

Abstract
Activation of phospholipase Cβ (PLCβ) by G proteins leads to a chain of events that result in an increase in intracellular calcium and activation of protein kinase C (PKC). It has been found that PKC phosphorylates PLCβ1 on S887 in vitro without affecting its enzymatic activity or its ability to be activated by Gα(q) proteins. To understand whether S887 phosphorylation affects the enzyme's activity in cells, we constructed two mutants that mimic the wild type and PKC-phosphoryated enzymes (S887A and S887D). We find that these constructs bind similarly to Gα(q) in vitro. When expressed in HEK293 cells, both mutants associate identically to Gα(q) in both the basal and stimulated states. Both mutants diffuse with similar rates and also interact identically with another known binding partner, Translin-Associated factor X (TRAX), which associates with PLCβ1 in the cytosol and nucleus. However, the two mutants localize differently in the cell. We find that S887A has a much higher nuclear localization than its S887D counterpart both in HEK293 cells and PC12 cells. Our studies suggest that PKC phosphorylation regulates the level of PLCβ1 cytosolic and nuclear activity by regulating its cellular compartmentalization.
1. Introduction
Phospholipase Cβ (PLCβ) enzymes are regulated by the Gαq family of heterotrimeric G proteins that are coupled to receptors such as acetylcholine and angiotensin II (see [1]). PLCs catalyze the hydrolysis of the lipid phosphatidylinositol(4,5)-bisphate (PIP2) to produce the second messengers inositol 1,4,5 trisphosphate (IP3) and diacylglyercol (DAG). These messengers in turn cause an increase in intracellular Ca2+ and activate protein kinase C (PKC), respectively. PKC enzymes have been shown to play roles in cellular processes such as cell proliferation, cell cycle progression, differentiation, tumorogenesis, apoptosis, cytoskeletal remodeling, ion channels, and secretion [2]. There are many families of PKC and in this study we focus on PKCα which is activated by an increased level of intracellular Ca2+ and by translocation to membrane driven by DAG, which both result from PIP2 hydrolysis catalyzed by phospholipase C (PLC). Interestingly, there is evidence that PLCβ, but not PLCγ or PLCδ, are PKC substrates [3] suggesting a feedback mechanism. However, in vitro studies have shown that phosphorylation of PLCβ1 on Ser887 does not affect the activity of the PLCβ1 or its ability to be activated by G proteins [4].
Gαq binds the C-terminal region of PLCβ1 resulting in activation of the enzyme (for review see [5]). Unlike Gαq which appears to be localized solely on the plasma membrane in many cell lines, PLCβ1 is localized on the plasma membrane, in the cytosol and in the nucleus [6, 7]. There is evidence that the activity of the nuclear population is regulated by PKC (see[8]). In Swiss 3T3 cells, generation of nuclear DAG by PLCβ1 causes movement of PKCα from the cytosol into the nucleus where it phosphorylates PLCβ1 resulting in reduced nuclear PLC activity [9]. The idea that nuclear PLCβ1 is regulated by PKC is interesting because Gαq, its only established regulator, has yet to be found in the nucleus.
Recently, we found that PLCβ1 interacts with the protein TRAX in the same region as Gαq [10]. TRAX shuttles from the cytosol to the nucleus where it participates in RNA silencing [11]. The cellular interaction between PLCβ1 and TRAX is unclear. It is tempting to speculate phosphorylation may modulate interactions between TRAX and PLCβ1 in the cytosol or in the nucleus.
In general, phosphorylation may provide an additional layer of regulation that would uncouple PLCβ from G protein regulation or TRAX association. We reasoned that although phosphorylation may not directly affect PLCβ activity, it may affect its localization in cells and its association with other proteins. Here, to investigate the role of phosphorylation of S887 of PLCβ1, we made a mutant that cannot be phosphorylated by PKC on S887 (S887A) and one that should mimic the phosphorylated state (S887D). We determined their cellular localization by attaching an eYFP tag on the N-terminus, which also allowed us to monitor their association with eCFP-Gαq by Förster Resonance Energy Transfer (FRET). Our studies suggest that phosphorylation does not affect the ability of PLCβ1 to interact with Gαq in cells, but does affect its cellular localization. Thus, phosphorylation is an important but indirect mechanism of nuclear PLCβ1 regulation.
