

Abstract
A high-resolution structure of a ligand-bound, soluble form of human monoglyceride lipase (MGL) is presented. The structure highlights a novel conformation of the regulatory lid-domain present in the lipase family as well as the binding mode of a pharmaceutically relevant reversible inhibitor. Analysis of the structure lacking the inhibitor indicates that the closed conformation can accommodate the native substrate 2-arachidonoyl glycerol. A model is proposed in which MGL undergoes conformational and electrostatic changes during the catalytic cycle ultimately resulting in its dissociation from the membrane upon completion of the cycle. In addition, the study outlines a successful approach to transform membrane associated proteins, which tend to aggregate upon purification, into a monomeric and soluble form.
Keywords: cannabinoid receptor, monoglyceride lipase, lid-domain, protein inhibitor complex
Introduction
While various medical properties of cannabis have been appreciated for millennia by many cultures around the world, the molecular and pharmacological substrates underlying these effects have only recently been elucidated. Δ9-Tetrahydrocannabinol, the main psychoactive substance found in the Cannabis plant was isolated in the early 1960s. Tetrahydrocannabinol activates two distinct G protein-coupled receptors, cannabinoid receptor 1 (CB1) and cannabinoid receptor 2 (CB2).1,2 CB1 is abundant in the central nervous system,3–5 whereas CB2 is particularly expressed in cells of the immune system and in peripheral tissues.2,6 Analgesic effects of cannabinoids have been well documented, with accumulating evidence that cannabinoids can produce antinociception through central as well as peripheral mechanisms and involving both CB1 and CB2.7–13
2-Arachidonoyl glycerol, an arachidonic acid derivative, is one of the two major and most well studied endogenous ligands for CB1 and CB2 acting as a potent agonist of CB1 and CB2 receptors.14–16 2-Arachidonoyl glycerol is produced by neurons on demand and acts near its site of synthesis. The in vivo effect of 2-arachidonoyl glycerol is local and transient due to its rapid degradation by intracellular enzymes. In the brain, the main enzyme responsible for the hydrolysis of 2-arachidonoyl glycerol is monoglyceride lipase (MGL).17,18
MGL is a membrane associated enzyme but has also been located in the cytosol.19,20 It is a member of the α/β-hydrolase family of enzymes21 and is thought to be related to microbial haloperoxidases and lysophospholipases.22 The classical serine hydrolase catalytic triad was identified to be Ser122, Asp239, and His269. MGL also contains the GlyXxxSerXxxGly consensus sequence present in most serine hydrolases.22 The physiological roles of MGL appear to be tissue specific. In lipid-metabolizing tissues, MGL is thought to catalyze the final step of the lipolytic cascade that releases fatty acids from triacylglycerol stores.23 In the central nervous system, MGL is the key enzyme modulating endocannabinoid signaling via hydrolysis of 2-arachidonoyl glycerol.
Recently, pharmacologic studies using selective and potent inhibitors of MGL have indicated that MGL may be a novel target for the treatment of pain. The recently published crystal structures of MGL, both apo and in complex with the covalent inhibitor SAR629, have revealed the α/β-hydrolase fold of MGL.24,25
In this report, a new crystal structure of a mutant MGL in complex with a potent inhibitor is presented. Two mutations (Leu169Ser and Leu176Ser) were engineered to increase the solubility of MGL and to allow purification without detergent. An additional lysine to alanine mutation (Lys36Ala) enabled the generation of high quality diffracting crystals. The new MGL structure reveals a hitherto unknown closed conformation of the regulatory lid-domain, which almost completely encloses the bound inhibitor. Analysis of the new conformation offers a model in which MGL undergoes conformational and electrostatic changes leading to its dissociation from the membrane during the catalytic cycle.
Results and Discussion
Engineering MGL–construct design
At the initiation of the project, the crystal structure of MGL was unknown and detergent was shown to be required for the isolation of MGL from natural sources and purification of recombinant MGL from baculovirus-infected insect cells.26–28 Without detergent the protein is prone to substantial aggregation, consistent with an enzyme that is associated with membranes. Based on the hypothesis that hydrophobic residues in MGL's regulatory lid-domain are likely to be involved in membrane association and in promoting aggregation, a strategy was employed to identify and mutate likely residues responsible for these effects. Sequence alignment and homology modeling were used to identify the boundaries of the lid-domain and potentially surface exposed residues.
A library of constructs was designed to increase the hydrophilicity of the MGL surface without altering the hydrolase fold and the function of MGL. Based on the homology model, five leucines (Leu167, Leu169, Leu171, Leu174, and Leu176) were identified to be part of the lid-domain and selected for mutation. The residues were replaced by serine or glutamine either independently or in pairs (Table I). In addition, five lysine residues were identified at the surface of the MGL homology model and were mutated to alanine. Mutations of surface lysine residues, the side chains of which have high conformational entropy, have been reported to stabilize crystal contacts, promote crystallization and improve crystal quality.29,30 The surface mutations were introduced into the MGL double lid-domain mutant construct (hMGL 1-303 Leu169Ser, Leu176Ser) for which crystallization leads had been obtained. Surface mutations were either introduced independently or in combination with other surface mutations (Table I). N-terminal and C-terminal truncation constructs were also designed (Table I) to eliminate potential flexible terminal amino acids that may be energetically unfavorable for crystal packing. The N-terminus was truncated at residue 9, 19, 26, and 33. The C-terminus was truncated at 297 and 292. The N-and C-terminal truncations were introduced independently or combined with other truncations and were introduced into the mutant MGL (hMGL 1-303 Leu169Ser, Leu176Ser) construct (Table I). All constructs, including wild-type MGL, were engineered with an N-terminal histidine tag followed by a TEV protease cleavage site. A total of 26 mutant MGL clones were generated by mixing and matching the lid-domain mutations, surface mutations, and truncations (Table I). All constructs were made in baculovirus and expressed in Sf9 cells as described under Material and Methods.
Table I.
Monoglyceride Lipase Constructs Generated and Purification Yields* in mg/L
| Yield mg/L | |
|---|---|
| Lid-domain mutations | |
| 1-303 Leu169Ser, Leu176Ser | 4.5 |
| 1-303 Leu167Gln | 2.3 |
| 1-303 Leu171Gln | 0.7 |
| 1-303 Leu174Gln | 0 |
| 1-303 Leu167Gln, Leu171Gln | 5 |
| 1-303 Leu167Gln, Leu174Gln | 7 |
| 1-303 Leu171Gln, Leu174Gln | 1.5 |
| 1-303 Leu167Gln, Leu171Gln, Leu174Gln | 0 |
| Lid-domain mutations + truncations | |
| 1-297 Leu169Ser, Leu176Ser | 2 |
| 1-292 Leu169Ser, Leu176Ser | 0.5 |
| 9-303 Leu169Ser, Leu176Ser | 2 |
| 9-297 Leu169Ser, Leu176Ser | 1 |
| 19-303 Leu169Ser, Leu176Ser | 0 |
| 19-297 Leu169Ser, Leu176Ser | 0 |
| 19-292 Leu169Ser, Leu176Ser | 0 |
| 26-303 Leu169Ser, Leu176Ser | 0 |
| 26-297 Leu169Ser, Leu176Ser | 0 |
| 26-292 Leu169Ser, Leu176Ser | 0 |
| 33-303 Leu169Ser, Leu176Ser | 0 |
| 33-297 Leu169Ser, Leu176Ser | 0 |
| 33-292 Leu169Ser, Leu176Ser | 0 |
| Lid-domain mutations + Surface mutations | |
| 1-303 Leu169Ser, Leu176Ser, Lys36Ala | 3.6 |
| 1-303 Leu169Ser, Leu176Ser, Lys160Ala | 0.7 |
| 1-303 Leu169Ser, Leu176Ser, Lys165Ala | 0.5 |
| 1-303 Leu169Ser, Leu176Ser, Lys226Ala | 2.3 |
| 1-303 Leu169Ser, Leu176Ser, Lys36Ala, Lys236Ala | 1.5 |
Purification yields are from one single experiment.
