

Abstract
The membrane-integral transcriptional activator CadC comprises sensory and transcriptional regulatory functions within one polypeptide chain. Its C-terminal periplasmic domain, CadCpd, is responsible for sensing of environmental pH as well as for binding of the feedback inhibitor cadaverine. Here we describe the crystal structure of CadCpd (residues 188–512) solved at a resolution of 1.8 Å via multiple wavelength anomalous dispersion (MAD) using a ReCl62− derivative. CadCpd reveals a novel fold comprising two subdomains: an N-terminal subdomain dominated by a β-sheet in contact with three α-helices and a C-terminal subdomain formed by an eleven-membered α-helical bundle, which is oriented almost perpendicular to the helices in the first subdomain. Further to the native protein, crystal structures were also solved for its variants D471N and D471E, which show functionally different behavior in pH sensing. Interestingly, in the heavy metal derivative of CadCpd used for MAD phasing a ReCl62− ion was found in a cavity located between the two subdomains. Amino acid side chains that coordinate this complex ion are conserved in CadC homologues from various bacterial species, suggesting a function of the cavity in the binding of cadaverine, which was supported by docking studies. Notably, CadCpd forms a homo-dimer in solution, which can be explained by an extended, albeit rather polar interface between two symmetry-related monomers in the crystal structure. The occurrence of several acidic residues in this region suggests protonation-dependent changes in the mode of dimerization, which could eventually trigger transcriptional activation by CadC in the bacterial cytoplasm.
Keywords: cadaverine, pH-sensing, periplasmic domain, ToxR-like protein, X-ray structure
Introduction
Bacteria adapt to changes of external pH by various physiological processes and alterations in gene expression. Thus, several genes are induced in the neutrophilic bacterium Escherichia coli upon exposure to acidic conditions to maintain the cytoplasmic pH in the physiological range between 7.6 and 7.8.1 In particular, the degradative amino acid decarboxylase systems adi, gad, and cad are among those genes.2 Under conditions of an acidic environment and in the presence of external lysine the membrane-integral transcriptional activator CadC is triggered and induces transcription of the cadBA operon, which encodes the cadaverine/lysine-antiporter CadB and the lysine decarboxylase CadA. CadA intracellularly decarboxylates lysine to cadaverine under consumption of one proton. The resulting cadaverine is exported by CadB, which in turn takes up lysine form the external medium.3
CadC is an integral membrane protein of 512 amino acids comprising an N-terminal cytoplasmic DNA-binding domain (residues 1–159), a transmembrane helix (residues 160–187), and a C-terminal periplasmic domain (residues 188–512).4 CadC belongs to the family of ToxR-like proteins, which includes CadC of Salmonella enterica serovar Typhimurium and Vibrio cholerae (regulating the cadBA operon),5 ToxR from V. cholerae (regulating cholera toxin, pilus, and outer-membrane protein expression),6 TcpP from V. cholerae (ToxR-coregulator of virulence gene expression),7 PsaE from Yersinia pestis (regulating fimbriae genes),8 and WmpR from Pseudoalteromonas tunicata (regulating iron uptake).9 Despite low sequence homology (outside the CadC orthologs) proteins of this family are characterized by a common three-domain topology and all combine sensory and transcriptional regulatory functions within one polypeptide chain. Therefore, these proteins represent the simplest known transmembrane signaling system that transduces information across the lipid bilayer without involving chemical modification.
The DNA-binding function of CadC is mediated by its cytoplasmic domain that contains a helix-turn-helix motif similar to the ROII subgroup of DNA-binding domains found in response regulators such as PhoP from Bacillus subtilis,10 VirG from Agrobacterium tumefaciens11 or OmpR from Escherichia coli.12 The DNA-binding sites were identified and the interaction between purified CadC and the promoter region was demonstrated in vitro.13 Expression of the cadBA operon is activated at low external pH and concomitantly available lysine. Recently, it was shown that CadC is not a direct sensor of the external lysine concentration. Instead, lysine is co-sensed via interplay with the lysine-specific permease LysP.14,15 Still, the pH-sensory function is assigned to the periplasmic domain of CadC.16,17 Several CadC variants with single amino acid replacements in the periplasmic domain affecting the pH-dependent cadBA expression were previously identified.16,17 For example, replacement of Asp471 against Asn resulted in a pH-independent activation of expression. In contrast, CadC with Glu at the same position was no longer able to respond to acidification of the environment. Thus, single amino acid replacements can fix CadC either in an ON or an OFF state, respectively.
A combination of biochemical experiments and computational simulations revealed that the transcriptional stimulating activity of CadC is further regulated via feedback inhibition by the product of the CadA decarboxylation reaction, namely cadaverine.18 Consistently, cadaverine binds to the periplasmic domain of CadC with moderate affinity (KD = 96 μM).14 Nevertheless, it is hardly understood how CadC adopts its active state after receiving the pH signal and, also, how cadaverine inactivates CadC. To gain insight into the structural mechanisms, we have carried out a crystallographic analysis of the periplasmic domain CadCpd and its two variants D471E and D471N.
Results
Three-dimensional structure of the periplasmic domain of CadC
The X-ray structure of CadCpd (residues 188–512) was solved via multiple wavelength anomalous dispersion (MAD) at a resolution of 2.3 Å by using a crystal soaked with K2ReCl6. Further crystal structures were solved for the native apo-protein at a resolution of 1.8 Å as well as for the CadCpd variants D471E and D471N at resolutions of 1.9 and 2.2 Å, respectively (Table I). CadCpd appears as an irregularly shaped protein with a maximal diameter of about 60 Å and a diameter of approximately 30 Å at its narrowest site. The entire periplasmic protein domain is essentially composed of two subdomains: the first subdomain comprises a mixed, parallel/antiparallel β-sheet packed on one side against two α-helices as part of a three-helix bundle, whereas the second subdomain is a pure α-helical bundle encompassing 11 helices (Fig. 1). Taken together, α-helices and β-strands account for 70% of the 314 ordered residues in the periplasmic domain of the wild type CadC apo-protein.
Table I.
