

Abstract
Calmodulin (CaM) is a Ca2+-binding protein that functions as a ubiquitous Ca2+-signaling molecule, through conformational changes from the “closed” apo conformation to the “open” Ca2+-bound conformation. Mg2+ also binds to CaM and stabilizes its folded structure, but the NMR signals are broadened by slow conformational fluctuations. Using the E104D/E140D mutant, designed to decrease the signal broadening in the presence of Mg2+ with minimal perturbations of the overall structure, the solution structure of the Mg2+-bound form of the CaM C-terminal domain was determined by multidimensional NMR spectroscopy. The Mg2+-induced conformational change mainly occurred in EF hand IV, while EF-hand III retained the apo structure. The helix G and helix H sides of the binding sequence undergo conformational changes needed for the Mg2+ coordination, and thus the helices tilt slightly. The aromatic rings on helix H move to form a new cluster of aromatic rings in the hydrophobic core. Although helix G tilts slightly to the open orientation, the closed conformation is maintained. The fact that the Mg2+-induced conformational changes in EF-hand IV and the hydrophobic core are also seen upon Ca2+ binding suggests that the Ca2+-induced conformational changes can be divided into two categories, those specific to Ca2+ and those common to Ca2+ and Mg2+.
Keywords: calmodulin, magnesium, conformational exchange, NMR, solution structure
Introduction
Calmodulin (CaM) is a ubiquitous intracellular Ca2+-binding protein that functions predominantly in the integration of Ca2+ in signal transduction processes. It is composed of two homologous globular domains (N- and C-terminal domains),1–4 each consisting of two Ca2+ binding motifs, called EF-hand motifs.5 The EF-hand possesses a helix-loop-strand-helix structure and binds to Ca2+ via a 12-residue binding sequence. The binding sequence, which is often referred to as the EF-hand loop, is structurally composed of a loop, a strand and the N-terminal part of an α-helix. Ca2+ is chelated by seven oxygens from six residues, at positions 1, 3, 5, 7, 9, and 12 in a pentagonal bipyramidal fashion. The residues at positions 1, 3, and 5 are located in the loop, those at positions 7 and 9 are located in the strand, and the residue at position 12 is located in the helix structure. Ca2+ binding to the EF-hand of CaM induces the exposure of the hydrophobic core that provides the interaction site for target molecules, including calcium channel, myosin light chain kinase, CaM kinase, calcineurin, and nitric oxide synthase.1–9
Mg2+ can also bind to all of the EF-hands of CaM, and shares the same binding site with Ca2+.10–14 The Mg2+ association constants range between 102 and 104 M−1.10 Since Mg2+ exists in the cytosol at a millimolar concentration,15 the high affinity sites (EF-hands I and IV) can be converted into the Mg2+-bound forms. The folded structure of CaM is stabilized by Mg2+ binding.13,16 The Mg2+-bound form of CaM is more stable than the apo form of CaM against denaturation by chemicals, such as urea and guanidine, and by thermodynamic effects.17,18 Mg2+ increases the denaturation temperature of each domain of CaM by more than 20°C18 (Supporting Information Table S1). Mg2+ binding seems to be important to stabilize the structure of CaM.
Ca2+ and Mg2+ cause distinct conformational changes of CaM.10,12,13 In the Ca2+-bound form, the hydrophobic core is exposed.1 However, it has been proposed that the hydrophobic core is not exposed in the Mg2+-bound form.10 The Mg2+-bound structure of the N-terminal domain of soybean calmodulin isoform 4 (sCaM4), which shares about 78% sequence identity with bovine CaM,19 has been investigated by the backbone residual dipolar coupling approach.16 The apo form of the N-terminal domain of sCaM4 is unfolded. In the presence of Mg2+, the N-terminal domain of sCaM4 folds into an α-helix-rich structure.16 This is an excellent example of stabilization by Mg2+ binding. The Mg2+-bound form of sCaM4 adopts a closed conformation. Since the structure was calculated by the backbone assignments and the backbone RDC restraints, only the information about the orientation of the EF-hand helices was experimentally determined. Other details, including the Mg2+ coordination, the side chain orientations and the arrangement of the hydrophobic core, have remained unknown. To further clarify the details of the Mg2+-induced conformational changes and the features of the Mg2+-bound state, the tertiary structure of the Mg2+-bound form at atomic resolution is needed.
