-infectious inflammatory diseases. How reticular cells in bone marrow orchestrate monocyte emigration from the bone marrow, beyond producing MCP1, remains unclear. It is possible, since the endothelium of the bone marrow is fenestrated (Tavassoli and Shaklai, 1979), that MSCs or CAR cells directly abut the vascular compartment and thus guide monocytes into the bloodstream. Alternatively, bone marrow reticular cells may express MCP1 on the abluminal side of endothelial cells, thus guiding monocytes to vascular sites at which point other trafficking mechanisms may be activated to enable entry into the bloodstream. Further studies will be required to distinguish between these possibilities. Deeper understanding of these processes may lead to novel therapeutic approaches to control monocyte emigration from bone marrow and monocyte frequencies in the circulation.
Methods
Mice
All mice used in this study were bred at Memorial Sloan-Kettering Research Animal Resources Center. Ccl2−/−, Ccr2−/− and CCR2 reporter mice were previously described (Hohl et al., 2009; Jia et al., 2008). Ifnar1−/− mice were purchased from B&K Universal. Myd88−/− mice were obtained from Dr. S. Akira (University of Osaka, Osaka, Japan). Ifnar1−/− and Myd88−/− mice strains were crossed to obtain Myd88−/−Ifnar1−/− mice. M1R mice were crossed with Ccl2−/− mice to obtain the M1R-Ccl2−/− strain, and CCR2 reporter mice were crossed with Ccr2−/− mice to obtain the CCR2 reporter-Ccr2−/− strain. All mice were either generated on the C57BL/6 background or backcrossed at least 10 generations onto the C57BL/6 background. Mice were housed in a specific pathogen-free facility and all animal studies were conducted in compliance with protocols approved by the animal care committee at Memorial Sloan-Kettering Cancer Center.
Generation of MCP1 reporter (M1R)
MCP1 reporter mice were generated by bacterial artificial chromosome (BAC)-mediated transgenesis using the recombineering strategy described by Heintz and colleagues (Gong et al., 2002). Briefly, the endogenous Ccl2 locus was identified on BAC clone RP23-328G11 (~220 kb in size; obtained from CHORI) derived from chromosome 11 by polymerase chain reaction (PCR) screening of candidate BACs mapped to this region. A 2.5-kb fragment that contained 500 bp upstream and 2.0 kb downstream of the Ccl2 gene start codon was amplified and modified immediately upstream of the stop codon in exon 3 of the Ccl2 gene by insertion of nucleotides encoding the HA peptide (-YPYDVPDYA-), followed by a 19 residue aphthovirus 2A cleavage site (-APVKQTLNFDLLKLAGDVESNPGP-) (Donnelly et al., 2001), a furin cleavage site (-RAKR-) (Thomas, 2002), and aa 2–239 of enhanced GFP (Tsien, 1998). The GFP coding sequence was followed by a stop codon. The resulting 3.3 Kb fragment was cloned into Asc I and Not I sites in the shuttle vector pLD53SC.AB, obtained from Dr. Dan R. Littman (New York University, New York, NY).
To modify BAC clone RP23-328G11 for transgenesis, the shuttle vector containing the 3.3 kb Ccl2.GFP insert was integrated into the BAC by homologous recombination and cointegrates were selected by chloramphenicol and ampicillin treatment. Resolution of cointegrates through a second homologous recombination event was achieved by negative selection on sucrose, resulting in the complete excision of the shuttle vector backbone that includes the SacB gene (For details, see reference (Gong et al., 2002)). The resulting modified BAC encoding GFP under control of the endogenous Ccl2 promoter and regulatory elements was analyzed by Southern blotting to verify GFP integration at the expected site, the Ccl2 gene was sequenced, purified by centrifugation through a cesium chloride gradient and injected into fertilized C57BL/6 oocytes. Two potential founder animals were identified among 41 offspring screened by PCR.