Crystallographic and Refinement Statistics
| Protein | CadCpd • ReCl62− | CadCpd | ||||
|---|---|---|---|---|---|---|
| Dataset | Peak | Inflection | Remote | Wild Type | D471E | D471N |
| Space group | P6122 | P6122 | P6122 | P3221 | ||
| Unit cell dimensions, a, b, c [Å], α = β = 90°, γ = 120° | 80.56 80.56 199.78 | 80.88 80.88 200.30 | 81.00 81.00 200.65 | 83.90 83.90 199.09 | 83.63 83.63 199.49 | 79.73 79.73 126.01 |
| Wavelength [Å] | 1.17652 | 1.17705 | 1.07813 | 0.91841 | 0.91841 | 0.91841 |
| Resolution range [Å]a | 69.77–2.30 (2.36–2.30) | 70.04–2.60 (2.74–2.60) | 70.15–2.90 (3.06–2.90) | 72.66–1.80 (1.85–1.80) | 72.43–1.90 (1.95–1.90) | 69.05–2.20 (2.26–2.20) |
| I/σIa | 6.2 (2.1) | 7.4 (2.2) | 9.0 (2.5) | 5.6 (2.7) | 5.2 (2.6) | 20.2 (4.6) |
| Rmergeab[%] | 9.5 (35.5) | 8.0 (34.1) | 6.7 (30.6) | 8.5 (27.6) | 8.6 (27.6) | 5.2 (36.7) |
| Unique reflections | 16,693 | 12,472 | 9,169 | 39,276 | 33,530 | 24,043 |
| Multiplicitya | 14.1 (14.5) | 13.9 (14.4) | 13.8 (14.3) | 14.3 (15.1) | 13.5 (13.7) | 5.6 (5.1) |
| Completenessa | 98.5 (97.9) | 99.2 (98.8) | 99.4 (99.2) | 99.9 (100.0) | 99.9 (100.0) | 99.6 (99.9) |
| Anomalous multiplicitya | 7.7 (7.7) | 7.7 (7.7) | 7.7 (7.7) | – | – | – |
| Anomalous completenessa | 99.5 (98.9) | 99.8 (99.5) | 99.9 (99.7) | – | – | – |
| Phasing and refinement: | ||||||
| Figure of merit centric, acentric reflections | 0.697, 0.419 | – | – | – | ||
| Rcryst, Rfreec[%] | 23.0, 27.1 | 19.1, 21.8 | 20.2, 23.1 | 22.8, 27.9 | ||
| Protein atoms | 2,268 | 2,497 | 2,507 | 2,362 | ||
| Ligand atoms | 112 | – | – | – | ||
| Water molecules | 34 | 237 | 234 | 114 | ||
| Average B factor [Å2] | 24.34 | 17.04 | 22.38 | 33.60 | ||
| Geometry: | ||||||
| R.m.s.d. bond lengths, angles [Å, °] | 0.018, 1.752 | 0.012, 1.217 | 0.014, 1.300 | 0.018, 1.513 | ||
| Ramachandran analysis: | ||||||
| Core, allowed, generously allowed, disallowed [%] | 91.3, 8.3, 0.0, 0.4 | 93.0, 6.7, 0.0, 0.4 | 93.4, 5.9, 0.3, 0.3 | 93.2, 6.1, 0.4, 0.4 | ||
Values in parentheses are for the highest resolution shell.
Rfree is the Rcryst with 5% of the reflections that were randomly selected and excluded from refinement: 
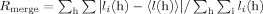
Figure 1.
Overview of the CadCpd crystal structure. (A) Cartoon representation of the apo-protein solved at 1.8 Å. The protein consists of two subdomains. Secondary structure elements are depicted in yellow (β-strands), blue (α-helices of the N-terminal subdomain), and salmon (α-helices of the C-terminal subdomain). The two short 310-helices 334–336 and 411–413 are also shown in salmon. The disordered loop 207–214 has been modelled and is depicted in light gray. The disulfide bond connecting residues Cys208 and Cys272 is shown as red sticks and labeled. (B) Superposition of the CadCpd apo-protein (yellow-orange) with the ReCl62− complex (raspberry) and the variants D471E (green) and D471N (blue). Disordered loops were modelled with plausible geometry and are depicted in light gray. (C) Electrostatic surface representation of the CadCpd apo-structure in an orientation identical to (B). Negatively charged areas are colored red (−10 kBT/e), positively charged areas are colored blue (10 kBT/e). (D) The molecule in the same representation rotated vertically by around 180° with respect to (C), illustrating a narrow entrance to the central cavity indicated by a dashed ellipse. Residues mentioned in the text are labeled. An interactive view is available in the electronic version of the article.
Following to an N-terminal loop at positions 191–194, the polypeptide chain first enters a five-stranded β-sheet at strand B. The chain then adopts a flexible loop not fully defined in the electron density, runs back through an α-helix (helix 1) and enters the β-sheet again as strand A, which is arranged parallel to strand B. After a long loop the chain proceeds back to the β-sheet as strand C, again parallel to B. Two more strands, D and E, connected by shorter loops, follow and are aligned antiparallel to strand C and to each other. The chain then enters an element of two antiparallel α-helices (helices 2 and 3) which, together with helix 1, form the three-helix bundle in the N-terminal subdomain. Notably, all secondary structure elements within this subdomain are oriented in the same direction. For residue Cys208, which is part of the flexible loop connecting strand A and helix 1, no electron density was observed with any dataset, indicating a presumably free (reduced) state in these crystal structures. Because of its proximity to the only other thiol side chain of the well resolved residue Cys272, however, a disulfide bond could be formed between these two amino acids and, for the purpose of illustration, has been modelled here [cf. Fig. 1(A)]. Indeed, the presence of such a disulfide bridge was detected for CadCpd in biochemical experiments (Tetsch et al., in preparation).
Separated by a loop that forms a sharp turn between residues Leu330 and Leu336, the polypeptide chain enters the C-terminal subdomain of CadCpd. Its altogether 11 α-helices (nos. 4–14) are organized as a bundle of five antiparallel α-helical pairs, whereby helix 13 comprises just one turn. These pairs are twisted in a clockwise direction, resulting in a spiral bundle such that the C-terminus of helix 4 is situated close to the N-terminal end of helix 12. The very short helix 13 together with helix 14 form the C-terminus of the structure. Notably, the bundle of α-helices in the second subdomain is roughly oriented perpendicular to the three-helix motif of the first subdomain.
Comparison between the CadCpd apo-structure and its rhenate complex
The crystal structures of the apo-form of CadCpd and its rhenate complex are very similar even though a rather large number of altogether 16 bound ReCl62− complex ions was identified: the root mean square deviation (r.m.s.d.) of the polypeptide chains was 0.79 Å upon superposition of the 282 Cα atoms of common ordered residues in all four models (192–204, 217–245, 251–261, 273–293, 298–394, and 399–509). However, three exposed regions showed significant structural differences [cf. Fig. 1(B)]. One deviation occurs at a stretch formed by residues 294–300, which are part of a loop at the N-terminus of α-helix 2. Second, the stretch 331–339 forms part of a loop at the N-terminus of α-helix 4 and is located close to a bound ReCl62− ion at the protein surface. Finally, the segment 388–399 forms a loop connecting α-helices 6 and 7, whose Cα positions are shifted up to 6.7 Å. Two ReCl62− ions were bound in the vicinity of this site.
Structures of the CadCpd variants D471E and D471N
Residue Asp471 was previously identified in a random mutagenesis study to be involved in pH-sensing.16 Recent site-directed mutagenesis experiments furthermore revealed that replacement of Asp471 with Glu (D471E) resulted in a CadC variant preventing cadBA expression, also under acidic conditions (OFF state), whereas the variant with an Asn side chain at this position (D471N) led to a pH-insensitive active protein (ON state).17 The X-ray structures of wild type CadCpd and its variant D471E were superimposable with an r.m.s.d. value of 0.11 Å using 282 Cα atoms, indicating a virtually identical conformation [Fig. 1(B)]. However, the deviations in the variant D471N, which was crystallized under acidic conditions in a different space group (see Table I), were more significant and reflected by a higher r.m.s.d. value of 0.95 Å.