Characterization of engineered MGL–protein aggregation
Purification of wild-type MGL (hMGL 1-303) required detergent. A pilot purification of wild-type MGL performed in the absence of detergent generated no protein (data not shown). A second purification of wild-type MGL was done with Bugbuster® lysis buffer, a proprietary lysis buffer containing detergent (Invitrogen). After lysis, the purification protocol and buffers were the same as described for mutant MGL in Experimental Procedures. An average 2.2 mg of wild-type MGL per liter of cell culture was obtained. Further analysis by size exclusion showed that this protein was aggregated [Fig. 1(A)]. Contrary to our experiences, two recent independent reports published after this work was initiated demonstrated successful production of a soluble wild-type human MGL that did not require detergent during purification and was not aggregated.24,25,31 The protein produced was the 313 aminoacids MGL isoform (GenBank AAH06230) versus the 303 aminoacid isoform (GenBank CAC43316) described in this manuscript. Production of the soluble wild-type long isoform of MGL was achieved in Escherichia coli BL21 and Rosetta strain while expression in the baculovirus expression system produced only insoluble, aggregated protein as described above. It is conceivable that expression of MGL in the baculovirus expression system produces a slightly different protein that is more prone to aggregation than when expressed in E. coli.
Figure 1.
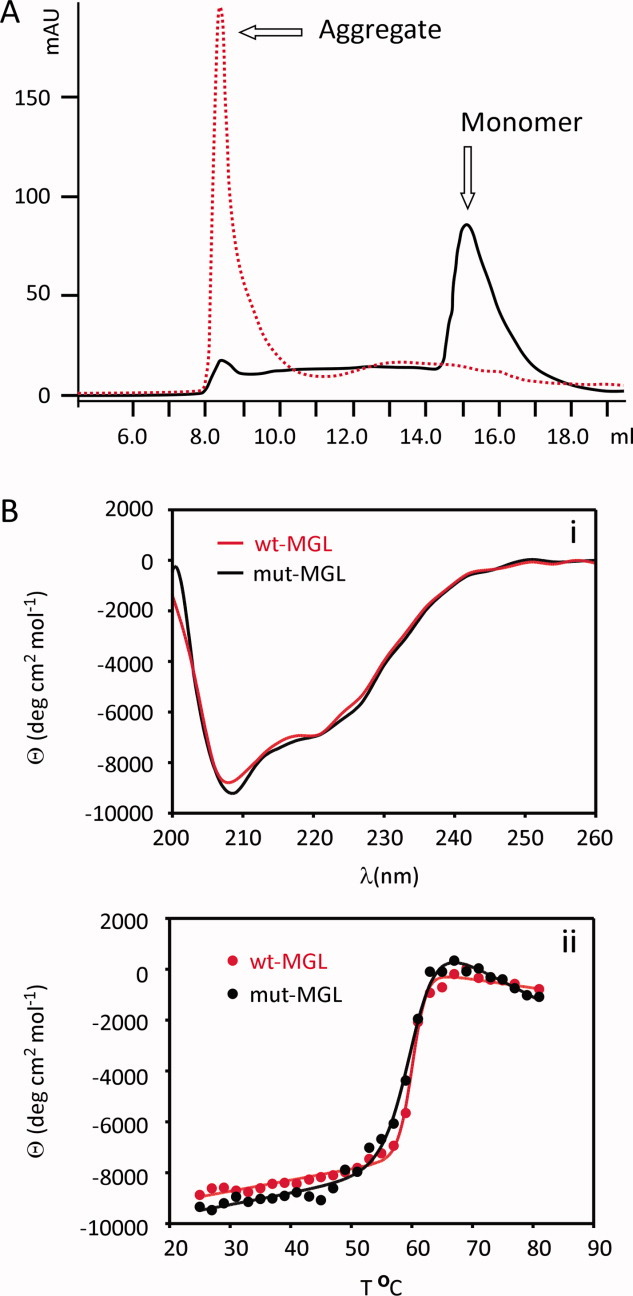
Size exclusion profiles and circular dichroism. A: Size exclusion elution profiles for wild-type MGL 1-303, red dotted line, and mutant MGL (hMGL 1–303 Leu169Ser, Leu176Ser), black solid line, purified in the absence of detergent showing 100% aggregation and 90% monomer, respectively. B: Circular dichroism structural analysis of wild-type MGL 1-303, red line, and mutant MGL (hMGL 1–303 Leu169Ser, Leu176Ser), black line. (i) Far UV scans from 200 to 260 nm. (ii) Temperature melts from 25 to 80C monitored at 210 nm.
The engineered mutant constructs were purified in the absence of detergent under the protocol described in Experimental Procedures. Purification yields are shown in Table I and are the result of one experiment. Constructs containing the lid-domain mutation, generated between 0.7 and 4.5 mg/L, except mutant MGL containing the Leu174Gln mutation and mutant MGL containing the triple mutation Leu167Gln, Leu171Gln, Leu174Gln, which upon purification did not yield any protein (Table I). Analysis by size exclusion chromatography showed mainly monomeric protein [Fig. 1(A)], indicating that the mutations significantly improved protein solubility. Constructs truncated at residues 19, 26, or 33 generated no protein. Construct with C-terminal truncations generated between 0.5 and 2 mg of soluble protein per liter of cell culture. Constructs with a combination of lid-domain and surface mutations generated between 0.5 and 3.6 mg/L and showed very low aggregation as well.
Characterization of engineered MGL–circular dichroism
Since preliminary crystals had been obtained with TEV protease cleaved mutant MGL (hMGL 1-303 Leu169Ser, Leu176Ser), this construct was selected for further characterization by circular dichroism to ensure that the mutations introduced did not adversely affect protein conformation. The circular dichroism scans for the wild-type MGL and the TEV protease cleaved mutant MGL (hMGL 1-303 Leu169Ser, Leu176Ser) were similar, indicating that the two enzymes held a similar fold [Fig. 1(B)].
Characterization of engineered MGL–kinetic analysis
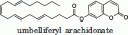
The catalytic activity of several MGL mutants was assayed, using two different substrates, and found to be comparable to wild-type MGL. A small fluorescent substrate, 4-methylumbelliferyl butyrate, was used to compare the activity of the engineered mutants to the activity of wild-type MGL. The catalytic efficiency (kcat/KM) for the hydrolysis of the 4-methylumbelliferyl butyrate was similar for wild-type MGL and all MGL mutants tested (Table II). A larger more aliphatic substrate – umbelliferyl arachidonate with a structure more closely related to the MGL natural substrate, 2-arachidonoyl glycerol—was used to compare the activity of wild-type MGL to TEV protease cleaved mutant MGL (hMGL 1-303 Leu169Ser, Leu176Ser). The catalytic efficiency for the hydrolysis of the umbelliferyl arachidonate substrate was found equivalent between wild-type MGL and the TEV protease cleaved mutant MGL (hMGL 1-303 Leu169Ser, Leu176Ser), confirming that the mutations introduced did not affect the catalytic activity of MGL (Table II).
Table II.
Kinetic Constants
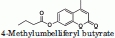 | |||
|---|---|---|---|
| Construct | KM (μM) | kcat (min−1) | kcat/KM (μM−1 min−1) |
| wild-type 1-303 | 162 | 68 | 0.42 |
| Lid-domain mutants | |||
| 1-303 Leu169Ser, Leu176Ser | 136 | 48 | 0.35 |
| 9-393 Leu169Ser, Leu176Ser | 88 | 27 | 0.31 |
| 9-297 Leu169Ser, Leu176Ser | 126 | 26 | 0.21 |
| 1-303 Leu171Gln | 105 | 51 | 0.48 |
| 1-303 Leu167Gln, Leu171Gln | 84 | 59 | 0.71 |
| 1-303 Leu167Gln, Leu174Gln | 84 | 70 | 0.83 |
| 1-303 Leu171Gln, Leu174Gln | 89 | 47 | 0.52 |
| Lid-domain + Surface mutants | |||
| 1-303 Leu169Ser, Leu176Ser, Lys36Ala | 124 | 51 | 0.41 |
| 1-303 Leu169Ser, Leu176Ser, Lys160Ala | 90 | 30 | 0.33 |
| 1-303 Leu169Ser, Leu176Ser, Lys165Ala | 137 | 27 | 0.2 |
| 1-303 Leu169Ser, Leu176Ser, Lys226Ala | 110 | 38 | 0.35 |
| 1-303 Leu169Ser, Leu176Ser, Lys36Ala, Lys226Ala | 123 | 30 | 0.25 |
 | |||
| wild-type 1-303 | * | * | 0.09 |
| mutant 1-303, Leu169Ser, Leu176Ser | * | * | 0.1 |
Kinetic constants of the various MGL constructs using 4-methylumbelliferyl butyrate or umbelliferyl arachidonate as substrates. Values for the 4-methylumbelliferyl butyrate substrate are the average of 2 or 4 separate assays. kcat/KM values for the umbelliferyl arachidonate substrate are the average values for the hydrolysis of five different substrate concentrations at [S] < KM.