Generation of Ccl2 targeted mice
The floxed Ccl2 mouse strain was generated using standard gene targeting protocol using Cre-lox and Flp-Frt recombination systems. Briefly, mouse genomic sequences of the Ccl2 gene were isolated from BAC clone RP23-328G11 (obtained from CHORI), and modified by tagging the 3′ end with a cleavable RFP (mcherry) as described in the methods of generating M1R. The targeting vector was constructed by cloning the modified sequences together with 3kb and 5kb homologous regions flanking the Ccl2 locus into plasmid pEZ-Frt-lox. Two LoxP sites were introduced and flank the second and third exons, which encode the functional domain of MCP1. The linearized targeting vector was transfected into embryonic stem cells derived from the C57BL/6 strain. Homologous recombinants were drug selected with G418 and ganciclovir and further identified by Southern-blot analysis. Clones carrying the mutated allele of the Ccl2 gene were injected into blastocysts and implanted into pseudopregnant foster mice. Chimeric progeny were bred to C57BL/6 mice, and the F1 generation was screened for germline transmission of the mutated allele. The neomycin resistance gene carried on the mutate allele and flanked with Frt sites was removed by breeding to transgenic mice carrying the gene encoding flippase. The resultant floxed MCP1 RFP mice were maintained and named Ccl2-RFPflox/flox. Mice with tissue specific, conditional deletion of Ccl2 were generated by breeding Ccl2-RFPflox/flox to transgenic strains carrying Nes-Cre, Tek-Cre, Lyz2-Cre, Alb-Cre, respectively (all purchased from Jackson Laboratories).
TLR stimulation and L. monocytogenes infection
LPS (lrl-pelps), Pam3Cys (tlrl-pms), Flagellin (tlrl-pstfla) and CpG (tlrl-modna) were purchased from InvivoGen (San Diego, CA). Mice were injected intraperitoneally (i.p.) with 20 pg-2 ug LPS, 200 ng Pam3Cys, 200 ng Flagellin, 20 mg CpG. Bacteria were grown to log phase (A600 of 0.1), and mice were infected intravenously (i.v.) with 3000 L. monocytogenes strain 10403S as described previously (Jia et al., 2008).
Bone marrow immunofluorescence
Mice were euthanized and perfused with 4% paraformaldehyde. Femurs were postfixed, cryoprotected, and snap frozen in OCT compound. Sections (12 μm) were blocked (5% donkey serum) and incubated at 2ug/ml in primary Rat anti-CD31 (MEC-13.3, BD 550274), Goat anti-VE-cadherin (R&D BAF1002), Rabbit anti-RFP-biotin (Abcam ab34771). After incubation in fluorophore-conjugated secondary antibodies at1 μg/ml, Donkey anti-rat IgG Alexa 594 (A21209, Invitrogen), Donkey anti-goat Cy3 (705-165-147, Jackson IR), and Streptavidin-Texas Red (200-072-211, Jackson IR), sections were counterstained with Hoechst 33342(H3570, Invitrogen). Images were acquired with Leica TCS SP2 AOBS laser scanning confocal microscope with 20× and 63× objective lens.
Flow cytometry
Single-cell blood, bone marrow and spleen samples were prepared as described before (Jia et al., 2008). Briefly, blood samples were harvested from mouse-tail and resuspended in heparin. Bone marrow cells were collected by flushing mouse femurs with ice-cold PBS. To obtain bone marrow endothelial cells and MSCs, bone marrow cells were further digested in 10mg/ml collagenase type IV (Worthington) and 20 U/ml DNase (Roche) at 37°C for 40 min. Spleens were harvested, minced in PBS, 5% FCS, 3 mg/ml collagenase type IV (Worthington), and 20 U/ml DNase (Roche) and were incubated at 37°C for 60 min. Lysis of red blood cells was performed and samples were resuspended in ice-cold PBS with 1% BSA. Cell suspensions were enumerated using a Z2 Coulter counter (Beckman Instruments) with a 6–15 μm window, and analyzed on a BD LSR II cytometer. All antibodies were purchased from BD Biosciences unless otherwise stated. The staining protocols included combinations of the following antibodies: anti-Ly6C (clone AL-21, FITC), anti-Ly6G (1A8, PE), anti-CD11b (M1/70, PerCP-Cy5.5), anti-CD45 (30F-11; APC), anti-CD31 (MEC-13.3, PE) and anti-VE-cadherin (BAF1002, R&D).