Considerable differences in the structures of wild type CadCpd and its variant D471N occur at residues 296–297 in a loop between β-strand E and α-helix 2 in the N-terminal subdomain. There is also discrepancy in a long stretch comprising residues 320–340, which connects the N- and the C-terminal subdomains. It includes α-helix 3, a loop followed by a 310-helix, and the N-terminal part of α-helix 4 [cf. Fig. 1(B)]. Another deviation is evident at the amino acid stretch 388–415 comprising the C-terminus of α-helix 6, a loop, α-helix 7, another loop, a 310-helix, and the connecting loop to α-helix 8. In contrast, α-helices 9–14 do not show significant deviations, in particular α-helix 11, which carries the mutation at position 471. Notably, among all those differences only two loops are involved in crystal packing contacts: stretch 293–298 with contacting residues Val293, Thr295, Asn296, and Asn298 and stretch 390–393 with contacting residues His390 and Pro391. Overall, it seems that wild type CadCpd was crystallized in an inactive state also mimicked by CadCpd(D471E), whereas CadCpd(D471N) should better represent the active state, which may also be reflected by the fact that the D471N mutation led to a different crystallization behavior (and altered space group) of CadCpd.
Crystal contacts of the CadCpd wild type protein and its variants D471E and D471N
The wild type CadCpd (both apo-protein and Re-complex) and the variant D471E isomorphously crystallized in the same space group P6122 and, hence, both proteins form the same crystal packing contacts with altogether five neighboring symmetry-equivalent molecules. Interestingly, a detailed analysis of these packing interactions using the PISA server19 indicated the formation of a functional dimer with one of the neighboring molecules, which was similarly detectable also in the P3221 crystal form of the D471N variant (Fig. 2). The putative dimers in all three crystal structures yielded a complexation significance score (CSS) of 1.000. Furthermore, the shape complementarity of the wild type CadC dimer was calculated as SC = 0.76 (using a 1.7 Å probe sphere radius), which is in the range of physiologically functional protein dimers.20 These structural findings were backed by biochemical results of size exclusion chromatography experiments, revealing dimerization of the periplasmic domain also in solution [cf. Fig. 2(D)].
Figure 2.

Analysis of CadCpd dimerization. (A) Stereo view of one CadCpd dimer related by a crystallographic symmetry axis. One monomer is colored as in Figure 1(A), the other one is uniformly colored cyan. The N- and C-termini are both directed towards the lipid bilayer indicated at the bottom. (B) Hydrophobic surface representation of the two CadCpd monomers rotated by 90° about a horizontal axis (with the N-termini directed toward the front) and then rotated by ±90° apart to reveal the dimer interface. The interface forming residues are colored according to increasing hydrophobicity from green (hydrophilic) over white (neutral, including the polypeptide backbone) to brown (hydrophobic).47 Residues Asp471 and Lys255, which form a salt bridge, are labeled. (C) The same view as in (B) with electrostatic surface representation as in Figure 1(C and D). (D) Analytical size exclusion chromatography of the recombinant CadCpd at pH 7.5. The protein eluted with a retention volume of 14.4 mL, which corresponds to a molecular size of 64.8 kDa (left) as determined from a plot of log MW of calibration proteins against their elution volumes (right): cytochrome c (12.4 kDa), carbonic anhydrase (29 kDa), ovalbumin (43 kDa), bovine serum albumin (66.3 kDa), alcohol dehydrogenase (150 kDa), β-amylase (200 kDa), apoferritin (443 kDa). The apparent size of CadCpd is considerably larger than the calculated mass of the proteolytic product after thrombin cleavage (41.8 kDa), indicating stable dimer formation in solution.
The dimer formation can be described as an extended contact between two more or less planar and surprisingly polar surfaces on both monomers with local C2 symmetry. In addition, each subunit forms a short arm with its N-terminal loop residues 190–196, which nestles in a crevice of the opposite subunit. Loop residues 248–252 form one side of this niche and residues 504–511 from the C-terminal α-helix 14 form the other side [cf. Fig. 2(A)]. The total dimer interface of the wild type apo-protein buries a solvent accessible area of 1,761 Å2 per monomer, which corresponds to 11.2% of the total molecular surface. The dimer interface comprises 53 residues from each monomer with an extensive network of 11 hydrogen bonds and 6 salt bridges, including 50 buried water molecules, but exhibits only few (20) hydrophobic side chains. In case of CadCpd(D471E) the interface area is slightly larger with 1,994 Å2 (12.6% of the total molecular surface) comprising 58 residues with 11 hydrogen bonds and 2 salt bridges. However, the interface area of CadCpd(D471N) is just marginally smaller, with 1,732 Å2 (10.8%), than the one of the wild type protein, comprising 51 residues with 9 hydrogen bonds and 2 salt bridges.
Interestingly, all the different side chains at position 471, that is Asp, Glu, or Asn, participate in a (pairwise symmetric) dimer contact. The wild type residue Asp471 forms several hydrogen bonds with its carboxylate group, both to two water molecules and to the NZ atom of Lys255 in the neighboring molecule. In the isomorphously crystallized variant D471E both carboxylate oxygens of the longer Glu471 side chain form direct hydrogen bonds to the NZ atom of Lys255. In the variant D471N the mutated residue Asn471 forms one direct hydrogen bond to Lys255 NZ and a second one to a water molecule. Apart from residue Asp471, there are up to seven other acidic amino acids involved in the dimer interface: Asp198, Asp248, Glu249, Asp280, Asp283, Asp445, and Glu468. Of these, Glu249 and Asp283 form salt bridges with residues Arg191 and Arg467, respectively. Such a polar and wet, loosely packed, and both-sided negatively charged interface is unusual for a stable protein dimer. In fact, these peculiar biophysical properties suggest a charge-driven sensing mechanism of the CadC transcriptional activator as will be further discussed below.
The putative ligand cavity and the ReCl62− complex of CadCpd
The two subdomains of CadCpd enclose a cylinder-shaped cavity in between, which is about 20 Å long and has a diameter of 13 Å. A narrow hole, lateral to the dimer interface described above, gives entrance to this cavity [Fig. 1(D)] from the solvent. Its diameter is about 5 Å and it is lined by residues Arg321, Glu324, Lys328, Asn414, and Asn415. Side chains of altogether 46 residues form the central cavity in CadCpd. Its internal surface is made by 20 aliphatic residues, of which 11 side chains (Leu347, Leu348, Leu355, Leu381, Val385, Val425, Leu428, Leu451, Val454, Leu455, and Ile493) form a hydrophobic pocket within the α-helical subdomain. Ten residues contribute polar side chains, of those Arg321 and Arg480 are positively charged and Asp225, Asp241, Glu378, and Glu447 are negatively charged [Fig. 3(A)]. Notably, in the heavy atom derivative that had been prepared for phase determination the cavity harbored one of the ReCl62− complex ions [Fig. 3(C)] while in the CadCpd apo-structure the cavity was filled with a cluster of 20 ordered solvent molecules arranged in typical hydrogen bonding distances.