Here, we performed a structural analysis of the Mg2+-bound form of the CaM C-terminal domain (residues 78–148, CaMC) by multidimensional NMR spectroscopy. The two domains (N-terminal domain and C-terminal domain) fold independently, and the Ca2+-binding affinity and the Ca2+-induced structural changes of these domains are both equivalent to those of intact CaM.18,20 Mg2+ preferentially binds to EF-hands I and IV of CaM.10 In EF-hand I, the Mg2+-induced chemical shift changes are localized in the N-terminal part of the binding sequence.14 However, in EF-hand IV, the Mg2+-induced changes occur throughout the binding sequence. The NMR signals and the fluorescence intensity of Y138, located on the C-terminal part of the EF-hand IV binding sequence, change upon Mg2+ binding.12,21 Therefore, larger structural changes are anticipated for CaMC.
In an NMR study, the HSQC spectra of the apo and Ca2+-bound forms of CaM displayed well-dispersed signals. On the other hand, the NMR spectra of the Mg2+-bound form of CaM showed signal broadening for most of the residues, indicating that a slow conformational fluctuation, which is an exchange between multiple conformations, occurs around the core of the molecule. NMR signal broadening upon Mg2+ binding is a common feature among EF-hand proteins.22,23 NMR is an appropriate method to monitor the exchange process. The signal broadening problem with the Mg2+-bound form of CaMC was solved by double point mutations. The solution structure of the Mg2+-bound form of CaMC was determined by multidimensional NMR spectroscopy, using the mutant. We observed the structural changes upon Mg2+ binding. Although the overall structure was the closed form, like the apo form, most of the structural changes in the binding site and the core region were similar to those induced by Ca2+.
Results
Mg2+ induces hydrogen bond interactions in the EF-hand IV loop structure
The glycine residue at position 6 of the binding sequence forms a hydrogen bond between its amide proton and the side chain of the first residue in the binding sequence [Fig. 1(A)].1 The hydrogen bond is thought to contribute to the formation of the chelating structure. However, the hydrogen bond of EF-hand IV is formed only in the Ca2+-bound state, and is absent in the apo state. Hydrogen bond formation can be monitored by the down-field shift of the amide proton signals in NMR spectra.24 The signal of G134 (position 6 in EF-hand IV) is detected in the normal shift region in the apo state and shows a large down-field shift upon Ca2+ binding.24 The large down-field shift indicates that Ca2+ binding enhances the hydrogen bonding.24
Figure 1.
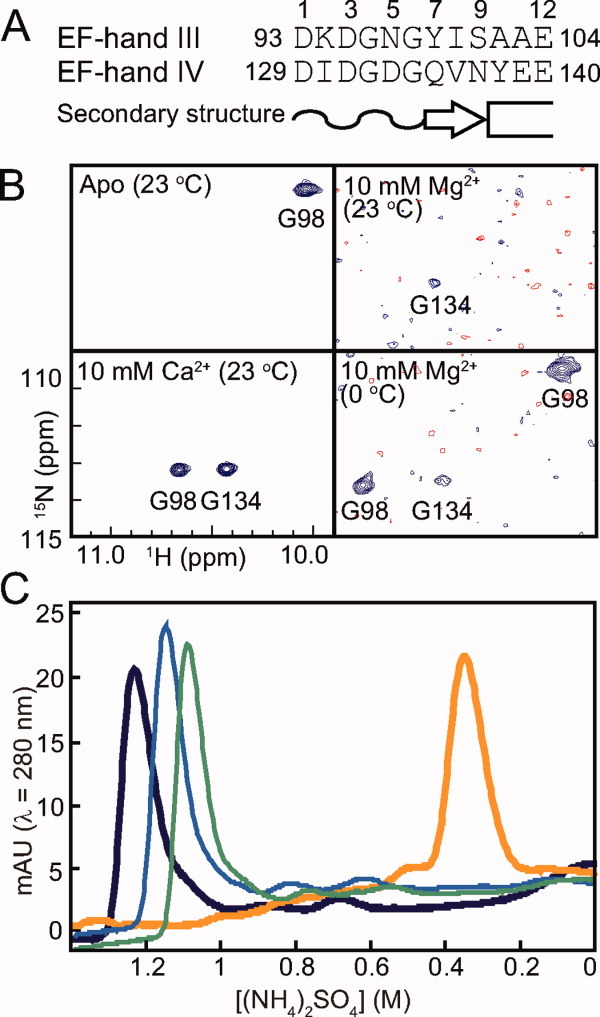
Mg2+-induced and Ca2+-induced changes of CaMC. (A) Primary structure of the EF-hand metal-binding sequences of CaMC (EF-hand III and EF-hand IV). The Ca2+ and Mg2+ coordinating ligands are indicated by the coordination numbers. The loop structure, the β-strand and the α-helix are indicated as a wavy line, an arrow and a box, respectively. (B) Selected regions of the 1H-15N HSQC spectra of CaMC without Ca2+ or Mg2+ at 23°C (upper left), with 10 mM Mg2+ at 23°C (upper right), with 10 mM Mg2+ at 0°C (lower right) and with 10 mM Ca2+ (lower left) at 23°C. (C) Hydrophobic interaction column chromatography. The samples were eluted by a concentration gradient of (NH4)2SO4 from 2M to 0M. 0 mM (no Mg2+ or Ca2+ added; green, thin trace), 10 mM Mg2+ (blue, thin trace), 100 mM Mg2+ (dark blue, thick trace), 5 mM Ca2+ (orange, thick trace).