CFU-F assay from sorted cells
CD45− GFP+ and CD45− GFP− cells were flow cytometrically-sorted from the BM of M1R transgenic mice 4 hr after LPS administration and cultured with red-phenol-free α-MEM medium supplemented with 15% FBS and 1% penicillin–streptomycin (Invitrogen) at 37° C under 5% CO2 in a water-jacketed incubator. Half-medium changes were performed every three-four days. Fibroblastic colonies were counted after 10 days in culture. Osteoblastic differentiation was induced by culturing cells for 3 weeks with 50 mg/ml L-ascorbic acid 2-phosphate, 10 mM glycerophosphate (Sigma) and 15% FBS in α-MEM with 1% penicillin-streptomycin (Invitrogen).
RNA isolation and quantitative real-time RT-PCR
Sorted cells were collected in lysis buffer and RNA isolation was performed using the Dynabeads mRNA DIRECT Micro Kit (Invitrogen). Conventional reverse transcription, using the Sprint PowerScript reverse transcriptase (Clontech), was performed in accordance with the manufacturers’ instructions. Quantitative real-time RT-PCR (Q-PCR) was performed with SYBR GREEN on an ABI PRISM 7900HT Sequence Detection System (Applied Biosystems). Primers were designed with the Primer Express software (Applied Biosystems) and when possible were selected to span introns to prevent the amplification of contaminating genomic DNA. A primer concentration of 300 nM was found to be optimal in all cases. The sequences of the oligonucleotides used are available upon request. The PCR protocol consisted of one cycle at 95 °C (10 min) followed by 40 cycles of 95 °C (15 s) and 60 °C (1 min). A dissociation curve analysis was included after each experiment to confirm the presence of a single product and the absence of primer dimers. Expression of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was generally used as a standard. The average threshold cycle number (Ct) for each tested mRNA was used to quantify the relative expression of each gene: 2^[−Ct(gene)−Ct(GAPDH)].
ELISA
Murine MCP1 was quantified using an ELISA kit from BD Biosciences. Blood was harvested and clotted to obtain serum for ELISA. To obtain organ lysates for chemokine assays, organs were harvested at the indicated times following infection, macerated in ice-cold PBS containing 0.01% Triton X-100, and centrifuged at 10,000 × g.
Statistical analyses
The unpaired Student’s t test was used for statistical analyses using GraphPad Prism 5.0 software. P values were calculated by t-test, * p<0.05, ** p<0.01, *** p<0.001. p < 0.05 was considered statistically significant. Error bars indicate SEM.
Supplementary Material
Acknowledgments
This work was supported by NIH grants 5R37AI039031 and 5P01CA023766-31 to EGP; K08AI071998 to TMH; RO1DK056638 and RO1HL097819 to PSF; Scholar Award from the American Society for Hematology to SM-F.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aldridge JR, Jr, Moseley CE, Boltz DA, Negovetich NJ, Reynolds C, Franks J, Brown SA, Doherty PC, Webster RG, Thomas PG. TNF/iNOS-producing dendritic cells are the necessary evil of lethal influenza virus infection. Proc Natl Acad Sci U S A. 2009;106:5306–5311. doi: 10.1073/pnas.0900655106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andonegui G, Bonder CS, Green F, Mullaly SC, Zbytnuik L, Raharjo E, Kubes P. Endothelium-derived Toll-like receptor-4 is the key molecule in LPS-induced neutrophil sequestration into lungs. J Clin Invest. 2003;111:1011–1020. doi: 10.1172/JCI16510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andonegui G, Zhou H, Bullard D, Kelly MM, Mullaly SC, McDonald B, Long EM, Robbins SM, Kubes P. Mice that exclusively express TLR4 on endothelial cells can efficiently clear a lethal systemic Gram-negative bacterial infection. J Clin Invest. 2009;119:1921–1930. doi: 10.1172/JCI36411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boring L, Gosling J, Cleary M, Charo IF. Decreased lesion formation in CCR2−/− mice reveals a role for chemokines in the initiation of atherosclerosis. Nature. 1998;394:894–897. doi: 10.1038/29788. [DOI] [PubMed] [Google Scholar]
- Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, Kazzaz Z, Bornstein E, Lambotte O, Altmann D, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med. 2006;12:1365–1371. doi: 10.1038/nm1511. [DOI] [PubMed] [Google Scholar]
- Clarke TB, Davis KM, Lysenko ES, Zhou AY, Yu Y, Weiser JN. Recognition of peptidoglycan from the microbiota by Nod1 enhances systemic innate immunity. Nat Med. 2010;16:228–231. doi: 10.1038/nm.2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane MJ, Hokeness-Antonelli KL, Salazar-Mather TP. Regulation of inflammatory monocyte/macrophage recruitment from the bone marrow during murine cytomegalovirus infection: role for type I interferons in localized induction of CCR2 ligands. J Immunol. 2009;183:2810–2817. doi: 10.4049/jimmunol.0900205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crisan M, Yap S, Casteilla L, Chen CW, Corselli M, Park TS, Andriolo G, Sun B, Zheng B, Zhang L, et al. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell. 2008;3:301–313. doi: 10.1016/j.stem.2008.07.003. [DOI] [PubMed] [Google Scholar]
- Donnelly ML, Hughes LE, Luke G, Mendoza H, ten Dam E, Gani D, Ryan MD. The ‘cleavage’ activities of foot-and-mouth disease virus 2A site-directed mutants and naturally occurring ‘2A-like’ sequences. J Gen Virol. 2001;82:1027–1041. doi: 10.1099/0022-1317-82-5-1027. [DOI] [PubMed] [Google Scholar]
- Dunay IR, Damatta RA, Fux B, Presti R, Greco S, Colonna M, Sibley LD. Gr1(+) inflammatory monocytes are required for mucosal resistance to the pathogen Toxoplasma gondii. Immunity. 2008;29:306–317. doi: 10.1016/j.immuni.2008.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eash KJ, Greenbaum AM, Gopalan PK, Link DC. CXCR2 and CXCR4 antagonistically regulate neutrophil trafficking from murine bone marrow. J Clin Invest. 2010;120:2423–2431. doi: 10.1172/JCI41649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erridge C, Attina T, Spickett CM, Webb DJ. A high-fat meal induces low-grade endotoxemia: evidence of a novel mechanism of postprandial inflammation. Am J Clin Nutr. 2007;86:1286–1292. doi: 10.1093/ajcn/86.5.1286. [DOI] [PubMed] [Google Scholar]
- Ghoshal S, Witta J, Zhong J, de Villiers W, Eckhardt E. Chylomicrons promote intestinal absorption of lipopolysaccharides. J Lipid Res. 2009;50:90–97. doi: 10.1194/jlr.M800156-JLR200. [DOI] [PubMed] [Google Scholar]
- Glass WG, Lim JK, Cholera R, Pletnev AG, Gao JL, Murphy PM. Chemokine receptor CCR5 promotes leukocyte trafficking to the brain and survival in West Nile virus infection. J Exp Med. 2005;202:1087–1098. doi: 10.1084/jem.20042530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass WG, McDermott DH, Lim JK, Lekhong S, Yu SF, Frank WA, Pape J, Cheshier RC, Murphy PM. CCR5 deficiency increases risk of symptomatic West Nile virus infection. J Exp Med. 2006;203:35–40. doi: 10.1084/jem.20051970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong S, Yang XW, Li C, Heintz N. Highly efficient modification of bacterial artificial chromosomes (BACs) using novel shuttle vectors containing the R6Kgamma origin of replication. Genome Res. 2002;12:1992–1998. doi: 10.1101/gr.476202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu L, Okada Y, Clinton SK, Gerard C, Sukhova GK, Libby P, Rollins BJ. Absence of monocyte chemoattractant protein-1 reduces atherosclerosis in low density lipoprotein receptor-deficient mice. Mol Cell. 1998;2:275–281. doi: 10.1016/s1097-2765(00)80139-2. [DOI] [PubMed] [Google Scholar]