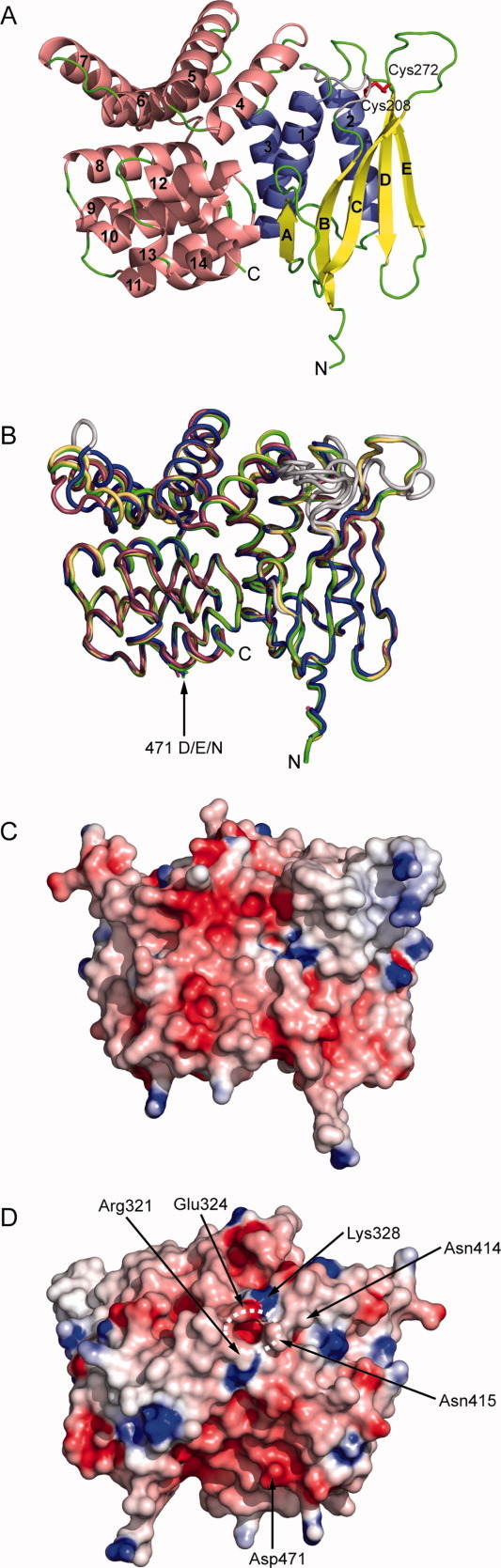
Figure 3.
Analysis of the cavity and the interface between the two subdomains of CadCpd. (A) Electrostatic surface properties (cf. Fig. 1) of the C-terminal α-helical subdomain (residues 333–509; left) and the N-terminal predominantly β-sheet subdomain (residues 190–332; right). Both moieties were rotated by ±90° about a vertical axis to visualize the internal cavity surfaces. Residues that contact the bound ReCl62− ion are labeled. (B) Hydrophobic surface representation colored as in Figure 2B. (C) Complexation of ReCl62− within the central cavity. Interacting side chains of CadC within a distance of 4 Å to the rhenate central ion are indicated. (D) Results from a docking simulation with cadaverine. The three cadaverine conformations with highest scores as well as the putative interacting residues in the cavity are depicted as sticks. Cadaverine carbon atoms are colored differently according to scoring rank order: 1, yellow; 2, salmon; 3, green. Oxygen atoms are shown in red, nitrogen atoms are shown in blue, hydrogen atoms are shown in white, and hydrogen bonds are indicated as dark green dashed lines.
Considering the in total doubly negative charge of the hexachlororhenate ion it is surprising that this ligand became accommodated in the mostly negatively charged cavity. Yet, the metal-coordinating chloride ions might be polarized by the electron-deficient ReIV central ion to such an extent that positive partial charges appear at the surface of this complex ion. Apart from the electrostatic aspect, the ReCl62− sterically fits well into the cavity and forms close contacts with five residues: Asp225, Thr229, His344, Glu447, and Trp450. In fact, their polar groups are in hydrogen bonding distance to the six chloride ions. In addition, there are three water molecules located in the cavity that contribute hydrogen bonds: one water molecule contacts Thr229, Asn232, and Met448, the second one contacts Ser417 and Ile418, and the third one forms hydrogen bonds to residues Gln421 and Glu447.
Docking studies with cadaverine
In principle, the cavity between the two subdomains of the periplasmic region would be nicely suited to accommodate the positively charged cadaverine ligand of CadC, both with regard to sterical fit and to electrostatic complementarity. Unfortunately, soaking experiments with this ligand followed by crystallographic analysis did not give rise to well defined electron density. To identify side chains that could possibly interact with cadaverine within the presumed binding site, we performed a computer simulation using the program AutoDock21 with cadaverine initially provided in a random orientation. All the 10 best scoring results yielded cadaverine in a mostly extended conformation and bound with similar orientation in the cavity [Fig. 3(D)]. Cadaverine was surrounded by residues Glu447, Gln421, and Tyr374 on one side and Asp225 as well as Thr229 on the other. The polar or negatively charged side chains were appropriately positioned to form hydrogen bonds to both positively charged amino groups of the ligand. AutoDock reported calculated dissociation constants for these 10 best CadCpd-cadaverine complexes in the range between 12 and 28 μM, which is in the same order of magnitude as the KD values from 96±18 μM previously measured for the wild type protein via fluorescence titration.14
Comparison between CadCpd and proteins with related folds
A search for structurally related proteins in the protein data bank (PDB) with DALI22 revealed several hits with Z-scores ranging from 11.8 to 9.5. Among the best 20 candidates no protein with a recognizable structural homology was found. Only parts of two structures (hit nos. 8 and 15) showed some local similarity with the α-helical subdomain of CadCpd (Fig. 4). The peroxisomal targeting signal 1 binding domain of Trypanosoma brucei peroxin 523 (PDB entry 3CVN; Z-score = 10.6) exhibits a twisted 18 α-helix bundle. A fragment of this structure formed by α-helices 3 to 12 shows some resemblance with CadCpd [Fig. 4(B)]. However, it is less compact and the mutual arrangement of the α-helices is more entwined in this section while one of the α-helices clearly has a different orientation. The transcription factor MalT domain III of Escherichia coli24 (PDB entry 1HZ4; Z-score = 9.9) likewise forms a twisted 18 α-helix structure. A fragment comprising α-helices 6–15 again shows similarity with the α-helical subdomain of CadCpd. Interestingly, this protein is also a transcriptional activator. The other 18 hits were predominantly all-α-helical proteins but did not exhibit significant similarity with CadCpd. Another search for structurally related proteins using the HHPred server25 revealed one additional protein with partial similarity to CadCpd: the tetratricopeptide repeat protein NlpI of Escherichia coli (PDB entry 1XNF).26 A fragment of this lipoprotein comprising α-helices 4 to 13 grossly resembles the α-helical architecture of CadCpd, yet the helix arrangement is clearly more open. Consequently, CadCpd exhibits a novel fold, both with the 11-α-helix bundle and the mixed β-sheet/α-helix assembly in its two subdomains.
Figure 4.