2. Materials and Methods
2.1 Reagents
Fully functional eCFP-Gαq [7] was from Dr. Catherine Berlot (Geisinger Clinic, Danville, PA) and fully functional eYFP-PLCβ1 was from Loren Runnels (Dept. of Cell Biology, Rutgers University) (see [6]). Purified TRAX and eCFP-TRAX was prepared as described [10].
2.2 Point Mutant Construction
eYFP-PLCβ1S887A and eYFP-PLCβ1-S887D point mutants were generated from the wild-type eYFP-PLCβ1 plasmid using the forward primers CAGCCTGCTCCAGGGGCTGTGAAGGCACCC for S887A and CAGCCTGCTCCAGGGGATGTGAAGGCACCC for S887D.
2.3 Instrumentation
Images, time-lapses, and z-stacks were taken on a Zeiss Axiovert 200M with an AxioCam MRm camera using Axiovision software (see [6]) or an Olympus Fluoview1000 (see [12]). Pixel analysis of confocal images was done using Image J software (NIH). Fluorescence spectra were taken on an ISS-PC (ISS, Urbana, IL).
2.4 Cell Culture and Transfection
PC12 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% equine serum, 5% fetal bovine serum, and 100mM sodium pyruvate and were incubated at 37° C with 5% CO2. Nerve growth factor (NGF, Sigma, St. Louis, MO) was added to a final concentration of 100ng/ml to induce differentiation. Plasmids were introduced into cells grown at 80-90% confluency by electroporation [6]. HEK293 cells were cultured in DMEM plus 10% FBS and 1% PenStrep at 37°C with 5% CO2 and transfected by calcium phosphate precipitation [13]. Cells were imaged ∼48 hours after transfection.
2.5 Activity Measurements
HEK293 cells were transfected with protein expression vectors for eYFP-PLCβ1(wt), eYFP-PLCβ1S887A (887A), eYFP-PLCβ1 (887D) and free eYFP. Isolation of the membrane and cytosolic fractions were based on established procedures (see [14, 15]). Cells were harvested from T25 flasks, washed twice with PBS, brought up in PBS containing 1mM PMSF and 10μg/ml aprotinin, placed on ice, and homogenized. Nuclei were pelleted at 750 × g for five minutes at 4° C and the supernatant spun at 10,000 × g for 35 minutes at 4° C to obtain the cytosolic fraction. The pellet or membrane fraction was brought up in PBS containing 1mM PMSF and 10μg/ml aprotinin. Assays were carried out using 5μg of material for 1 minute incubation at 37°C.
2.6 Single cell FRET measurements
FRET measurements were performed by collecting images through a FRET filter which isolates wavelengths of CFP excitation and YFP emission, along with CPF and YFP filters (Chroma, Inc.) to correct for bleed-through [6]. NetFRET and normalized FRET images were calculated using a plug-in for ImageJ software (N.I.H.) based on the method of [16].
To analyze the intensity distribution of a protein within a z-stack, the images were run through a program which lists the pixel intensity value at a user specified x,y coordinate for a 3 × 3 pixel area (9 pixels through the cell) for all images within the stack and the values for each pixel per slice were then averaged.
2.7 FRAP Studies
Fluorescence recovery after photobleaching (FRAP) measurements were carried out using an N2 laser (Spectra Physics) of 100-milliwatt power to photobleach a circular region of 2 μ diameter. The time dependence of intensity recovery was fit to a single exponential curve.