To investigate the hydrogen-bond formation in EF-hand IV upon Mg2+ binding, the 1H-15N HSQC spectra of CaMC with and without 10 mM Mg2+ were recorded. Only the resonance of G98 was detected in the down-field region in the apo state [Fig. 1(B), upper left]. At 10 mM Mg2+, the G134 signal of the apo-form disappeared, and a weak down-field signal appeared at a different position from G98 in the apo state [Fig. 1(B), upper right]. The chemical shift of the signal was almost identical to that of G134 in the Ca2+-bound form [Fig. 1(B), lower left]. Therefore, the down-field signal was assigned to G134. This indicates that the hydrogen bond involving the amide proton of G134 is formed upon Mg2+ binding. It seems that EF-hand IV is mostly converted to the Mg2+-bound form, although the signal is very weak. (The signal broadening is described below.) In contrast, the signal from G98 disappeared [Fig. 1(B), upper right], suggesting that EF-hand III underwent a conformational exchange between the Mg2+-free and Mg2+-bound states. As previously reported by Ohki et al.,10 EF-hand IV was preferentially occupied by Mg2+, when compared with EF-hand III. In the spectra obtained at a lower temperature, to reduce the exchange rate, two down-field signals appeared in addition to the weak down-field signal of G134 [Fig. 1(B), lower right]. One was the apo-form of G98, and the other was the Mg2+-bound form. This confirmed the assignment of G134 and suggested that the Mg2+ is incorporated at least into the position 1 site bonded to position 6. The formation of the hydrogen bond is one of the stabilizing effects of Mg2+-binding.
The hydrophobicity of CaMC is not affected by Mg2+ binding
Ca2+ binding to CaM induces the exposure of the hydrophobic core on the protein surface, thus increasing the hydrophobicity of the molecule. The effects of Mg2+ and Ca2+ on the hydrophobicity of CaMC were evaluated by hydrophobic interaction chromatography. As shown in Figure 1(C), the apo form was eluted at [(NH4)2SO4] = 1.08M, and the Ca2+-bound form of CaMC displayed a peak at [(NH4)2SO4] = 0.35M in the presence of 5 mM Ca2+, indicating that Ca2+ binding increased the hydrophobicity of the protein. CaMC was eluted from the column at (NH4)2SO4 concentrations of 1.14M and 1.2M in the presence of 10 mM and 100 mM Mg2+, respectively [Fig. 1(C)]. Since the Mg2+ binding did not increase the hydrophobicity of the protein, the hydrophobic core is probably not exposed by the Mg2+ binding.
Conformational exchange among the Mg2+-bound forms of CaMC
The two-dimensional 1H-15N HSQC spectra of the apo and Ca2+-bound forms of CaMC displayed well-dispersed peaks with sharp line widths at 23°C (Fig. 2, upper panels). With 10 mM Mg2+, the spectrum showed broadened peaks and fewer signals (Fig. 2, lower left). In the presence of excess (100 mM) Mg2+, although some signals were detected, about 30 percent of the signals were missing in the spectrum (Fig. 2, lower right). These results reflect the exchange process between the apo and Mg2+-bound forms, and the additional conformational exchange among the Mg2+-bound structures. It should be noted that the Mg2+-induced broadening of the NMR signals does not represent the instability of the Mg2+-bound form of CaM. As indicated by the thermal unfolding experiment (Supporting Information Table S1), Mg2+ binding stabilizes the folding of CaM. The NMR signals of the Mg2+-bound form became sharper at higher temperatures [Fig. 3(A)]. Elevating the temperature increases the exchange rate and provides favorable conditions for observing the Mg2+-bound form by NMR. Although the NMR signals of the Mg2+-bound form became sharper at 50°C, the number of peaks in the HSQC spectrum was still slightly smaller than the expected number, and most of the signals were somewhat broadened.