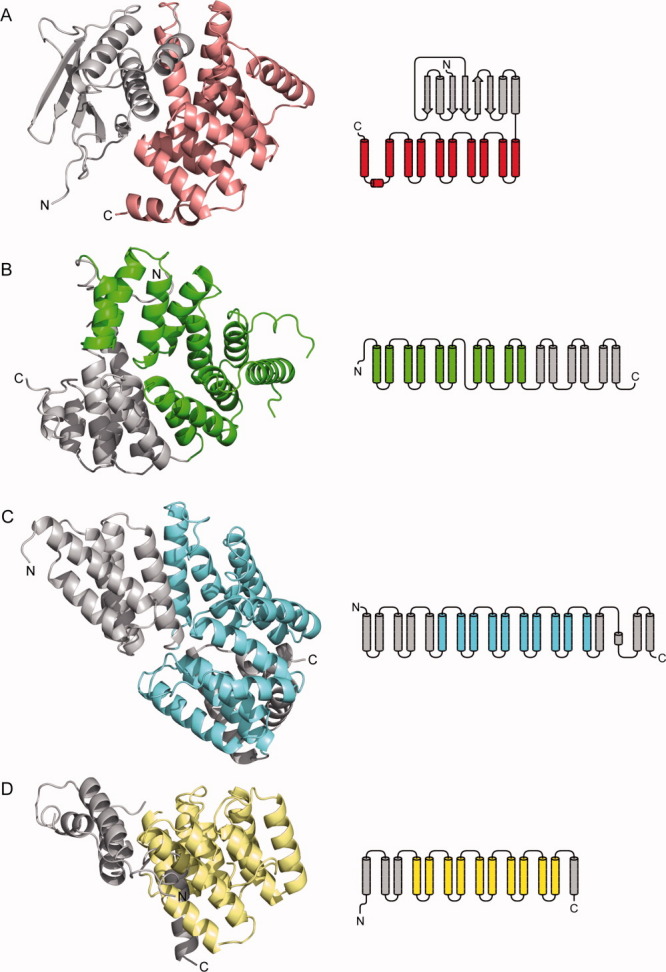
Comparison of the crystal structure and secondary structure topology of CadCpd with its most closely related α-helical bundle proteins. Subdomains to be considered for the comparison are colored whereas other structural elements are shown in gray. (A) CadCpd with its C-terminal subdomain (residues 333–512) comprising 11 α-helices (salmon). (B) The peroxisomal targeting signal 1 binding domain of Trypanosoma brucei peroxin 5 (PDB entry 3CVN ) forms a twisted 16 α-helix bundle; helices 3–12 are colored green. (C) The transcription factor MalT domain III protein of Escherichia coli (1HZ4 ) forms another twisted 18 α-helix bundle; helices 6–15 are colored blue. (D) The tetratricopeptide repeat protein NlpI of Escherichia coli (1XNF ) with α-helices 4–13 colored yellow.
Homologous proteins with similar sequence motifs
A BLAST27 search with the amino acid sequence of CadCpd in the NCBI nonredundant protein sequences database28 revealed homologous CadC proteins from 24 different bacterial species with sequence identities ranging from 91 to 21%, namely (with GI numbers of the NCBI database given in parentheses) Escherichia albertii (170766700), Escherichia fergusonii (218549242), Klebsiella variicola (288936734), Klebsiella pneumoniae (206578436), Salmonella enterica (161502288), Yersinia ruckeri (238756479), Salmonella typhimurium (16765877), Serratia proteamaculans (15737200), Serratia odorifera (293394724), Edwardsiella tarda (294635142), Edwardsiella ictaluri (238918793), Vibrio cholerae (147674140), Vibrio orientalis (261250472), Vibrio harveyi (156972567), Vibrio shilonii (149190651), Vibrio metschnikovii (260774390), Vibrio vulnificus (27365392), Vibrio mimicus (262163818), Vibrio parahaemolyticus (28899667), Vibrio fischeri (59712667), Vibrio alginolyticus (91228716), Aeromonas hydrophila (117620838), Aliivibrio salmonicida LFI1238 (209695912), and Acidobacteria bacterium Ellin345 (94968198). Apart from these hits there were only two shorter sequences with similarity to CadCpd, yet corresponding to proteins of other classes: the putative serine/threonine kinase of Psychroflexus torquis (91215656) with 24% identity to the CadC sequence 266–381 and the TPR repeat-containing serine/threonine protein kinase of Acidobacteria bacterium (94969422) with 27% identity to the CadC sequence 248–387. Interestingly, several CadC orthologs revealed conserved residues at the positions that line the putative cadaverine cavity described above (Fig. 5).
Figure 5.

Sequence alignment of CadCpd orthologs (residues 188–512) from a representative set of γ-proteobacteria. Amino acids lining the characteristic cavity of CadCpd are labeled as small vertical arrows above the sequences while residues involved in the dimer interface are indicated by arrows underneath, many of which involve fully conserved positions. Secondary structure elements are shown as horizontal arrows (β-strands) and rods (α-helices), respectively, above the alignment according to the crystal structure of the Escherichia coli CadCpd protein determined in this study. Amino acid Asp471, which is important for pH-sensing, is marked by an asterisk. GI numbers (http://www.ncbi.nlm.nih.gov/Sitemap/sequenceIDs.html) are given in parentheses: Escherichia coli MG1655 (16131959), Escherichia fergusonii ATCC 35469 (218549242), Salmonella enterica serovar typhimurium LT2 (16765877), Serratia proteamaculans 568 (157323768), Yersinia ruckeri ATCC 29473 (238756479), Klebsiella pneumoniae NTUH-K2044 (218117876), Edwardsiella tarda EIB202 (267983771).
Discussion
CadC belongs to the ToxR-like regulators that encompass biochemically non-modified one-component systems with similar gross topology, including several low pH-induced transcription regulators. Because of the important role of these proteins for bacterial virulence, for example ToxR of Vibrio cholerae, there is elevated interest in their biomolecular mechanism. Albeit there is no clear correlation between the primary structures of the periplasmic domains of CadC and ToxR, even in terms of size (CadCpd: 325 amino acids; ToxRpd: 100 amino acids), it is conceivable that the general mechanism of signal perception and transduction across the inner bacterial membrane should be related.
The X-ray analysis of CadCpd reported here provides the first known tertiary structure for a sensory domain of a (pH-activated) ToxR-like regulator. A characteristic feature of the CadCpd structure is its composition of two subdomains with a cavity at their interface that is suited to accommodate cadaverine, the feedback inhibitor of the Cad system. The docking results demonstrate that the side chains of the residues Glu447, Asp225, Thr229, Tyr374, and Gln421 within this pocket are appropriately oriented to bind the positively charged ligand. This finding is in agreement with the binding of the hexachlororhenate ion in almost the same place. In this case, the side chains of residues Asp225, Thr229, His344, Glu447, and Trp450 are involved. Notably, the predicted dissociation constants for the CadCpd-cadaverine complex from the docking experiment of 12–28 μM are in the same order of magnitude as the experimentally determined value of 96 ± 18 μM.14 The lower estimated value may be caused by incorrect assumptions for the electrostatic contribution to the binding interaction. However, the measured moderate affinity explains the lack of success in our soaking experiments with this ligand. Generally, the flexibility of the cadaverine ligand and the preponderance of electrostatic interactions should facilitate adaptation of the mode of binding to partly different charged and/or polar residues that line the ligand pocket in other species (cf. Fig. 5). For example, it seems that in bacteria that carry Gly instead of Asp at position 225 the role of the charged side chain could be taken over by the adjacent Asp229 (instead of Thr).