(*)The solubility limit of the umbelliferyl arachidonate substrate did not allow for the determination of KM and kcat.
Characterization of engineered MGL–thermal shift assay
A thermal denaturation assay, ThermoFluor®32,33 was used to characterize the wild-type and mutated MGL constructs. The thermal shift assay detects small changes in the intrinsic melting temperature of proteins based on binding of ligands. The technique monitors changes in the fluorescent intensity of dyes such as 1-anilinonaphthalene-8-sulfonic acid (1,8-ANS). Fluorescence of the dye is quenched in aqueous environments but increases upon binding to the hydrophobic core revealed upon denaturation of proteins.
Wild-type MGL exhibited very poor melting characteristics, typical for aggregated or unfolded proteins (Fig. 2). While the midpoint of the melting transition (Tm) was calculated to be 58°C for wild-type MGL, the increased initial fluorescence indicates a highly aggregated protein and the minute transition results in a high level of inaccuracy for the calculated Tm. In contrast the calculated Tm for the mutant MGL (hMGL 1–303 Leu169Ser, Leu176Ser) was consistently measured to ∼56.7°C, and the lower baseline and steeper transition suggest a more stable and cooperatively folded protein, with better properties for high throughput screening and crystallization experiments.
Figure 2.
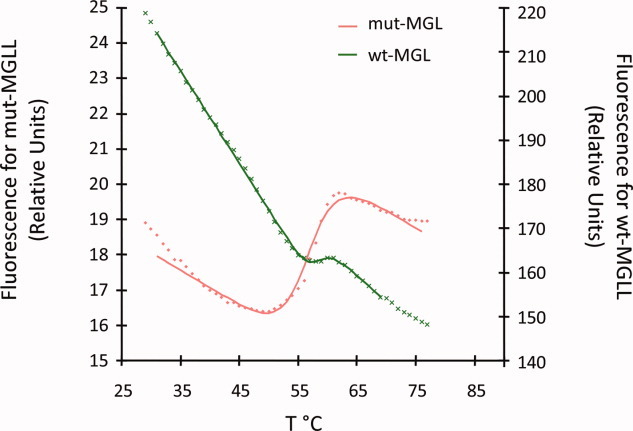
Thermal shift assay. Thermal shift data showing melting transitions for wild-type MGL 1–303 (green line) and mutant MGL (hMGL 1–303 Leu169Ser, Leu176Ser), red line.
Crystallization of MGL
All mutants were prepared as described under Experimental Procedure and subjected to crystallization trials. Purity greater than 95% as determined by SDS-PAGE was achieved for all proteins. Crystals generated of all apo proteins diffracted only between 8.0 Å and 9.0 Å, despite extensive optimization efforts. Co-crystallization with methyl arachidonyl fluorophosphonate, an irreversible active-site inhibitor of MGL and serine hydrolases in general, did not significantly improve diffraction.
Co-crystallization of TEV protease cleaved mutant MGL (hMGL 1-303 Leu169Ser, Leu176Ser, Lys36Ala) with a medium affinity ∼1 μM ligand (data not shown) generated crystals that diffracted to 2.3 Å but with diffused scattering along one axis. Further optimization experiments did not improve diffraction quality. High quality diffracting crystals were obtained by co-crystallization of TEV protease cleaved mutant MGL (hMGL 1-303 Leu169Ser, Leu176Ser, Lys36Ala) with a ligand (Compound 1) that was 10-fold more potent (Fig. 3 inset). Selected crystals diffracted to 1.3 Å and allowed the collection of a high quality dataset, which was processed to 1.35 Å.
Figure 3.

Overview of structure. Ribbon representation of the MGL structure. Color-coding is according to secondary structure (α-helices: magenta, β-sheets: yellow). The ligand (Compound 1) is drawn in ball-and-stick representation (green carbons). The protein adopts a typical α/β hydrolase fold21 comprised of 8 β-sheets, with β2 being antiparallel to the other sheets and six canonical α-helices [α1 or αA, α2(αB), α3(αC), α6(αD), α7(αE), α8(αF)] and two additional α-helices comprising the lid sub-domain [α4( ), α5(
), α5( )]. The inhibitor is located in the active-site, which is capped by loops connecting helices α4–α6. Inset: Structure of Compound 1.
)]. The inhibitor is located in the active-site, which is capped by loops connecting helices α4–α6. Inset: Structure of Compound 1.
Overall structure of MGL
MGL is part of the subfamily of lipid hydrolases, which in turn is part of a larger family of α/β-hydrolases with diverse catalytic functions. Members of this superfamily include: ester hydrolases, lipid hydrolases, thioester hydrolases, peptide hydrolases, haloperoxidases, dehalogenases, epoxide hydrolases, and C–C bond breaking enzymes.34 All these enzymes share a common folding motif called the α/β-hydrolase fold.21,35 This fold is characterized by eight β-sheets flanked on both sides by α-helices. β-sheet 2 is antiparallel to the other sheets and the first and last canonical helix (α1, or αA and α6, or αF) are located on one side of the sheets, whereas the remainder of the helices are present on the opposite side. The α/β-hydrolase fold tolerates a wide variety of inserts without losing the core folding motif. These inserts serve to modify and regulate the catalytic activity of the respective proteins. They can occur in several locations, but are mostly located in a loop region between strand β6 and helix α6.
Here, we describe the X-ray structure of the inhibitor-bound form of human MGL. This structure is determined with use of molecular replacement to a resolution of 1.35 Å (Fig. 3). The structure of MGL conforms very closely to the canonical α/β-hydrolase fold and is characterized by eight β-sheets, which form a partial β-barrel adorned on both sides with eight α-helices. MGL contains two additional helices [α4 () and α5 ()], which are part of the “lid” sub-domain (residues 151–225) and are inserted in the protein sequence between sheet β6 and helix α6 (αD). Helices α1 (αA) and α8 (αF) are located on the concave side of the barrel and helices α2 (αB), α3 (αC), α6(αD), and α7(αE) are on the convex side. Both “lid”-helices are oriented in front of the molecule perpendicular to the plane of the β-barrel.
MGL binding pocket
Compound 1 (Fig. 3) is bound in an extended and closed binding pocket, which is located between helices α4, α6, α7, and α5 [Fig. 4(A)]. Even though the solvent accessible surface area of the compound (712 Å2) is fairly large, it is almost completely enclosed by the protein. The protein accomplishes this by employing a so-called “lid,” “cap,” or “flap” -domain, which regulates access to the binding site based on the membrane-bound state of the protein. The lid-domain is comprised of residues 151–225 from helices α4–α6 (also referred to as and throughout the literature). The catalytic triad of MGL, consisting of residues Ser122, Asp239, and His269, is located in the center of the binding pocket. The catalytic nucleophile Ser122 resides on a tight turn between strand β5 and helix α3, which is also commonly referred to as the “nucleophilic elbow.” The structurally conserved network of hydrogen-bond donors, which comprises the nucleophilic elbow and the loop connecting α1 and β3 (Gly50, Ala51, Met123, and Gly124) is called the oxyanion hole and serves to stabilize the anionic transition state of the catalytic reaction. The amide carbonyl of Compound 1 points into the oxyanion hole and forms a critical hydrogen bond with the backbone amide nitrogen of Met123 adjacent to the catalytic serine. The azetidine-piperazine-pyrimidine part of the ligand projects into a narrow amphiphilic pocket and fills the available space almost completely [Fig. 4(B)]. This portion of the ligand does not participate in hydrogen bond interactions with the protein, but one of the pyrimidine nitrogens forms a hydrogen bond to a water-network involving two buried water molecules and the side-chains of residues Glu53, Arg57, and His272. A face-to-face π-stacking interaction of the pyrimidine ring with Tyr194 provides further interaction energy.
Figure 4.