Figure 2.
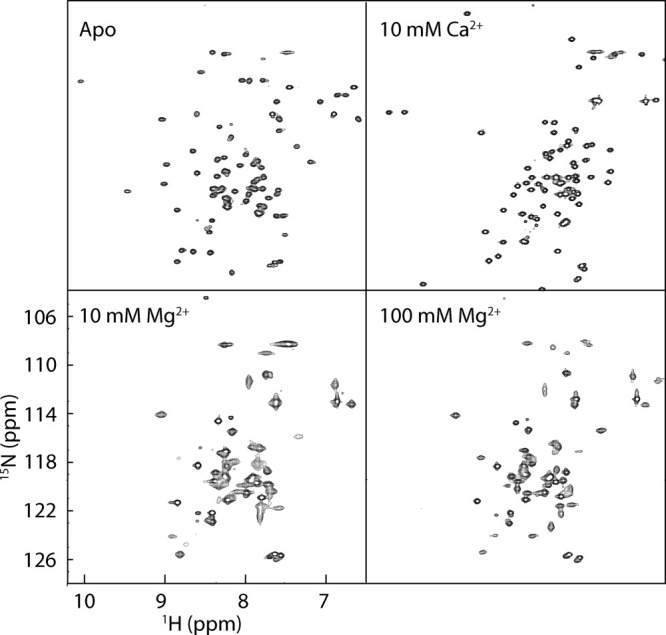
Mg2+-induced exchange broadening. The 1H-15N HSQC spectra of CaMC alone (upper left), with 10 mM Ca2+ (upper right), with 10 mM Mg2+ (lower left), and with 100 mM Mg2+ (lower right) at 23°C.
Figure 3.

1H-15N HSQC NMR spectra of Mg2+-bound forms of CaMC wild type and DD mutant. (A) HSQC spectra of CaMC wild type (left) and DD mutant (right) with 100 mM Mg2+ at 50°C. I130, V136, N137, and Y138 are labeled. (B) Secondary shifts of I130, V136, N137, and Y138 calculated with Cα (left) and CO (right) chemical shifts. Open bars correspond to the shift of the apo form of the wild type, blue bars represent the shift of the Mg2+-bound form of the wild type and green bars indicate the shift of the Mg2+-bound form of the DD mutant. Asterisks show residues with uncalculated secondary shifts, due to the lack of assignment.
Conformational exchange converged by point mutations
To improve the behavior of the Mg2+-bound state of CaMC, the E104D, E140D, and E104D/E140D (DD) mutants were constructed. The rationale for introducing these particular mutations was based on the Ca2+/Mg2+ binding properties of the EF-hands. E104 and E140 are located at position 12 in each of the EF-hand III and IV binding sequences, respectively [(Fig. 1(A)]. The glutamate is highly conserved at position 12 of the binding sequences. The substitution of aspartate for glutamate in other EF-hand proteins is known to diminish the binding selectivity of Ca2+ versus Mg2+.25–27
The 1H-15N HSQC spectra of all three mutants are similar to that of the wild type in the apo state at 23°C (data not shown), indicating that the proteins share a similar conformation. The stabilizing effect by Mg2+ was also conserved among the mutants (Supporting Information Table S1). Unexpectedly, the HSQC spectra of the apo forms of the E104D and DD mutants were well dispersed even at 50°C, whereas those of the wild type and the E140D mutant were crowded around the middle of the spectrum (Supporting Information Fig. S1). In the apo state, the S101 signals of the E104D and DD mutants were shifted down-field in the 1H-15N HSQC spectra (Supporting Information Fig. S2). The down-field shift can be interpreted as the strengthening of the hydrogen bonding between the backbone amide of S101 and the side chain of E104D. S101 is located just before the helix and is involved in capping of helix F.2 The N-terminus of helix F is fitted with the capping box motif.28 The side chain of S101 forms a hydrogen bond with the backbone of E104 and, reciprocally, the side chain of E104 forms a hydrogen bond with the backbone of S101. The capping structure may stabilize helix F, which is known as a fragile helix in the apo state.18
The HSQC spectra of the Mg2+-bound form of the DD mutant at 50°C exhibited the best-resolved peaks among the wild type and mutant proteins. The backbone resonances of the Mg2+-bound forms of the wild type and the DD mutant were assigned, and the chemical shifts were compared. The amide chemical shift differences of I130, V136, N137, and Y138 in the EF-hand IV binding sequence were relatively larger than those of the other residues [Fig. 3(A) and Supporting Information Table S2]. To assess whether the DD mutation changes the backbone structures of these residues, the secondary shifts were calculated from the resonances of 13Cα, 13CO, and compared between the Mg2+-bound forms of the wild type and the DD mutant [Fig. 3(B) and Supporting Information Table S2]. These resonances were consistent between the wild type and the DD mutant, indicating that the backbone structures of these residues were essentially retained. The differences in the amide chemical shifts between the wild type and the DD mutant may be explained by the effects of the side chains. In the apo form of the wild type,3 these resonances differed significantly from those of the Mg2+-bound form, suggesting that Mg2+ binding caused the changes in the backbone structures of these residues in