Another remarkable feature of the CadCpd crystal structure is the formation of a biological dimer as revealed by PISA and CS analysis and experimentally backed by size exclusion chromatography. The quaternary structure of this dimer would allow an arrangement with respect to the lipid bilayer that is compatible with a preceding transmembrane segment leading into the N-terminal subdomain dominated by the five-stranded β-sheet. Thus, it is conceivable that pH-dependent protonation of negatively charged side chains, including residue Asp471, should affect the interface of the dimer, resulting in an altered mutual orientation of the periplasmic domains and, thus, to a general conformational effect that becomes transduced via the transmembrane segment and ultimately triggers transcriptional activity on the cytoplasmic side. The observation that the mutation D471N (representing the ON state) both has a pronounced effect on the solubility of the protein (see Materials and Methods) and leads to a different crystal packing supports the notion of minute conformational changes at the dimer interface that should functionally influence transcriptional activity. This would also be consistent with an additional role of cadaverine binding at the cavity within each periplasmic domain on the mode of dimer association, apart from the pH-sensing effect. Notably, the complex between CadCpd and the rhenate ligand showed a significantly smaller interface area of 1368 Å2 per monomer with fewer hydrogen bonds (4) and salt bridges (2).
In conclusion, the three-dimensional structure of CadCpd elucidated in this study establishes a new protein superfamily as there is no significant structural similarity to any other protein in the PDB. Only sub-structures of three large α-helical bundle proteins show certain resemblance to the C-terminal α-helical subdomain of CadCpd. In particular, there is no structurally related protein with the N-terminal β-sheet subdomain of CadCpd. The results from a BLAST search underline these findings because there is no independent protein in the database that shows significant sequence similarity to CadCpd, including members of the ToxR-like protein family itself. However, CadC homologues with high sequence similarity are found in several γ-proteobacterial genera like the Salmonella, Serratia, and Klebsiella. Thus, it can be expected that these CadC-related proteins form a novel structural family with similar functions in bacterial cell physiology. Mutagenesis experiments are currently under way to further study the structural mechanisms that underly the pH- and ligand-dependent transcriptional activation of this interesting bacterial membrane sensor protein.
Materials and Methods
Overexpression and purification of wild type CadCpd and its variants D471E and D471N
CadCpd was produced as a fusion protein consisting of N-terminal thioredoxin, a double His6-tag interspersed with thrombin and enterokinase cleavage sites, and the CadC fragment comprising residues 188–512 (Swiss-Prot entry P23890). This hybrid protein was overproduced in Escherichia coli Origami B (DE3)/pLysS using the plasmid pET32a-CadC188-512 as described before.14 The structural genes for the CadCpd variants D471E and D471N were obtained by site-directed mutagenesis and expressed like the wild type protein.17 Briefly, the fusion proteins were purified from the bacterial whole cell extract by Ni-NTA affinity chromatography. After that, thioredoxin was cleaved off using the Thrombin Cleavage Capture Kit (Merck, Darmstadt, Germany). The reaction product was dialyzed against 20 mM Tris/HCl pH 8.0, 130 mM NaCl and applied to a Q Sepharose Fast Flow anion exchange column (GE Healthcare, Munich, Germany), followed by elution with a gradient ranging from 130 mM to 1 M NaCl in 20 mM Tris/HCl pH 8.0. Fractions containing CadCpd were pooled and concentrated to 11 mg/mL by ultrafiltration. The wild type protein was finally purified by size exclusion chromatography using an S75 analytic HR 10/30 column (GE Healthcare) in the presence of 10 mM Tris/HCl pH 7.5, 150 mM NaCl. CadCpd was then concentrated with 10 kDa cut-off Vivaspin tubes (Vivascience, Hannover, Germany) yielding two batches with concentrations of 8.8 and 13 mg/mL, respectively, and filtrated with 0.45 μm Spin-X centrifuge tubes (Corning Inc., Corning, NY) before crystallization. To measure the apparent size of the wild type CadCpd protein, a size exclusion chromatography was performed on a calibrated S200 analytic HR 10/30 column (GE Healthcare) in the presence of 150 mM NaCl, 10 mM Tris/HCl pH 7.5. The CadCpd variant D471E was purified in the same way and finally concentrated to 14 mg/mL. The variant D471N, however, formed a gel-like precipitate after the first dialysis against 20 mM Tris/HCl pH 8.0, 130 mM NaCl, resulting in an initial protein concentration of merely 2.8 mg/mL. Following centrifugation and sterile filtration, the protein was purified via anion exchange chromatography as described above, and, after adjusting the NaCl concentration to 500 mM, subjected to ultrafiltration. The concentrated sample, which turned opaque after resting on ice for an hour, was cleared by filtration through a Spin-X tube, resulting in a final protein concentration of 6.9 mg/mL, and used for crystallization trials without size exclusion chromatography.
Protein crystallization and preparation of heavy atom derivatives
CadCpd was crystallized using the hanging drop vapor diffusion technique, yielding crystals within 1 week at 20°C. The precipitants were 1.4 to 1.7 M (NH4)2SO4 in combination with 4.5−11% (v/v) 2-propanol (without additional buffer except for the 10 mM Tris/HCl pH 7.5 from the original protein solution; cf. above). Crystals of native CadCpd were harvested from their drops, dipped into the reservoir solution supplemented with 30% (v/v) glycerol, and flash-frozen. Crystals of the variant D471E were grown under the same conditions, whereas crystals of the variant D471N were obtained in an acidic milieu with 20% (w/v) PEG 6000, 100 mM Na-citrate pH 5. In the latter case, the droplets were set up with a 2:1 protein to precipitant volume ratio to compensate for the lower concentration of the original protein solution. A search for heavy atom compounds (Hampton Research, Aliso Viejo, CA) that bind to wild type CadCpd was performed by means of native polyacrylamide gel electrophoresis (PAGE).29 To this end, 5 μL of each heavy atom compound with a concentration of 10 mM in aqueous solution was added to 5 μL of a 2 mg/mL CadCpd solution in 10 mM Tris/HCl pH 7.5, 150 mM NaCl and the mixture was applied to 6.5% (w/v) PAGE in the presence of 750 mM Tris/HCl pH 7.5 using 190 mM glycine, 10 mM Tris/HCl pH 8.3 as running buffer. A detectable band shift of the protein-heavy atom complex led to the identification of K2ReCl6 as a promising compound. To prepare the heavy atom derivative for X-ray data collection, crystals of CadCpd were first transferred from the mother liquor described above to 2.5 M Li2SO4 to avoid the formation of metal-ammonia complexes. Then, a solution of 30 mM K2ReCl6 and 1.7 M Li2SO4 was cautiously added to the drop such that the final ReCl62− concentration reached 5 mM. Within about 10 min the initially pale yellow color of the droplet changed to dark violet. On the next day, the deeply colored crystals were washed in 2.5 M Li2SO4 to remove precipitated rhenium salt. Then, the soaking was repeated for 4 h, again followed by washing of the crystals with 2.5 M Li2SO4. Finally, crystals were transferred to 2.5 M Li2SO4, 30% (v/v) glycerol, using MicroMount loops (MiTeGen, Ithaca, NY) and flash-frozen in liquid nitrogen.