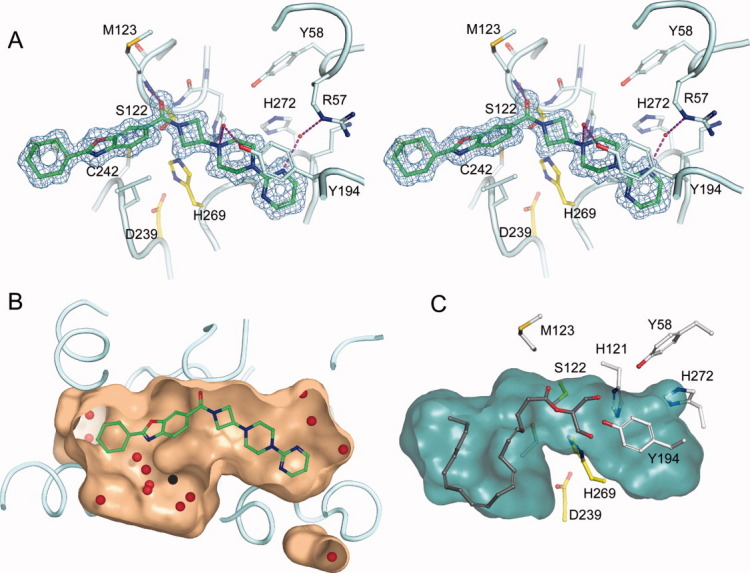
The active-site of MGL. A: Stereo view of a close-up of the MGL active-site. The azetidine amide carbonyl of Compound 1 forms a critical salt bridge with the backbone amide nitrogen of Met123, in addition a π-π stacking interaction with Tyr194 and a water-mediated hydrogen bonding network (magenta lines) with Arg57 and Tyr194 is evident. The inhibitor (green carbons) extends deeply into the active-site and is almost completely buried by the lid-domain, which has been partially omitted from display for clarity. The electron density 2fo-fc map surrounding Compound 1 (blue) is contoured at 1.0 σ and has been limited in its display to 1.5 Å from the nearest atom of Compound 1. Residues of the catalytic triad are depicted in yellow carbons. B: Close-up view of Compound 1 in a cut-away surface depiction of the binding site. The surface was generated using the program “Hollow” (http://hollow.sourceforge.net), by filling the voids of the binding site with waters on a 0.2 Å grid and casting a surface around the waters. Waters found in the active-site are depicted as red spheres. C: Docking model of 2-arachidonoyl glycerol into the closed form of MGL. The surface of the binding site cavity was created using the cavity detection algorithm implemented in PyMOL.
The binding pocket close to the benzoxazole-cyclohexane part of the ligand is less occluded than its counterpart. The benzoxazole portion of the ligand is located in a hydrophobic environment constituted mainly from side chains of aliphatic residues. The cyclohexane portion projects into a more spacious void. Along with the benzoxazole, the cyclohexane portion is the only part of the inhibitor that is accessible by solvent in the protein bound state [Fig. 5(B), left panel]. These parts of the ligand form mostly van der Waals interactions with the protein. The cyclohexane part of the molecule is less well ordered and shows elevated average B-factors (22 Å2) than the remainder of the ligand (9 Å2). Concomitantly, a region of the lid-domain (α4 and part of the loop connecting to α5; residues 151–179) with which the inhibitor interacts displays significantly higher average temperature factors (23 Å2, based on Cα) as compared to the rest of the protein (12 Å2) as well. The elevated temperature factors signify the inherent flexibility of this region, which probably facilitates its displacement from the surface of the protein during ligand binding and release.
Figure 5.

Enabling mutations and electrostatics. A: Location of the enabling mutations. Leu169Ser and Leu176Ser are located in the lid sub-domain (magenta) and prevent protein aggregation. Lys36Ala is located on the loop between β2 and β4 and would make interactions with a symmetry mate if present in the wild-type protein. B. Electrostatic potential of MGL contoured at ±5 kT (red, negative potential; blue, positive potential). Orientation of the protein is identical to the one in panel A. MGL's lid-domain in the closed conformation (B, left) shows a somewhat reduced hydrophobic character as compared to the apo form of MGL (PDB ID 3JHU ) in the open conformation (B, right). The reduced hydrophobic character and the presence of the acidic patch on the right side of the molecule could point to a potential mechanism for dissociation from the membrane during catalysis. Remarkably, replacement of the enabling mutations with wild-type residues (B, middle) does not appear to have a discernable effect on the electrostatic potential of the protein.
To address the question whether the closed form of MGL can accommodate the substrate 2-arachidonoyl glycerol, we performed a docking study using Glide from Schrodinger, Inc. The docking model of MGL in the closed form shows that the protein can accommodate 2-arachidonoyl glycerol completely [Fig. 4(C)]. The substrate, which is folded onto itself, is completely buried in the active-site and displays the proper orientation for catalysis.
Enabling mutations and electrostatic potential
One of the two mutations in the lid-domain that helped increase solubility, Leu169Ser, is located at the C-terminal end of helix α4 (). The other mutation that helped prevent aggregation, Leu176Ser, is located on a loop connecting α4 to α5 [Fig. 5(A)]. The lid-domain in the engineered protein still contains quite a few surface exposed aliphatic residues, but the two mutations are as demonstrated sufficient to change the inherent lipophilic character of the protein enough to prevent aggregation and transform the protein into a more soluble form. Interestingly, a comparison of the experimentally determined structure of the mutant MGL and a wild-type model constructed by replacing the mutated amino acids with the native amino acids, reveals that the enabling mutations do not appear to have a discernable effect on the electrostatic surface of the protein [Fig. 5(B), left and middle].
The Lys36Ala surface mutation, inspired by a series of reports indicating that the replacement of flexible residues on the surface of proteins helps to promote crystallization,36 is present on a loop connecting sheets β2 and β3. This loop interacts with the lid-domain of a neighboring symmetry-related molecule between Val170 and Pro172. Analysis of this packing interaction reveals that a limited number of conformations of the lysine would have fit snuggly into this packing interface, so the reason that Lys36Ala promotes crystallization via prevention of clashes is not obvious. We hypothesize that the mutation is nevertheless beneficial, because this particular part of the lid-domain exhibits relatively high temperature factors and is less well ordered than other parts of the molecule. It is conceivable that this high dynamic mobility would cause the lid to clash with Lys36 in certain portions of the conformational pool. The Lys36Ala mutation would eliminate this potential for clashes and may thus contribute to the successful crystallization of the molecule and improved diffraction quality of the crystals.
Comparison to other MGL structures
Recently, three other crystal structures of MGL in both apo and liganded form were reported by two different laboratories.24,25 In both cases, the structure was solved in the space group I222 with two molecules per asymmetric unit. Superposition (based on backbone Cα positions) of the previously published MGL structures with the one in this report reveals that the majority of the Cα atoms can be superimposed with very little difference [Fig. 6(A)]. However, a subset of the lid-domain (151–225) namely residues in the range of 151–173 display large variations. A more detailed comparison against the apo structure (PDB ID 3HJU)24 reveals that the lid-domain undergoes a substantial rearrangement upon ligand binding [Fig. 6(B,C)]. The transition from the open apo-form to the closed ligand bound form can be best described as a rolling motion of α-helix 4 over the active site opening resulting in an almost 180° counter-clockwise rotation (viewed from C to N-terminus). During the process of the transition α-helix 4 (residues 158–170 in the open conformation) also incorporates residues 153–157 at its N-terminus. The end effect of this inward coiling action is that the entrance to the active-site is substantially more occluded, almost completely burying the compound inside the protein. The small opening at the side of the active-site, which has been proposed to be the exit for the cleaved glycerol moiety,24,25 is also completely closed by this structural rearrangement.
Figure 6.

Flexible region in MGL and ligand binding induced conformational changes. A: Variability of structural elements in MGL as defined by the average distance of Cα positions across all published MGL structures. Average distances of superimposed and matching Cα-atoms are mapped onto a “putty” cartoon representation of MGL. The average distances are color coded from blue (lowest average distance) to red (highest average distance) and represent values from 0.086 Å to 4.5 Å. The same values are also encoded in the radius of the cartoon. B: Overlay of the mutant MGL (cyan) in complex with Compound 1 and apo MGL (dark yellow, PDB ID 3HJU , chain A). The superposition is almost identical for large parts of the molecule, except for a region surrounding α-helix 4 with its connecting loops and the loop linking α-helix 5 and α-helix 6. The arrow indicates the approximate viewpoint assumed in panel C. C: Panel illustrating the rearrangement of α-helix 4. The movement of α-helix 4 from the open to the closed position is characterized by a concomitant rolling motion over the active site opening and an almost 180° counter-clockwise rotation (viewed from C to N-terminus). The movements are indicated by colored arrows, corresponding residues in both structures representing the start and endpoint of the transition are indicated as well. Less pronounced changes are observed in the flanking loop regions during the transition. α-Helix 5, which is also part of the lid-domain, displays only a modest amount of movement.