3. Results
3.1 S887A and S887D bind similarly to Gαq
To determine whether phosphorylation affects the interaction between PLCβ1 and Gαq, we measured the binding of the two mutants to Gαq in vitro. For these studies, we expressed the eYFP-tagged enzymes in HEK293 cells and purified the cytosolic fractions. We then titrated purified, activated Gαq that was labeled with the fluorescence resonance energy transfer (FRET) acceptor (Alexa546) to the cytosolic fractions. We measured the association between eYFP-PLCβ1 to Gαq (GTPγS)-Alexa546 acceptors by their increase in fluorescence intensity when exciting at the donor fluorescence wavelength in the presence and absence of donors. We found that the binding curves of the wild type and the two S887 mutants to Alexa - Gαq (GTPγS) were identical to those previously obtained [10, 17](data not shown) suggesting that phosphorylation of S887 does not affect its association to Gα(q).
We have previously measured the association between eCFP-- Gαq and eYFP-PLCβ1 in living HEK293 cells and PC12 cells [6]. Those studies were carried out by transiently transfecting the cells with the fluorescent-tagged proteins at low levels and measuring the FRET by collecting images through a FRET filter which excites eCFP donors and collects emission from eYFP acceptors. Comparing these images to simultaneous images taken through CFP and YFP filters to correct for bleed-through allowed us to determine the value of FRETof each pixel in real time in the basal state and during stimulation states (for details see [6]). We found that a large fraction of eCFP-Gαq and eYFP-PLCβ1 are associated on the plasma membrane and that the amount of associated protein does not change with stimulation in both PC12 and HEK293 cells (Fig. 1b) [6].
Figure 1.
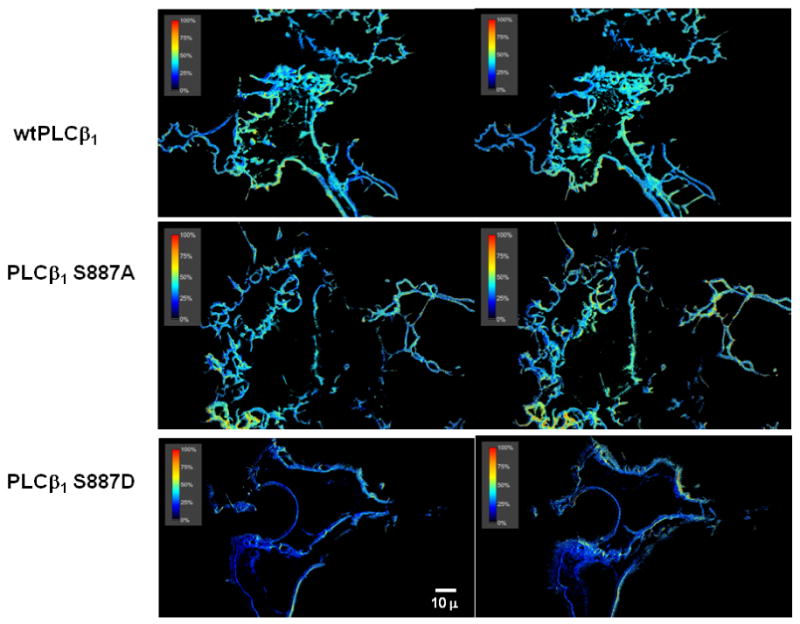
Representative images showing the FRET between eCFP-Gαq and eYFP-PLCβ1 enzymes in transiently transfected HEK293 cells. Note that FRET is only seen on the plasma membrane where Gαq is localized. The FRET averages for several cells are listed in the text. Shown are images for the wt (top) S887A (middle) and S887D (bottom) 0s (left panels) and 140s after (right panels) viewing.