Data collection and processing, model building, and refinement
For the structure solution of CadCpd via MAD, three datasets from a single Re-soaked crystal of space group P6122 were collected at BESSY beam line 14.2. Diffraction up to a resolution of 2.3 Å (peak), 2.6 Å (inflection), and 2.9 Å (high energy remote) was measured at 100 K (Table I). Data were processed with MOSFLM, scaled with SCALA,30 and heavy atom sites were identified with SHELXD.31 Refinement of the Re sites, MAD phasing, solvent flattening, and calculation of a native electron density map were carried out with SHARP.32 The initial atomic model of the Re-complexed CadCpd was built using ARP/wARP30 and manually corrected with Coot.33 Altogether 16 ReCl62− sites were verified by inspecting the anomalous density map, and their occupancy was adjusted to yield B-factors similar to surrounding protein atoms. The geometry of the ReCl62− complex ion itself was obtained from the Inorganic Crystal Structure Database,34 revealing Re-CI distances in the range from 2.353 to 2.367 Å, consistent with literature data.35 Hence, restraints during refinement were set for Re-Cl distances to 2.36 Å and for Cl-Re-Cl angles to 90°. After initial refinement of the protein-rhenate complex the crystal structures of the native protein and its variants were solved by molecular replacement with Phaser36 using corresponding data sets collected at BESSY beam lines 14.1 and 14.2 (Table I). In the case of the D471N crystal, diffraction data were processed with XDS.37 Water molecules were added to all models using ARP/wARP, rotamers of Asn and Gln residues were adjusted with NQ-Flipper38 and, subsequently, all protein models were refined with Refmac530 in iterative cycles interspersed with manual correction.
The electron density map of native CadCpd at a resolution of 1.8 Å (Table I) allowed model building for 314 of the 362 residues present in the recombinant protein, namely Ser190–Lys206 and Tyr215–Ser511. For the ReCl62− derivative of CadCpd, there was electron density for altogether 286 residues: Ile192–Val204, Leu217–Tyr245, Ser251–Phe261, Phe273–Val293, and Asn298–Leu509. For the CadCpd variant D471E the electron density map at 1.9 Å resolution covered residues Lys189–Lys206 and Tyr215–Ser511, while in the case of D471N there was electron density at 2.2 Å resolution for residues Ser190–Val204, Gln216–Asn263, Cys272–Glu394, and Ala399–Ser511. The C-terminal residue Glu512 as well as the N-terminal His6-tag, including the remaining upstream linker of the processed fusion protein (47 residues), were invisible in all structures. The models were validated with PROCHECK39 and WHAT_CHECK.40 Secondary structure elements were assigned using DSSP41 and secondary structure topologies were depicted with TopDraw.42 Crystal packing contacts and protein interfaces were analyzed with PISA19 and with SC20 from CCP4.30 Graphics were prepared with PyMOL43 and GRASP.44 Structural superposition was carried out with PyMOL while docking studies were performed with AutoDock 4.21 Evolutionary related CadCpd sequences were searched with BLAST27 in the NCBI non-redundant protein sequences database.28 Sequence alignments were subsequently made with ClustalW245 and edited using Aline46 with a low similarity cut off value of 0.241 (similarity groups DE FWY HKR ILMV NQ ST). Structurally related proteins were searched with DALI22 and HHPred.25 The atomic coordinates and structure factors of the CadCpd models have been deposited at the RCSB Protein Data Bank under the accession codes 3LY7 (wild type protein), 3LYA (Re-derivative), 3LY8 (variant D471E), and 3LY9 (variant D471N).
Acknowledgments
The authors wish to thank Korinna Burdack and Josef Danzer for technical assistance and Eberhardt Herdtweck and Martin U. Schmidt for database searches. The synchrotron measurements at BESSY and Free University Berlin at BESSY beamlines 14.1 and 14.2 were supported by the BMBF (project funding reference number 05 ES3XBA/5); we thank Jörg Schulze, Georg Zocher, and Sandra Pühringer for assistance. I.H. received a research scholarship from the Elite Network of Bavaria.
References
- 1.Padan E, Zilberstein D, Rottenberg H. The proton electrochemical gradient in Escherichia coli cells. Eur J Biochem. 1976;63:533–541. doi: 10.1111/j.1432-1033.1976.tb10257.x. [DOI] [PubMed] [Google Scholar]
- 2.Gale EF, Epps HM. The effect of the pH of the medium during growth on the enzymic activities of bacteria (Escherichia coli and Micrococcus lysodeikticus) and the biological significance of the changes produced. Biochem J. 1942;36:600–618. doi: 10.1042/bj0360600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Soksawatmaekhin W, Kuraishi A, Sakata K, Kashiwagi K, Igarashi K. Excretion and uptake of cadaverine by CadB and its physiological functions in Escherichia coli. Mol Microbiol. 2004;51:1401–1412. doi: 10.1046/j.1365-2958.2003.03913.x. [DOI] [PubMed] [Google Scholar]
- 4.Watson N, Dunyak DS, Rosey EL, Slonczewski JL, Olson ER. Identification of elements involved in transcriptional regulation of the Escherichia coli cad operon by external pH. J Bacteriol. 1992;174:530–540. doi: 10.1128/jb.174.2.530-540.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Merrell DS, Camilli A. Regulation of Vibrio cholerae genes required for acid tolerance by a member of the “ToxR-like” family of transcriptional regulators. J Bacteriol. 2000;182:5342–5350. doi: 10.1128/jb.182.19.5342-5350.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miller VL, Taylor RK, Mekalanos JJ. Cholera toxin transcriptional activator toxR is a transmembrane DNA binding protein. Cell. 1987;48:271–279. doi: 10.1016/0092-8674(87)90430-2. [DOI] [PubMed] [Google Scholar]
- 7.Häse CC, Mekalanos JJ. TcpP protein is a positive regulator of virulence gene expression in Vibrio cholerae. Proc Natl Acad Sci USA. 1998;95:730–734. doi: 10.1073/pnas.95.2.730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang Y, Isberg RR. Transcriptional regulation of the Yersinia pseudotuberculosis pH6 antigen adhesin by two envelope-associated components. Mol Microbiol. 1997;24:499–510. doi: 10.1046/j.1365-2958.1997.3511719.x. [DOI] [PubMed] [Google Scholar]