- Handel TM, Johnson Z, Crown SE, Lau EK, Proudfoot AE. Regulation of protein function by glycosaminoglycans--as exemplified by chemokines. Annu Rev Biochem. 2005;74:385–410. doi: 10.1146/annurev.biochem.72.121801.161747. [DOI] [PubMed] [Google Scholar]
- Hohl TM, Rivera A, Lipuma L, Gallegos A, Shi C, Mack M, Pamer EG. Inflammatory monocytes facilitate adaptive CD4 T cell responses during respiratory fungal infection. Cell Host Microbe. 2009;6:470–481. doi: 10.1016/j.chom.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DR, Wang J, Kivisakk P, Rollins BJ, Ransohoff RM. Absence of monocyte chemoattractant protein 1 in mice leads to decreased local macrophage recruitment and antigen-specific T helper cell type 1 immune response in experimental autoimmune encephalomyelitis. J Exp Med. 2001;193:713–726. doi: 10.1084/jem.193.6.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamieson T, Cook DN, Nibbs RJ, Rot A, Nixon C, McLean P, Alcami A, Lira SA, Wiekowski M, Graham GJ. The chemokine receptor D6 limits the inflammatory response in vivo. Nat Immunol. 2005;6:403–411. doi: 10.1038/ni1182. [DOI] [PubMed] [Google Scholar]
- Jia T, Leiner I, Dorothee G, Brandl K, Pamer EG. MyD88 and Type I interferon receptor-mediated chemokine induction and monocyte recruitment during Listeria monocytogenes infection. J Immunol. 2009;183:1271–1278. doi: 10.4049/jimmunol.0900460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia T, Serbina NV, Brandl K, Zhong MX, Leiner IM, Charo IF, Pamer EG. Additive roles for MCP-1 and MCP-3 in CCR2-mediated recruitment of inflammatory monocytes during Listeria monocytogenes infection. J Immunol. 2008;180:6846–6853. doi: 10.4049/jimmunol.180.10.6846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung H, Bhangoo S, Banisadr G, Freitag C, Ren D, White FA, Miller RJ. Visualization of chemokine receptor activation in transgenic mice reveals peripheral activation of CCR2 receptors in states of neuropathic pain. J Neurosci. 2009;29:8051–8062. doi: 10.1523/JNEUROSCI.0485-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurihara T, Warr G, Loy J, Bravo R. Defects in macrophage recruitment and host defense in mice lacking the CCR2 chemokine receptor. J Exp Med. 1997;186:1757–1762. doi: 10.1084/jem.186.10.1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin C, Burdon PC, Bridger G, Gutierrez-Ramos JC, Williams TJ, Rankin SM. Chemokines acting via CXCR2 and CXCR4 control the release of neutrophils from the bone marrow and their return following senescence. Immunity. 2003;19:583–593. doi: 10.1016/s1074-7613(03)00263-2. [DOI] [PubMed] [Google Scholar]
- Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature. 2007;449:819–826. doi: 10.1038/nature06246. [DOI] [PubMed] [Google Scholar]
- Mendez-Ferrer S, Michurina TV, Ferraro F, Mazloom AR, Macarthur BD, Lira SA, Scadden DT, Ma’ayan A, Enikolopov GN, Frenette PS. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature. 2010;466:829–834. doi: 10.1038/nature09262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omatsu Y, Sugiyama T, Kohara H, Kondoh G, Fujii N, Kohno K, Nagasawa T. The essential functions of adipo-osteogenic progenitors as the hematopoietic stem and progenitor cell niche. Immunity. 2010;33:387–399. doi: 10.1016/j.immuni.2010.08.017. [DOI] [PubMed] [Google Scholar]
- Ping D, Jones PL, Boss JM. TNF regulates the in vivo occupancy of both distal and proximal regulatory regions of the MCP-1/JE gene. Immunity. 1996;4:455–469. doi: 10.1016/s1074-7613(00)80412-4. [DOI] [PubMed] [Google Scholar]