To determine whether point mutations at residue 887 affected the association of PLCβ1 with Gαq, we measured the amount of FRET between eCFP-Gαq and eYFP-PLCβ1 S887A or S887D in living HEK293 cells (Fig. 1). As expected, FRET is seen only on the plasma membrane since this is where all detectable Gαq is localized. We find that the overall values of FRET efficiencies for the mutants are within error of the value determined for wild-type (38±4% wt, 30±6% S887A, 39±3% S887D). These values can be compared to the negative FRET control composed of cells transfected with free eCFP and eYFP (15±2%) and to the positive FRET control composed of eCFP and eYFP attached to opposite ends of a dodecapeptide (70±4%) [6, 18]. Our results show that a high and similar amount of the wild type and mutant enzymes are associated to Gαq. We have previously found that the addition of 1μM carbachol, a stable form of acetylcholine, which causes the activation of Gαq did not significantly affect the cellular distribution of FRET values for the wild type [6] and similarly, we could not detect changes in FRET for the mutant enzymes (see below). Our data suggest that phosphorylation of S887 does not affect cellular interactions between PLCβ1 - Gαq.
3.2 Comparison of diffusion of wild type and mutant PLCβ1
If mutations at S887 disrupt interactions that normally hold PLCβ1 in a complex with other proteins, we would expect that their cellular diffusion would differ from wild type. We tested this idea using fluorescence recovery after photobleaching (FRAP) (see [19]). Here, diffusion is assessed by bleaching a small spot in a cell with a high intensity laser and measuring the rate of fluorescence recovery due to diffusion of fluorophores into the bleached area. We found that both mutants showed almost identical diffusion behavior as wild type (Table 1), which is within error of the parameters obtained for the wild type enzyme. Thus, any differences in cellular localization and environment are not reflected in diffusion.
TABLE 1. Fluorescence Recovery After Photobleaching Results of PLCβ1 enzymes expressed in HEK293 Cells.
| MobileFraction | t1/2 (sec) | n | |
|---|---|---|---|
| eYFP-PLCβ1 | 69 ± 9% | 8.5±1.8 | 5 |
| eYFP-LCβ1S887A | 61 ± 8% | 10.6±2.7 | 6 |
| eYFP-PLCβ1S887D | 60 ± 9% | 9.7±1.9 | 6 |
where enzyme was transiently transfected in HEK293 cells and viewed using L15 medium. In this study, a 2 μ spot was bleached by a 100mW laser and the recovery curves were fit to a single exponential. The mobile fraction represents the percent of the fluorescence that recovers as compared to a spot in the cell that was not bleached. The value of t 1/2 is the midpoint of the recovery.
3.3 Comparison of Cellular Activities of the Mutants
Previous studies suggested that PKC phosphorylation affected the cellular activity of PLCβ enzymes but not the activity of the purified enzyme [3]. To determine whether the cellular activity of the mutants differed, we isolated the cytosolic and membrane fractions of HEK293 cells expressing S887A or S887D mutants (see Methods). We then compared the ability of these fractions to hydrolyze PIP2 by following the amount of 3H-Ins(1,4,5)P3 produced after the addition of small, unilamellar vesicles doped with 3H-PIP2 as previously described [20]. We find that the activities of the two mutants in the membrane fractions were similar, but the activity of S887A in the cytosol was higher (Fig 2). A similar behavior was seen when the mutants were expressed in PC12 cells (data not shown). We note that we did not assay the purity of the samples and it is possible that cross-contamination occurred. However, all samples were prepared identically and if cross-contamination occurred, it would be expected to effect the relative magnitude of the values reducing their differences. Nevertheless, these results suggest that phosphorylation reduces the activity of the cytosolic population of the enzyme, or alternately, that some cytosolic factors enhances the activity of the non-phosphorylated enzyme.
Figure 2.