This report represents the second structure of MGL in complex with a ligand. In the previously reported structure,24 in one copy of the dimer present in the asymmetric unit a single molecule of the inhibitor SAR629 is covalently bound to the terminal oxygen of the catalytic Ser122, whereas in another copy two inhibitor molecules are detected. One inhibitor in the aforementioned covalently bound conformation and another one stacked on top of the covalent inhibitor. The authors deem the additionally bound inhibitor to be an artifact due to the high concentrations of compound present in the experiment. The protein itself is on the open conformation in contrast to the closed structure in this report, which has implications for the shape and expanse of the binding site. Compound 1 spans the whole active site and is confined on the right by the closed off cavity [Figs. 4(B) and 7], leaving little room for optimization of the inhibitor. On the left side, α-helix 4 in the closed conformation, walls off the active site at the cyclohexyl moiety. In contrast SAR629 is located mainly at the “left” part of the binding cavity (Fig. 7), with both fluoro-phenyl groups being partially solvent exposed due to the open conformation of the protein. An essential interaction for Compound 1 is the hydrogen bond of its amide carbonyl with the backbone of Met123. The carbamate carbonyl of SAR629 is located very close to the amide carbonyl of Compound 1, but points into a different direction and interacts with the backbone of Ala51 and Met123 simultaneously. Aside from this similarity both compounds share very little interaction space. Closer analysis of the superposition of both protein-inhibitor complexes reveals that SAR629 would be unable to bind in the closed conformation since both the piperazine and the upper left fluoro-phenyl group would clash with the protein, whereas Compound 1 would most likely be able to bind in the open conformation with minor adjustments (data not shown).
Figure 7.
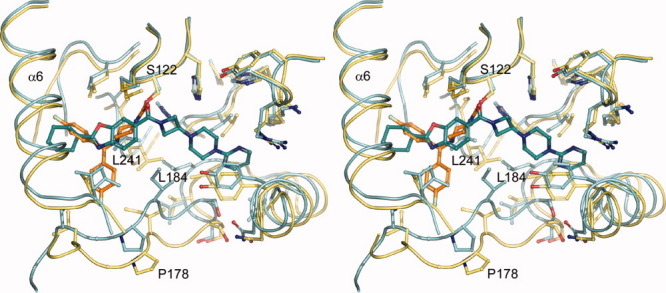
Comparison of ligand binding mode between non-covalent and covalent inhibitor. Stereo depiction of the differences in binding mode between the non-covalent reversible inhibitor Compound 1 and the covalent inhibitor SAR62924 bound to MGL. Selected regions of both proteins in the foreground and α-helix 4 were omitted for clarity reasons. Compound 1 (teal colored carbons) spans the entire active site cavity of MGL (cyan), whereas SAR629 (orange carbons), which is covalently bound to the hydroxyl group of Ser122, solely occupies the “left” section of the binding cavity (yellow). Selected residues are depicted in ball-and-stick representations and were labeled to highlight the differences in the sidechain orientation during the transition from the closed (Compound 1) to the open conformation (SAR629).
The binding site itself changes from the open to the closed conformation, especially the loop region from residues 174 to 182, which connects α-helix 4 to α-helix 5, moves “upwards” and closer to Compound 1 (Fig. 7), therefore narrowing the active site tunnel. Flanking loop regions also show variations, notably leucines 184 and 241 are shifted and present in different rotamers, substantially changing the shape of the binding pocket. Catalytically relevant residues (Ser122, Asp239, His269) and flanking helices (α5 and α6) on the other hand superimpose very well.
Aside from the obvious structural differences between the apo and ligand-bound forms of MGL, differences in the electrostatic potential between these two forms can also be observed [Fig. 5(B)]. The hydrophobic character of the lid-domain in the closed, complexed conformation is somewhat reduced as compared to the open apo conformation. Furthermore, an acidic patch, associated with the region surrounding Met88 (G79, H80, R87, M88, C201, M123, F209) in the closed conformation, is transformed into a more basic character during the transition from the closed to the open conformation.
Due to a lack of biological data with respect to the functional significance of α-helix 4, the implications of these transitions are not completely understood. Nevertheless, it is reasonable to postulate that in the open conformation of MGL the hydrophobic character of α-helix 4, together with the basic patch, serves to position the protein close to the negatively charged membrane to allow the diffusion of a substrate molecule into the wide open active-site funnel. Upon substrate binding, α-helix 4 would close over the bound substrate, catalysis would commence, and MGL could potentially dissociate from the membrane due to charge-charge repulsion of the acidic patch with the membrane. After dissociation, the enzyme would once again assume the open position, release the formed products, and reassociate with the membrane. The physiological reasons behind this proposed detachment from the membrane during the catalysis cycle are not known up to this point.
Conclusion
Directed mutagenesis was employed to engineer a soluble, monomeric, and crystallizable form of the membrane-associated lipase MGL. The crystal structure of MGL in complex with a reversible and pharmacologically relevant ligand has been solved to 1.35 Å. This ligand bound MGL is presented in a novel conformation. The structure displays the typical α/β hydrolase fold, but in contrast to previously solved structures of MGL, the structure reported here displays a ligand-induced closed conformation of the regulatory important lid-domain or cap-domain. The α-helix 4 of the lid-domain folds over the bound ligand and almost completely buries it within the active-site. Analysis of the electrostatic properties of the mutated protein against its wild-type counterpart reveals that the mutations (Lys36Ala, Leu169Ser, and Leu176Ser) do not significantly alter the electrostatic potential of the molecule. However, the transition from the open to the closed state of the protein induces a change in the electrostatic potential of MGL, which we hypothesize could lead to detachment of the protein from the lipid membrane during the catalytic cycle.
Two mutations (Leu169Ser and Leu176Ser) in α-helix-4, which is part of the lid-domain of MGL, increase the solubility enough to allow for the purification of monomeric protein in the absence of detergent. Interestingly, the lid-domain in the engineered protein still contains a significant portion of hydrophobic surface residues, but the mutations are able to modify the hydrophobic character of the protein enough to prevent aggregation in solution. The mutations also vastly improve the melting characteristics of the protein to the extent that the engineered protein was used successfully in a thermal shift based screening campaign. An additional lysine to alanine mutation at residue 36 enables the generation of crystals, which after co-crystallization with a high-affinity ligand, diffract to 1.30 Å, consistent with other reports of using lysine mutations to promote crystallization.29,30,36 In addition to providing insight into different conformations of MGL, this structure also offers guidance for efforts to develop inhibitors of MGL with desirable antinociceptive or other properties.
Material and Methods
Molecular modeling
A BLAST sequence search37 of the human MGL protein sequence reveals that human MGL has very little sequence homology with other mammalian lipases but shows a distant relationship to esterases, lysophospholipases, and haloperoxidases.22 Up until recently, the closest relative to MGL found in a search of sequences of structures deposited in the Protein Data Bank (PDB)38 is RsbQ, a stress-response regulator in Bacillus subtilis.39 RsbQ displays the highest sequence identity (16%) and smallest insertions and deletions relative to MGL of any structure available in the PDB. A lid or cap-domain composed of four helices is located at the surface of RsbQ surrounding a channel leading to the active-site. The MGL sequence (residues 158–188), which aligns with the lid-domain of RsbQ (residues 137–188) is amphipathic. Sixteen out of the thirty one residues are hydrophobic, seven of which are leucines. Fifteen of the residues are hydrophilic, four of which are electropositive lysines and arginines.
A homology model of MGL has been created using RsbQ (PDB ID 1WOM)39 as a template and the “quick refine” option in GeneMine.40,41 The model shows a α/β hydrolase domain and a lid-domain. As predicted by sequence alignment, the amphiphatic region of MGL (161–185) forms a helix similar to the lid-domain of RsbQ. We postulate that the high number of hydrophobic residues in this surface exposed helix defines a region that is involved in association of MGL with the membrane. The lysine and arginine residues presumably could interact with the electronegative phospholipid heads.