- 9.Egan S, James S, Kjelleberg S. Identification and characterization of a putative transcriptional regulator controlling the expression of fouling inhibitors in Pseudoalteromonas tunicata. Appl Environ Microbiol. 2002;68:372–378. doi: 10.1128/AEM.68.1.372-378.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seki T, Yoshikawa H, Takahashi H, Saito H. Cloning and nucleotide sequence of phoP, the regulatory gene for alkaline phosphatase and phosphodiesterase in Bacillus subtilis. J Bacteriol. 1987;169:2913–2916. doi: 10.1128/jb.169.7.2913-2916.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Melchers LS, Thompson DV, Idler KB, Schilperoort RA, Hooykaas PJ. Nucleotide sequence of the virulence gene virG of the Agrobacterium tumefaciens octopine Ti plasmid: significant homology between virG and the regulatory genes ompR, phoB and dye of E. coli. Nucleic Acids Res. 1986;14:9933–9942. doi: 10.1093/nar/14.24.9933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Comeau DE, Ikenaka K, Tsung KL, Inouye M. Primary characterization of the protein products of the Escherichia coli ompB locus: structure and regulation of synthesis of the OmpR and EnvZ proteins. J Bacteriol. 1985;164:578–584. doi: 10.1128/jb.164.2.578-584.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuper C, Jung K. CadC-mediated activation of the cadBA promoter in Escherichia coli. J Mol Microbiol Biotechnol. 2005;10:26–39. doi: 10.1159/000090346. [DOI] [PubMed] [Google Scholar]
- 14.Tetsch L, Koller C, Haneburger I, Jung K. The membrane-integrated transcriptional activator CadC of Escherichia coli senses lysine indirectly via the interaction with the lysine permease LysP. Mol Microbiol. 2008;67:570–583. doi: 10.1111/j.1365-2958.2007.06070.x. [DOI] [PubMed] [Google Scholar]
- 15.Tetsch L, Jung K. The regulatory interplay between membrane-integrated sensors and transport proteins in bacteria. Mol Microbiol. 2009;73:982–991. doi: 10.1111/j.1365-2958.2009.06847.x. [DOI] [PubMed] [Google Scholar]
- 16.Dell CL, Neely MN, Olson ER. Altered pH and lysine signalling mutants of cadC, a gene encoding a membrane-bound transcriptional activator of the Escherichia coli cadBA operon. Mol Microbiol. 1994;14:7–16. doi: 10.1111/j.1365-2958.1994.tb01262.x. [DOI] [PubMed] [Google Scholar]
- 17.Haneburger I, Eichinger A, Skerra A, Jung K. New insights into the signaling mechanism of the pH-responsive, membrane-integrated transcriptional activator CadC of Escherichia coli. J Biol Chem. 2011 doi: 10.1074/jbc.M110.196923. in press. [doi: 10.1074/jbc.M110.196923] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fritz G, Koller C, Burdack K, Tetsch L, Haneburger I, Jung K, Gerland U. Induction kinetics of a conditional pH stress response system in Escherichia coli. J Mol Biol. 2009;393:272–286. doi: 10.1016/j.jmb.2009.08.037. [DOI] [PubMed] [Google Scholar]
- 19.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 20.Lawrence MC, Colman PM. Shape complementarity at protein/protein interfaces. J Mol Biol. 1993;234:946–950. doi: 10.1006/jmbi.1993.1648. [DOI] [PubMed] [Google Scholar]
- 21.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. Automated docking using a Lamarckian genetic algorithm and and empirical binding free energy function. J Comp Chem. 1998;19:1639–1662. [Google Scholar]
- 22.Holm L, Kaariainen S, Rosenstrom P, Schenkel A. Searching protein structure databases with DaliLite v.3. Bioinformatics. 2008;24:2780–2781. doi: 10.1093/bioinformatics/btn507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sampathkumar P, Roach C, Michels PA, Hol WG. Structural insights into the recognition of peroxisomal targeting signal 1 by Trypanosoma brucei peroxin 5. J Mol Biol. 2008;381:867–880. doi: 10.1016/j.jmb.2008.05.089. [DOI] [PubMed] [Google Scholar]
- 24.Steegborn C, Danot O, Huber R, Clausen T. Crystal structure of transcription factor MalT domain III: a novel helix repeat fold implicated in regulated oligomerization. Structure. 2001;9:1051–1060. doi: 10.1016/s0969-2126(01)00665-7. [DOI] [PubMed] [Google Scholar]
- 25.Söding J, Biegert A, Lupas AN. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005;33:W244–248. doi: 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wilson CG, Kajander T, Regan L. The crystal structure of NlpI. A prokaryotic tetratricopeptide repeat protein with a globular fold. FEBS J. 2005;272:166–179. doi: 10.1111/j.1432-1033.2004.04397.x. [DOI] [PubMed] [Google Scholar]
- 27.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 28.Bleasby AJ, Akrigg D, Attwood TK. OWL–a non-redundant composite protein sequence database. Nucleic Acids Res. 1994;22:3574–3577. [PMC free article] [PubMed] [Google Scholar]
- 29.Boggon TJ, Shapiro L. Screening for phasing atoms in protein crystallography. Structure. 2000;8:R143–149. doi: 10.1016/s0969-2126(00)00168-4. [DOI] [PubMed] [Google Scholar]
- 30.CCP4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 31.Schneider TR, Sheldrick GM. Substructure solution with SHELXD. Acta Crystallogr D Biol Crystallogr. 2002;58:1772–1779. doi: 10.1107/s0907444902011678. [DOI] [PubMed] [Google Scholar]
- 32.Bricogne G, Vonrhein C, Flensburg C, Schiltz M, Paciorek W. Generation, representation and flow of phase information in structure determination: recent developments in and around SHARP 2.0. Acta Crystallogr D Biol Crystallogr. 2003;59:2023–2030. doi: 10.1107/s0907444903017694. [DOI] [PubMed] [Google Scholar]
- 33.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 34.Bergerhoff G, Brown ID. Inorganic Crystal Structure Database. In: Allen FH, Bergerhoff G, Sievers R, editors. Crystallographic Databases. Chester: IUCr; 1987. [Google Scholar]
- 35.Barbour LJ, MacGillivray LR, Atwood JL. Crystal and molecular structure of [H3O•18-crown-6]2-[ReCl6] isolated from a liquid clathrate medium. J Chem Cryst. 1996;26:59–61. [Google Scholar]
- 36.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kabsch W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J Appl Cryst. 1993;26:795–800. [Google Scholar]
- 38.Weichenberger CX, Sippl MJ. NQ-Flipper: recognition and correction of erroneous asparagine and glutamine side-chain rotamers in protein structures. Nucleic Acids Res. 2007;35:W403–406. doi: 10.1093/nar/gkm263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Laskowski RA, MacArthur MW, Mos DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–291. [Google Scholar]
- 40.Hooft RW, Vriend G, Sander C, Abola EE. Errors in protein structures. Nature. 1996;381:272. doi: 10.1038/381272a0. [DOI] [PubMed] [Google Scholar]
- 41.Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 42.Bond CS. TopDraw: a sketchpad for protein structure topology cartoons. Bioinformatics. 2003;19:311–312. doi: 10.1093/bioinformatics/19.2.311. [DOI] [PubMed] [Google Scholar]
- 43.DeLano WL. The PyMOL Molecular Graphics System. California: DeLano Scientific; 2002. [Google Scholar]
- 44.Nicholls A, Sharp KA, Honig B. Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins. 1991;11:281–296. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]
- 45.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 46.Bond CS, Schüttelkopf AW. ALINE: a WYSIWYG protein-sequence alignment editor for publication-quality alignments. Acta Crystallogr D Biol Crystallogr. 2009;65:510–512. doi: 10.1107/S0907444909007835. [DOI] [PubMed] [Google Scholar]
- 47.Eichinger A, Nasreen A, Kim HJ, Skerra A. Structural insight into the dual ligand specificity and mode of high density lipoprotein association of apolipoprotein D. J Biol Chem. 2007;282:31068–31075. doi: 10.1074/jbc.M703552200. [DOI] [PubMed] [Google Scholar]