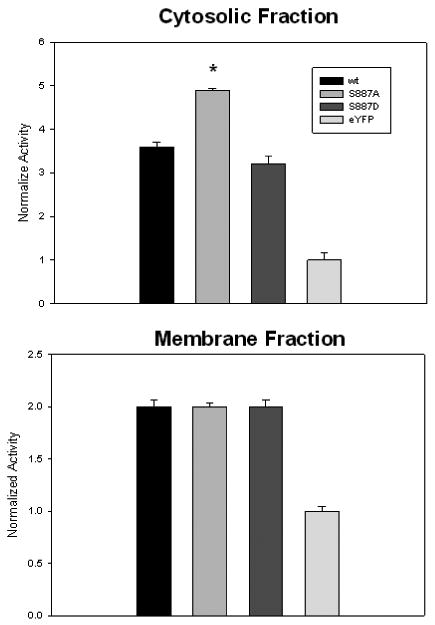
Relative activities of cytosolic (top) and membrane fractions (bottom) of HEK293 cells transfected with eYFP-PLCβ1wt, eYFP-PLCβ1 S887A, eYFP-PLCβ1 S887D or free eYFP. Membrane fractions were prepared as described in the methods. Enzyme concentrations were normalized by eYFP fluorescence and 60 μL of solution was incubated at 30°C with 2mM small unilamellar vesicles consisting of POPC:POPS:POPE:PIP2 31:32:32:5 doped with 3H-PIP2 at 60 μM free Ca2+, The amount of soluble 3H-Ins(1,4,5)P3 product was then measured after 3 minutes, where n=6-9 and S.D.is shown.
3.4 Interaction of the Mutants with TRAX
We have previously found that PLCβ1 binds strongly to TRAX [10]. TRAX and Gαq interact with PLCβ1in the same region. Association between TRAX and PLCβ1 occurs both in the cytosol and nucleus but not on the plasma membrane where PLCβ1 exclusively interacts with Gαq and where TRAX is absent. While TRAX has little effect on the basal activity of PLCβ1, it is possible that TRAX regulates its cellular localization through phosphorylation of S887. To determine whether this is the case, we measured the degree of FRET between eCFP-TRAX and eYFP-PLCβ1 in the cytosol and nucleus under basal and stimulated conditions (Fig. 3) in which the compartments were isolated by confocal imaging We find that the amount of FRET for the mutants and wild type enzyme is within error in both compartments and these values are unaltered under stimulated conditions. These studies suggest phosphorylation does not affect the cellular interaction between PLCβ1 and TRAX.
Figure 3.
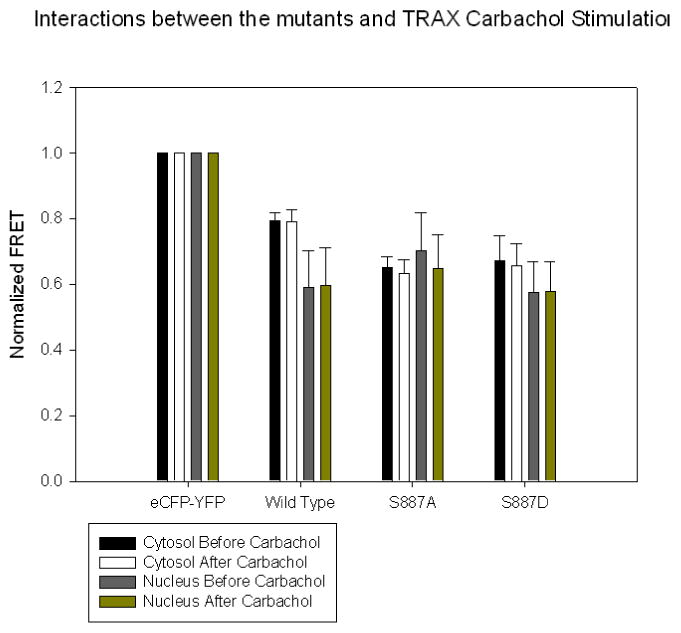
A compilation of FRET values comparing the association between eCFP-TRAX and eYFP-PLCβ1 enzymes in HEK293 cells under basal conditions and 5 minutes after stimulation with 1 μM carbachol where n=6 and SD is shown. FRET values were scaled to values determined for the positive control (eCFP-GLLAPALLGLLA-eYFP, 0.7) and the negative control (free eCFP and free eYFP, 0.15), respectively.