Cloning
The cDNA for human MGL was cloned from a human brain cDNA library and used as a template to generate a PCR fragment of MGL corresponding to amino acids 1-303 (5′ primer gagaatttggtattttcaaggtatgccagaggaaagttcccc; 3′ primer tggatgtgtatgtttctatcagggtggggacgaagttcc). The PCR product was purified (GENECLEAN SPIN kits, Qbiogene), treated with T4 polymerase (New England Biolabs), ligated into the modified pENTR.11cLIC vector and transformed into TOP10 one shot competent cells (Invitrogen). After sequence confirmation, the mutations were added by Quickchange mutagenesis, (Stratagene). The sequence-confirmed plasmid was purified for transfection into insect cells using the BaculoDirect Baculovirus Expression System (Invitrogen). The viral stock was propagated for two more amplifications at a low multiplicity of infection (MOI) to render a P2 virus stock.
Recombinant production of MGL
Large-scale expression was carried out in 2-liter shake flasks or WAVE bioreactors. The P2 virus was expanded to generate a high-titer P3 stock by infecting Sf9 cells in suspension at a MOI of 0.3 and harvesting the virus after 72 h. Cell paste for MGL was obtained by infecting Sf9 cells at a density of 1.5 × 106 cells/mL with a MOI of 1. Infected cultures were maintained at 27°C under constant shaking at 140 rpm. Cells were harvested 65–72 h post-infection by centrifugation at 1000g-force for 10 min at 4°C. Cell viability was determined by Guava ViaCount or Trypan Blue and routinely was between 60 and 80% at time of harvest. Cell pellets were washed once in phosphate-buffered saline with broad-range protease inhibitors and stored at –80°C.
Purification
The ÄKTA Xpress System (GE Healthcare) was used for all purification processes. All operations were carried out at 4°C.
Purification of mutant MGL
Frozen cell pellets were thawed and resuspended in buffer A [50 mM HEPES buffer pH 7.5, 400 mM NaCl, 5% glycerol, 0.05% BME, 1× Complete EDTA-free protease inhibitor cocktail tablets (Roche)], dounce homogenized, and mechanically lysed with a microfluidizer processor (Microfluidics). The extract was clarified by centrifugation at 40,000g-force for 1 h. The cleared lysate was loaded on a 1 mL His-Trap FF Crude column (GE-Healthcare) or a 5 mL His-Trap column for large-scale purifications at 4°C. The column was washed with 10–15 column volumes of buffer A containing 30 mM imidazole and MGL was eluted with 5 column volume of buffer A containing 400 mM imidazole. The elution peak was directly loaded on a Superdex 200 HR 16/60 column pre-equilibrated with 50 mM HEPES pH 7.5 buffer containing 200 mM NaCl, 2% glycerol, 2 mM dithiothreitol, 2 mM EDTA. Fractions were analyzed by SDS-PAGE. Fractions containing MGL were pooled. Expression yields were determined by Bradford assay using the protein assay kit from BioRad according to manufacturers' instruction with BSA as a standard.42
Purification of wild-type MGL
Frozen cell pellets for MGL wild-type were thawed, resuspended, and lysed in Bugbuster® lysis buffer (Invitrogen) for 1 h at 4°C. The extract was clarified by centrifugation at 40,000g-force for 1 h and the purification was carried following the protocol used for the mutant MGL, without further addition of detergent.
Tag cleavage
A total of 0.2 units of TEV Protease for each μg of MGL were added to the MGL pool to remove the histidine tag. The reaction was performed overnight at 4°C. Cleavage of the histidine tag was monitored by SDS-PAGE.
Circular dichroism
Circular dichroism experiments were performed on a circular dichroism spectrometer model 202 from Aviv Instruments Inc. The circular dichroism scans of wild-type MGL and mutant MGL (hMGL 1-303 Leu169Ser, Leu176Ser) (5 μM in 10 mM cacodylic acid, pH 7.0, and 140 mM NaCl) were measured from 200 to 260 nm. Temperature melts were monitored at 210 nm. The circular dichroism spectra were converted to molar ellipticity.
Kinetic analysis
The Michaelis-Menten parameters for the hydrolysis of 4-methylumbelliferyl butyrate (Sigma 19362) and umbelliferyl arachidonate (Sigma U0383) were determined using 4–5 nM of MGL in 20 mM PIPES, pH 7.0, and 150 mM NaCl at 37°C. The change in fluorescence due to substrate hydrolysis was monitored using excitation/emission wavelengths of 335/440 nm in a Safire II instrument from Tecan. The hyperbolic rates versus substrate concentration curves for the hydrolysis of 4-methylumbelliferyl butyrate were fit to the Michaelis-Menten equation using Excel.
The solubility limit of the umbelliferyl arachidonate substrate did not allow for the determination of KM and kcat. The kcat/KM for the hydrolysis of umbelliferyl arachidonate was determined at [S] < KM. The KM for umbelliferyl arachidonate was determined to be >30 μM. The kcat/KM values reported are the average from independent values determined from five substrate concentrations ranging from 40 to 700 nM.
Thermal stability
Three microliters of protein at a concentration of 0.05 mg/mL in 50 mM PIPES pH 7.0, 200 mM NaCl, 100 μM 1,8-ANS, and 0.001% Tween 20 were added in 384-well plates. Wells were overlaid with silicone oil (1 μL, Fluka, type DC 200) to prevent evaporation. Assay plates were heated at a rate of 1°C /min for all experiments over a temperature range sufficient to measure protein unfolding. Fluorescence was measured by continuous illumination with UV light (Hamamatsu LC6) supplied via fiber optic and filtered through a custom band-pass filter (380–400 nm; >6 OD cutoff). Fluorescence emission was detected by measuring light intensity using a CCD camera (Sensys, Roper Scientific) filtered to detect emission at 500 ± 25 nm, resulting in simultaneous and independent readings of all 384 wells. One or more images were collected at each temperature, and the sum of the pixel intensity in a given area of the assay plate was recorded versus temperature, and integrated to calculate the melting temperatures.
Crystallization
TEV protease cleaved mutant MGL construct (hMGL 1–303, Leu169Ser, Leu176Ser, Lys36Ala) at 1 mg/mL in 50 mM HEPES, pH 7.5, 200 mM NaCl, 2% glycerol, 2 mM EDTA, 2 mM dithiothreitol, was mixed with 2 fold excess of Compound 1, incubated overnight at 4°C and concentrated to 5.7 mg/mL.
Initial crystals were obtained by incubating 0.6 μL of complexed protein in a 1:1 ratio with well solution (18.4% PEG MME 5000, 50 mM Na-citrate pH 4.5, 0.2% glucopyranoside) in a proprietary 1536 plate modified to accommodate vapor-diffusion experiments and equilibrated at room temperature against 60 μL well solution. These crystals were used as a seed stock for the standard crystallization experiments. Under standard conditions, 1 μL of protein is mixed with 0.2 μL seed stock and 0.5 μL of 8–12% PEGMME 5000, 100 mM Na-MES pH 6.0, 0.2% glucopyranoside and equilibrated against 1 mL reservoir of 6–10% PEGMME 5000, 100 mM Na-MES pH 6.0, 0.2% glucopyranoside in a hanging drop vapor diffusion experiment at room temperature.
Structure determination and refinement
Crystals were harvested, transferred to 16% PEGMME 5000, 100 mM Na-MES pH 6.0, 25% glycerol and flash frozen in liquid nitrogen. A complete initial dataset was collected on a Rigaku M007HF generator at 100°K, data was processed in the HKL2000 suite43 or d*trek.44 All attempts to solve the structure via heavy atom soaks were unsuccessful due to the introduction of high nonisomorphism during the procedure; therefore a molecular replacement approach was pursued. Due to the low sequence similarity between MGL and other lipases, multiple search models were identified via a PSI-BLAST45 search against the PDB repository. The search models were prepared in CHAINSAW46 by either truncating each residue to Cα atoms or truncating nonidentical residues to Cγ atoms. An automated procedure using PHASER47 as the molecular replacement engine yielded a good solution with a single molecule of “Non-haem bromoperoxidase BPO-A1” (PDB ID 1A8Q) as search model. The initial rebuilding was performed using the default protocol in the AutoBuild Wizard in PHENIX,48,49 refinement and automated water picking was carried out in PHENIX.refine; Coot50 was employed for model building, ligand placement and manual assignment of water molecules. Ligand restraints were generated in PHENIX.elbow.48
The high resolution dataset was collected at the IMCA ID-17 beamline at the APS, processed with HKL2000 and structure refined in PHENIX.refine. Datacollection and refinement statistics are summarized in Table III. The final model was validated using tools implemented in Coot; all crystallographic figures were generated in PyMOL®.51
Table III.