- Poon M, Hsu WC, Bogdanov VY, Taubman MB. Secretion of monocyte chemotactic activity by cultured rat aortic smooth muscle cells in response to PDGF is due predominantly to the induction of JE/MCP-1. Am J Pathol. 1996;149:307–317. [PMC free article] [PubMed] [Google Scholar]
- Randolph GJ, Beaulieu S, Lebecque S, Steinman RM, Muller WA. Differentiation of monocytes into dendritic cells in a model of transendothelial trafficking. Science. 1998;282:480–483. doi: 10.1126/science.282.5388.480. [DOI] [PubMed] [Google Scholar]
- Rot A. Contribution of Duffy antigen to chemokine function. Cytokine Growth Factor Rev. 2005;16:687–694. doi: 10.1016/j.cytogfr.2005.05.011. [DOI] [PubMed] [Google Scholar]
- Rot A, von Andrian UH. Chemokines in innate and adaptive host defense: basic chemokinese grammar for immune cells. Annu Rev Immunol. 2004;22:891–928. doi: 10.1146/annurev.immunol.22.012703.104543. [DOI] [PubMed] [Google Scholar]
- Sacchetti B, Funari A, Michienzi S, Di Cesare S, Piersanti S, Saggio I, Tagliafico E, Ferrari S, Robey PG, Riminucci M, Bianco P. Self-renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell. 2007;131:324–336. doi: 10.1016/j.cell.2007.08.025. [DOI] [PubMed] [Google Scholar]
- Serbina NV, Hohl TM, Cherny M, Pamer EG. Selective expansion of the monocytic lineage directed by bacterial infection. J Immunol. 2009;183:1900–1910. doi: 10.4049/jimmunol.0900612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serbina NV, Jia T, Hohl TM, Pamer EG. Monocyte-mediated defense against microbial pathogens. Annu Rev Immunol. 2008;26:421–452. doi: 10.1146/annurev.immunol.26.021607.090326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serbina NV, Kuziel W, Flavell R, Akira S, Rollins B, Pamer EG. Sequential MyD88-independent and -dependent activation of innate immune responses to intracellular bacterial infection. Immunity. 2003;19:891–901. doi: 10.1016/s1074-7613(03)00330-3. [DOI] [PubMed] [Google Scholar]
- Serbina NV, Pamer EG. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat Immunol. 2006;7:311–317. doi: 10.1038/ni1309. [DOI] [PubMed] [Google Scholar]
- Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. 2006;25:977–988. doi: 10.1016/j.immuni.2006.10.016. [DOI] [PubMed] [Google Scholar]
- Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, Figueiredo JL, Kohler RH, Chudnovskiy A, Waterman P, et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325:612–616. doi: 10.1126/science.1175202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavassoli M, Shaklai M. Absence of tight junctions in endothelium of marrow sinuses: possible significance for marrow cell egress. Br J Haematol. 1979;41:303–307. doi: 10.1111/j.1365-2141.1979.tb05863.x. [DOI] [PubMed] [Google Scholar]
- Thomas G. Furin at the cutting edge: from protein traffic to embryogenesis and disease. Nat Rev Mol Cell Biol. 2002;3:753–766. doi: 10.1038/nrm934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien RY. The green fluorescent protein. Annu Rev Biochem. 1998;67:509–544. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- Tsou CL, Peters W, Si Y, Slaymaker S, Aslanian AM, Weisberg SP, Mack M, Charo IF. Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J Clin Invest. 2007;117:902–909. doi: 10.1172/JCI29919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuboi N, Yoshikai Y, Matsuo S, Kikuchi T, Iwami K, Nagai Y, Takeuchi O, Akira S, Matsuguchi T. Roles of toll-like receptors in C-C chemokine production by renal tubular epithelial cells. J Immunol. 2002;169:2026–2033. doi: 10.4049/jimmunol.169.4.2026. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.