3.5 Distribution of S887A and S887D in differentiated PC12 and HEK293 cells
We viewed the localization of the two mutants overexpressed in HEK293 cells. We find that S887A has a substantially higher nuclear localization as compared to wild type or S887D mutant (Fig. 4). By collecting confocal images of over 10 cells and measuring the intensities of the cytosolic and nuclear fractions in 6-12 regions of each cell, we find that the nuclear/cytosolic ratio of S887A – eYFP is 0.43 ±0.04 n= 242, and S887D-eYFP is 0.23±0.03, n=198. This result suggests that phosphorylation regulates the localization of the enzyme. A similar difference in localization is also found in differentiated PC12 cells (data not shown),
Figure 4.
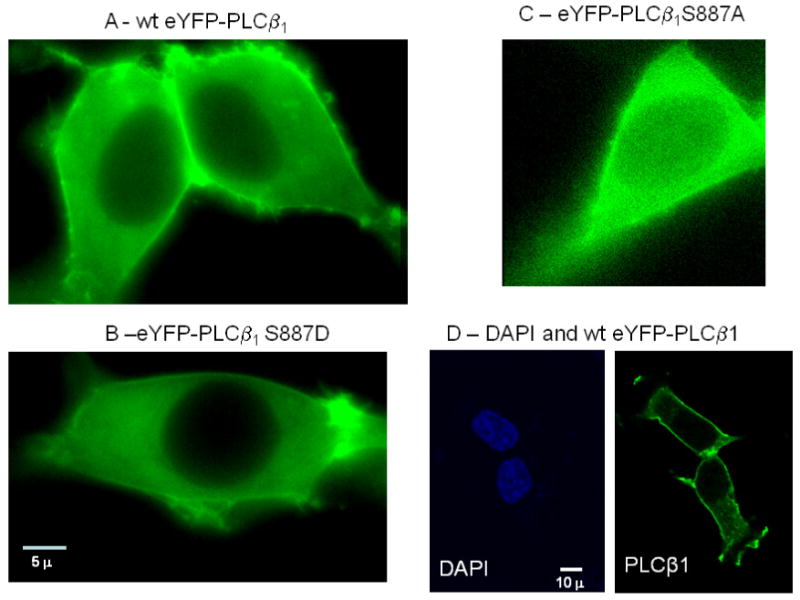
Representative images of HEK293 cells transfected with eYFP-wtPLCβ1 (A), eYFP-S887D (B) and eYFP-S887D where the cells were imaged 48 hours after transfection and viewed through a YFP filter and a 60× objective (see [6] for details). The images shown are from a 0.2μ slice in the middle of the cell so that the nuclear region can clearly be seen. Note that only the S887A mutant had a significant nuclear population. D – A side-by-side comparison of fixed HEK293 cells stained with DAPI to view the nuclei (left panel) of cells expressing eYFP-wtPLCβ1 (right panel),
4. Discussion
Activation of PKC, which is directly downstream of PLCβ, is critical for many cellular processes and the idea that it provides its own feedback loop through PLCβ is enticing However, phosphorylation by PKC of residue 887 does not decrease PLCβ1 activity or its ability to be activated by Gαq in vitro even though residue 887 resides in the same enzymatic region that confers Gαq activation [4]. To more directly determine the effects of PKC phosphorylation of the enzyme, we constructed two point mutants; one that cannot be phosphorylated (S887A) and a phosphorylation mutant (S887D). These mutations are commonly used to probe the role of phosphorylation. There are only small molecular differences between S and A side chains in regards to size and polarity although the S mutant is capable of hydrogen bonding. The charge/mass ratio of D side chains is somewhat smaller than phosphate and may, in limited cases, result in some differences in affinity with cellular components. PLCβ1 is a large protein (140 kDa), and the differences between S887A and S887D and their unphosphorylated and phosphorylated counterparts should be minor. In this regard, the behavior of these mutants are expected to be a good indicator of the role of phosphorylation.