Datacollection and Refinement Statistics
| MGL complex with 1 | |
|---|---|
| Data collection | |
| Wavelength (Å) | 1.0 |
| Resolution (Å)a | 1.35 (1.45–1.35) |
| Space group | C2221 |
| Unit cell parameters (Å) | a = 94.94, b = 128.14, c = 60.60 |
| No. of reflections | 327,079 |
| No. of unique reflections | 74,919 |
| Redundancy | 4.1 (2.1) |
| Completeness (%) | 93.2 (80.2) |
| Rmerge (%) | 6.2 (35.9) |
| I/σ(I) | 13.6 (3.3) |
| Refinement | |
| Molecules per ASUb | 1 |
| Resolution (Å) | 30.4 – 1.35 |
| No. of reflectionsc | 71,433 |
| No. of reflections in Rfree set | 1794 |
| Total No. of non H atoms | 2727 |
| No. of protein atoms | 2334 |
| No. of ligand atoms | 33 |
| No. of solvent molecules | 360 |
| R-factor (%) | 11.4 |
| Rfree (%) | 14.6 |
| R.M.S. Deviation from ideal geometry | |
| Bonds (Å) | 0.014 |
| Angles (°) | 1.435 |
| B-factors (Å2) | |
| Protein | 17.6 |
| Ligand | 13.6 |
| Ramachandran Plot | |
| Preferred Regions (%) | 97.7 |
| Allowed regions (%) | 2.3 |
| Disallowed regions (%) | 0 |
Values for the highest resolution shell are given in parenthesis.
ASU, asymmetric unit.
After conversion of I-obs to F-obs and rejections of F < 0.
The X-ray structure of monoglyceride lipase has been deposited into the Protein Data Bank. The ID number is 3PE6.
Electrostatic calculations
For electrostatic calculations solvent, ligands, hydrogen atoms, and alternate conformations were removed from the structure. The protein was charged at pH 7 using the procedure implemented in PDB2PQR v.1.5.52 Electrostatic potential was calculated at T = 298 K and 150 mM single charged ionic strength as implemented in APBS v.1.2.1.53 For visualization the potential at the solvent accessible surface was mapped onto the molecular protein surface as implemented in PyMOL.
Acknowledgments
Use of the IMCA-CAT beamline 17-ID (or 17-BM) at the Advanced Photon Source is supported by the companies of the Industrial Macromolecular Crystallography Association through a contract with Hauptman-Woodward Medical Research Institute. Use of the Advanced Photon Source is supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357.
Glossary
Abbrevations
- 1,8-ANS
1-anilinonaphthalene-8-sulfonic acid
- CB1
cannabinoid receptor 1
- CB2
cannabinoid receptor 2
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- MES
2-(N-morpholino)ethanesulfonic acid
- MGL
monoglyceride lipase
- PEG MME 5000
polyethylene glycol monomethyl ether 5 kDa average molecular weight
- PIPES
1,4-piperazinediethanesulfonic acid
- RsbQ
Bacillus subtilis stress-response regulator
- TEV
tobacco etch virus
- Tm
melting temperature
References
- 1.Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346:561–564. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- 2.Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- 3.Hohmann AG, Herkenham M. Cannabinoid receptors undergo axonal flow in sensory nerves. Neuroscience. 1999;92:1171–1175. doi: 10.1016/s0306-4522(99)00220-1. [DOI] [PubMed] [Google Scholar]
- 4.Walczak JS, Pichette V, Leblond F, Desbiens K, Beaulieu P. Behavioral, pharmacological and molecular characterization of the saphenous nerve partial ligation: a new model of neuropathic pain. Neuroscience. 2005;132:1093–1102. doi: 10.1016/j.neuroscience.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 5.Farquhar-Smith WP, Egertova M, Bradbury EJ, McMahon SB, Rice AS, Elphick MR. Cannabinoid CB(1) receptor expression in rat spinal cord. Mol Cell Neurosci. 2000;15:510–521. doi: 10.1006/mcne.2000.0844. [DOI] [PubMed] [Google Scholar]
- 6.Galiegue S, Mary S, Marchand J, Dussossoy D, Carriere D, Carayon P, Bouaboula M, Shire D, Le Fur G, Casellas P. Expression of central and peripheral cannabinoid receptors in human immune tissues and leukocyte subpopulations. Eur J Biochem. 1995;232:54–61. doi: 10.1111/j.1432-1033.1995.tb20780.x. [DOI] [PubMed] [Google Scholar]
- 7.Hohmann AG. Spinal and peripheral mechanisms of cannabinoid antinociception: behavioral, neurophysiological and neuroanatomical perspectives. Chem Phys Lipids. 2002;121:173–190. doi: 10.1016/s0009-3084(02)00154-8. [DOI] [PubMed] [Google Scholar]
- 8.Hanus L, Breuer A, Tchilibon S, Shiloah S, Goldenberg D, Horowitz M, Pertwee RG, Ross RA, Mechoulam R, Fride E. HU-308: a specific agonist for CB(2), a peripheral cannabinoid receptor. Proc Natl Acad Sci USA. 1999;96:14228–14233. doi: 10.1073/pnas.96.25.14228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hohmann AG, Farthing JN, Zvonok AM, Makriyannis A. Selective activation of cannabinoid CB2 receptors suppresses hyperalgesia evoked by intradermal capsaicin. J Pharmacol Exp Ther. 2004;308:446–453. doi: 10.1124/jpet.103.060079. [DOI] [PubMed] [Google Scholar]
- 10.Ibrahim MM, Rude ML, Stagg NJ, Mata HP, Lai J, Vanderah TW, Porreca F, Buckley NE, Makriyannis A, Malan TP., Jr CB2 cannabinoid receptor mediation of antinociception. Pain. 2006;122:36–42. doi: 10.1016/j.pain.2005.12.018. [DOI] [PubMed] [Google Scholar]
- 11.Malan TP, Jr., Ibrahim MM, Deng H, Liu Q, Mata HP, Vanderah T, Porreca F, Makriyannis A. CB2 cannabinoid receptor-mediated peripheral antinociception. Pain. 2001;93:239–245. doi: 10.1016/S0304-3959(01)00321-9. [DOI] [PubMed] [Google Scholar]
- 12.Malan TP, Jr., Ibrahim MM, Vanderah TW, Makriyannis A, Porreca F. Inhibition of pain responses by activation of CB(2) cannabinoid receptors. Chem Phys Lipids. 2002;121:191–200. doi: 10.1016/s0009-3084(02)00155-x. [DOI] [PubMed] [Google Scholar]
- 13.Quartilho A, Mata HP, Ibrahim MM, Vanderah TW, Porreca F, Makriyannis A, Malan TP., Jr Inhibition of inflammatory hyperalgesia by activation of peripheral CB2 cannabinoid receptors. Anesthesiology. 2003;99:955–960. doi: 10.1097/00000542-200310000-00031. [DOI] [PubMed] [Google Scholar]
- 14.Gonsiorek W, Lunn C, Fan X, Narula S, Lundell D, Hipkin RW. Endocannabinoid 2-arachidonyl glycerol is a full agonist through human type 2 cannabinoid receptor: antagonism by anandamide. Mol Pharmacol. 2000;57:1045–1050. [PubMed] [Google Scholar]
- 15.Maresz K, Carrier EJ, Ponomarev ED, Hillard CJ, Dittel BN. Modulation of the cannabinoid CB2 receptor in microglial cells in response to inflammatory stimuli. J Neurochem. 2005;95:437–445. doi: 10.1111/j.1471-4159.2005.03380.x. [DOI] [PubMed] [Google Scholar]
- 16.Sugiura T, Kondo S, Kishimoto S, Miyashita T, Nakane S, Kodaka T, Suhara Y, Takayama H, Waku K. Evidence that 2-arachidonoylglycerol but not N-palmitoylethanolamine or anandamide is the physiological ligand for the cannabinoid CB2 receptor. Comparison of the agonistic activities of various cannabinoid receptor ligands in HL-60 cells. J Biol Chem. 2000;275:605–612. doi: 10.1074/jbc.275.1.605. [DOI] [PubMed] [Google Scholar]
- 17.Dinh TP, Freund TF, Piomelli D. A role for monoglyceride lipase in 2-arachidonoylglycerol inactivation. Chem Phys Lipids. 2002;121:149–158. doi: 10.1016/s0009-3084(02)00150-0. [DOI] [PubMed] [Google Scholar]
- 18.Dinh TP, Kathuria S, Piomelli D. RNA interference suggests a primary role for monoacylglycerol lipase in the degradation of the endocannabinoid 2-arachidonoylglycerol. Mol Pharmacol. 2004;66:1260–1264. doi: 10.1124/mol.104.002071. [DOI] [PubMed] [Google Scholar]