Here, our studies suggest that PKC phosphorylation indirectly regulates PLCβ activity by regulating its cellular localization. We find that the S887A mutant has a much higher nuclear population then the S887D mutant. This latter observation correlates well with the finding that PKC phosphorylation reduces nuclear PLC activity [21, 22] and suggests that this reduction may be due to translocation of phosphorylated PLCβ1 out of the nucleus into the cytosol. We show that this difference in cellular localization is not due to changes in binding to the two known protein cellular partners of PLCβ 1, Gαq and TRAX. Additionally, the differences in cellular localization is not expected to be due to changes in membrane binding since membrane interactions are primarily driven by the N-terminal PH domain of the enzymes [23]. The much higher nuclear localization of S887A as compared to wild type of S887D is not due to a much larger over-expression since the expression of the proteins appeared comparable as determined by comparison of their overall fluorescence intensities.
It was surprising that both S887 mutants interacted similarly with Gαq and TRAX even though this site is located in the C-terminal region where these other proteins interact. We find that both mutants bind similarly to these proteins in the basal and stimulated states suggesting that phosphorylation does not play a role in G protein regulation or TRAX association. Interestingly, even though the activities of the membrane populations of the enzymes are similar, the cytosolic population of S887A displayed a higher cytosolic activity then either the wild type of S887D mutant. The reason for these differing activities is unclear especially since TRAX, PLCβ1's only established cytosolic binding partner, does not affect PLCβ1 activity [10]. The role of the enzyme in the cytosol is unclear. It may serve to regulate the PIP2 levels on internal membranes although its unstimulated activity is low. Alternately, it may serve as a reservoir for the nuclear population.
The role of PLCβ in the nucleus is also unclear. The nucleus has a phosphoinositol lipid signaling pathway that is independent of the one found on the plasma membrane (see [24]) and PLCβ1 is one of the two major PLCs in the nucleus. The second, PLCδ1, lacks the C-terminal extension of the PLCβ family. PLCδ1 binds strongly and specifically to PIP2 and is driven into the nucleus upon elevated PIP2 levels in a cell cycle specific manner [25, 26]. Since PLCβ1 does not bind strongly to PIP2, then other factors must be responsible for its nuclear localization. It has been found that cell stimulation by integrin growth factor results in the nuclear transit of PLCβ1. This in turn promotes nuclear entry of protein kinase C-α by generating DAG. Nuclear PKCα may phosphorylate PLCβ1 at S887 promoting its exit from the nucleus [21]. Our observation that S887A has a far greater nuclear localization correlates well with this model. It is possible that a negative charge on S887 alters the interaction between the enzyme and a cellular component promoting nuclear entry/exit. Alternately, phosphorylation of S887 may alter the exposure of the nearby nuclear localization signal in the C-terminal region of the enzyme.
5. Conclusions
Our results imply that PKC phosphorylation provides a secondary mechanism for PLCβ1 regulation through cellular localization. While the role of PLCβ1 in the nucleus or in the cytosol is not yet known, our studies demonstrate that its nuclear activity can be regulated by the amount of nuclear-localized enzyme via PKC phosphorylation rather than direct activation.