- 19.Blankman JL, Simon GM, Cravatt BF. A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem Biol. 2007;14:1347–1356. doi: 10.1016/j.chembiol.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dinh TP, Carpenter D, Leslie FM, Freund TF, Katona I, Sensi SL, Kathuria S, Piomelli D. Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc Natl Acad Sci USA. 2002;99:10819–10824. doi: 10.1073/pnas.152334899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ollis DL, Cheah E, Cygler M, Dijkstra B, Frolow F, Franken SM, Harel M, Remington SJ, Silman I, Schrag J, Sussman JL, Verschueren KHG, Goldman A. The alpha/beta hydrolase fold. Protein Eng. 1992;5:197–211. doi: 10.1093/protein/5.3.197. [DOI] [PubMed] [Google Scholar]
- 22.Karlsson M, Contreras JA, Hellman U, Tornqvist H, Holm C. cDNA cloning, tissue distribution, and identification of the catalytic triad of monoglyceride lipase. Evolutionary relationship to esterases, lysophospholipases, and haloperoxidases. J Biol Chem. 1997;272:27218–27223. doi: 10.1074/jbc.272.43.27218. [DOI] [PubMed] [Google Scholar]
- 23.Chon SH, Zhou YX, Dixon JL, Storch J. Intestinal monoacylglycerol metabolism: developmental and nutritional regulation of monoacylglycerol lipase and monoacylglycerol acyltransferase. J Biol Chem. 2007;282:33346–33357. doi: 10.1074/jbc.M706994200. [DOI] [PubMed] [Google Scholar]
- 24.Bertrand T, Auge F, Houtmann J, Rak A, Vallee F, Mikol V, Berne PF, Michot N, Cheuret D, Hoornaert C, Mathieu M. Structural basis for human monoglyceride lipase inhibition. J Mol Biol. 2010;396:663–673. doi: 10.1016/j.jmb.2009.11.060. [DOI] [PubMed] [Google Scholar]
- 25.Labar G, Bauvois C, Borel F, Ferrer JL, Wouters J, Lambert DM. Crystal structure of the human monoacylglycerol lipase, a key actor in endocannabinoid signaling. Chem Biochem. 2010;11:218–227. doi: 10.1002/cbic.200900621. [DOI] [PubMed] [Google Scholar]
- 26.Karlsson M, Tornqvist H, Holm C. Expression, purification, and characterization of histidine-tagged mouse monoglyceride lipase from baculovirus-infected insect cells. Protein Expr Purif. 2000;18:286–292. doi: 10.1006/prep.1999.1194. [DOI] [PubMed] [Google Scholar]
- 27.Somma-Delpero C, Valette A, Lepetit-Thevenin J, Nobili O, Boyer J, Verine A. Purification and properties of a monoacylglycerol lipase in human erythrocytes. Biochem J. 1995;312(Pt 2):519–525. doi: 10.1042/bj3120519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tornqvist H, Belfrage P. Purification and some properties of a monoacylglycerol-hydrolyzing enzyme of rat adipose tissue. J Biol Chem. 1976;251:813–819. [PubMed] [Google Scholar]
- 29.Longenecker KL, Garrard SM, Sheffield PJ, Derewenda ZS. Protein crystallization by rational mutagenesis of surface residues: Lys to Ala mutations promote crystallization of RhoGDI. Acta Crystallogr D Biol Crystallogr. 2001;57:679–688. doi: 10.1107/s0907444901003122. [DOI] [PubMed] [Google Scholar]
- 30.Mateja A, Devedjiev Y, Krowarsch D, Longenecker K, Dauter Z, Otlewski J, Derewenda ZS. The impact of Glu->Ala and Glu->Asp mutations on the crystallization properties of RhoGDI: the structure of RhoGDI at 1.3 A resolution. Acta Crystallogr D Biol Crystallogr. 2002;58:1983–1991. doi: 10.1107/s090744490201394x. [DOI] [PubMed] [Google Scholar]
- 31.Labar G, Bauvois C, Muccioli GG, Wouters J, Lambert DM. Disulfiram is an inhibitor of human purified monoacylglycerol lipase, the enzyme regulating 2-arachidonoylglycerol signaling. ChemBiochem. 2007;8:1293–1297. doi: 10.1002/cbic.200700139. [DOI] [PubMed] [Google Scholar]
- 32.Matulis D, Kranz JK, Salemme FR, Todd MJ. Thermodynamic stability of carbonic anhydrase: measurements of binding affinity and stoichiometry using ThermoFluor. Biochemistry. 2005;44:5258–5266. doi: 10.1021/bi048135v. [DOI] [PubMed] [Google Scholar]
- 33.Pantoliano MW, Petrella EC, Kwasnoski JD, Lobanov VS, Myslik J, Graf E, Carver T, Asel E, Springer BA, Lane P, Salemme FR. High-density miniaturized thermal shift assays as a general strategy for drug discovery. J Biomol Screen. 2001;6:429–440. doi: 10.1177/108705710100600609. [DOI] [PubMed] [Google Scholar]
- 34.Holmquist M. Alpha/Beta-hydrolase fold enzymes: structures, functions and mechanisms. Curr Protein Pept Sci. 2000;1:209–235. doi: 10.2174/1389203003381405. [DOI] [PubMed] [Google Scholar]
- 35.Heikinheimo P, Goldman A, Jeffries C, Ollis DL. Of barn owls and bankers: a lush variety of alpha/beta hydrolases. Structure. 1999;7:141–146. doi: 10.1016/s0969-2126(99)80079-3. [DOI] [PubMed] [Google Scholar]
- 36.Derewenda ZS, Vekilov PG. Entropy and surface engineering in protein crystallization. Acta Crystallogr D Biol Crystallogr. 2006;62:116–124. doi: 10.1107/S0907444905035237. [DOI] [PubMed] [Google Scholar]
- 37.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 38.Sussman JL, Lin D, Jiang J, Manning NO, Prilusky J, Ritter O, Abola EE. Protein Data Bank (PDB): database of three-dimensional structural information of biological macromolecules. Acta Crystallogr D Biol Crystallogr. 1998;54:1078–1084. doi: 10.1107/s0907444998009378. [DOI] [PubMed] [Google Scholar]
- 39.Kaneko T, Tanaka N, Kumasaka T. Crystal structures of RsbQ, a stress-response regulator in Bacillus subtilis. Protein Sci. 2005;14:558–565. doi: 10.1110/ps.041170005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee C, Irizarry K. The GeneMine system for genome/proteome annotation and collaborative data mining. IBM Syst J. 2001;40:592–603. [Google Scholar]
- 41.Levitt M. Accurate modeling of protein conformation by automatic segment matching. J Mol Biol. 1992;226:507–533. doi: 10.1016/0022-2836(92)90964-l. [DOI] [PubMed] [Google Scholar]
- 42.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 43.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. In: Carter CW, Sweet RM, editors. Methods in Enzymology. New York: Academic Press; 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 44.Pflugrath JW. The finer things in X-ray diffraction data collection. Acta Crystallogr D Biol Crystallogr. 1999;55:1718–1725. doi: 10.1107/s090744499900935x. [DOI] [PubMed] [Google Scholar]
- 45.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Collaborative Computational Project No 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 47.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallography. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 49.Terwilliger TC, Grosse-Kunstleve RW, Afonine PV, Moriarty NW, Zwart PH, Hung LW, Read RJ, Adams PD. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr D Biol Crystallogr. 2008;64:61–69. doi: 10.1107/S090744490705024X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 51.DeLano WL. The PyMOL Molecular Graphics System. Palo Alto, California: 2002. [Google Scholar]
- 52.Dolinsky TJ, Czodrowski P, Li H, Nielsen JE, Jensen JH, Klebe G, Baker NA. PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007;35:522–525. doi: 10.1093/nar/gkm276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]