Acknowledgments
This work was supported by GM053132.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rebecchi MJ, Pentyala SN. Physiol Rev. 2000;80:1291–1335. doi: 10.1152/physrev.2000.80.4.1291. [DOI] [PubMed] [Google Scholar]
- 2.Hug H, Sarre TF. Biochem J. 1993;291:329–343. doi: 10.1042/bj2910329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ryu S, Kim UM, Wahl MI, Brown AB, Carpenter G, Huang KP, Rhee SG. J Biol Chem. 1990;265:17941–17945. [PubMed] [Google Scholar]
- 4.Ryu SH, Kim UH, Wahl MI, Brown AB, Carpenter G, Huang KP, Rhee SG. J Biol Chem. 1990;265:17941–17945. [PubMed] [Google Scholar]
- 5.Drin G, Scarlata S. Cellular Signalling. 2007;19:1383–1392. doi: 10.1016/j.cellsig.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dowal L, Provitera P, Scarlata S. J Biol Chem. 2006;281:23999–24014. doi: 10.1074/jbc.M512330200. [DOI] [PubMed] [Google Scholar]
- 7.Hughes TE, Zhang H, Logothetis DE, Berlot CH. J Biol Chem. 2001;276:4227–4235. doi: 10.1074/jbc.M007608200. [DOI] [PubMed] [Google Scholar]
- 8.Cocco L, Martelli AM, Fiume R, Faenza I, Billi AM, Manzoli FA. Adv Enzyme Regul. 2006;46:2–11. doi: 10.1016/j.advenzreg.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 9.Xu A, Wang Y, Xu LY, Gilmour RS. J Biol Chem. 2001;276:14980–14986. doi: 10.1074/jbc.M009144200. [DOI] [PubMed] [Google Scholar]
- 10.Aisiku OR, Runnels LW, Scarlata S. PLoS ONE. 2010;5:e15001. doi: 10.1371/journal.pone.0015001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu Y, Ye X, Jiang F, Liang C, Chen D, Peng J, Kinch LN, Grishin NV, Liu Q. Science. 2009;325:750–753. doi: 10.1126/science.1176325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guo Y, Golebiewska U, D'Amico S, Scarlata S. Journal of Biological Chemistry. 2010;285:24999–25008. doi: 10.1074/jbc.M110.132654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guo Y, Rebecchi M, Scarlata S. J Biol Chem. 2005;280:1438–1427. doi: 10.1074/jbc.M407593200. [DOI] [PubMed] [Google Scholar]
- 14.Medina R, Grishina G, Meloni EG, Muth TR, Berlot CH. J Biol Chem. 1996;271:24720–24727. doi: 10.1074/jbc.271.40.24720. [DOI] [PubMed] [Google Scholar]
- 15.Lamb ME, De Weerd WF, Leeb-Lundberg LM. Biochem J. 2001;355:741–750. doi: 10.1042/bj3550741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xia Z, Yuechueng L. Biophys J. 2001;81:2395–2402. doi: 10.1016/S0006-3495(01)75886-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Runnels LW, Scarlata S. Biochemistry. 1999;38:1488–1496. doi: 10.1021/bi9821519. [DOI] [PubMed] [Google Scholar]
- 18.Philip F, Sengupta P, Scarlata S. J Biol Chem. 2007;282:19203–19216. doi: 10.1074/jbc.M701558200. [DOI] [PubMed] [Google Scholar]
- 19.Lakowicz J. Principles of Fluorescence Spectroscopy. Second. Plenum; New York: 1999. [Google Scholar]
- 20.Runnels LW, Scarlata SF. Biochemistry. 1998;37:15563–15574. doi: 10.1021/bi9811258. [DOI] [PubMed] [Google Scholar]
- 21.Xu A, Suh PG, Marmy-Conus N, Pearson RB, Seok OY, Cocco L, Gilmour R. Molec Cell Biol. 2001;21:2981–2990. doi: 10.1128/MCB.21.9.2981-2990.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu A, Wang YP, Xu LY, Gilmour RG. J Biol Chem. 2001;276:14980–14986. doi: 10.1074/jbc.M009144200. [DOI] [PubMed] [Google Scholar]
- 23.Wang T, Pentyala S, Rebecchi MJ, Scarlata S. Biochemistry. 1999;38:1517–1524. doi: 10.1021/bi982008f. [DOI] [PubMed] [Google Scholar]
- 24.Gonzales ML, Anderson RA. Journal of Cellular Biochemistry. 2006;97:252–260. doi: 10.1002/jcb.20655. [DOI] [PubMed] [Google Scholar]
- 25.Stallings JD, Tall EG, Pentyala S, Rebecchi MJ. J Biol Chem. 2005;280:22060–22069. doi: 10.1074/jbc.M413813200. [DOI] [PubMed] [Google Scholar]
- 26.Stallings J, Zhang Y, Naravez F, Rebecchi MJ. J Biol Chem -submitted. 2007 [Google Scholar]