the wild type, which were also retained in the DD mutant [Fig. 3(B)]. In other regions, the average chemical shift difference of the Mg2+-bound forms between the wild type and the DD mutant was 0.08, based on the equation described in Figure 4. This is a much smaller difference, when compared with 0.23 for the average chemical shift difference between the apo form and the Mg2+-bound form of the DD mutant. In the wild type, Mg2+-induced down-field shifts of G98 and G134 were observed [Fig. 1(B)]. These down-field shifts were also observed in the DD mutant (data not shown), suggesting that the Mg2+ ions were similarly coordinated in EF-hands III and IV of the DD mutant and the wild type. In the 13C NOESY-HSQC spectrum of the wild type, the NOEs were insufficient for the internal residues, including I100 and V136 on the β-sheet. Less broadening of the main chain and side chain signals of I100 and V136 and the surrounding residues was detected for the DD mutant. More NOE interactions were found, and the total number of NOEs was increased by 35%. A comparison of the 13C NOESY-HSQC spectra of the wild type and the DD mutant revealed that the NOE patterns of the wild type were conserved in the mutant, indicating that the conformation of the mutant predominantly resembled that of the wild type. The wild type may also adopt a minor conformation, and the exchange between them may cause the NMR signal broadening.
Figure 4.

Mg2+ binding to the CaMC DD mutant. (A) Titration with Mg2+. Superimposition of the 1H-15N HSQC spectra of the CaMC DD mutant with Mg2+ concentrations ranging from 0 to 100 mM. Each spectrum was acquired at 50°C. (B) Titration curve for Mg2+ binding to the CaMC DD mutant. Amide proton chemical shifts assigned to G96 (EF-hand III, circles) and G132 (EF-hand IV, squares), plotted as a function of the Mg2+ concentration. (C) Calculated populations of the different states of the protein, as a function of the Mg2+ concentration. The populations were calculated for KIII = 29.4 mM, KIV = 2.67 mM, and 1 mM protein. (D) Backbone amide chemical shift changes induced by Mg2+ binding. Black bars represent the chemical shift changes between 0 and 10 mM Mg2+, and magenta bars indicate the chemical shift changes between 10 and 100 mM Mg2+. The values were calculated with the following equation: 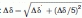 . The signals of E83, I85, N111, E114, V142, and Q143 were overlapped. The chemical shift changes of the D95, N97, G98, I100, V108, R126, A128, D131, D133, G134, and V136 residues were too broad, and are not included in the plot. The binding sequences of the EF-hands are indicated with a line. The helices are indicated by boxes with their names.
. The signals of E83, I85, N111, E114, V142, and Q143 were overlapped. The chemical shift changes of the D95, N97, G98, I100, V108, R126, A128, D131, D133, G134, and V136 residues were too broad, and are not included in the plot. The binding sequences of the EF-hands are indicated with a line. The helices are indicated by boxes with their names.
The substitution of glutamate with aspartate affects the Ca2+ binding potential
The binding potential of the DD mutant for Ca2+ was far less than that of the wild type. The Ca2+ titration, monitored by NMR, indicated that the DD mutant had a Ca2+-binding constant in the submillimolar range (data not shown). Considering that the wild type binds to Ca2+ with a Kd of ∼10−6 M, the substitution of glutamate with aspartate decreases the binding potential for Ca2+. The substitution affected the Ca2+-induced change in the hydrophobicity of the protein. The hydrophobic interaction chromatography revealed that the DD mutant with 5 mM Ca2+ eluted at a similar concentration of (NH4)2SO4 to that with either Mg2+ or EDTA (Supporting Information Fig. S3). This indicates that the hydrophobic cavity of the DD mutant is not exposed at Ca2+concentrations below 5 mM.
Mg2+ binding to EF-hand IV has a major impact on the conformational changes of CaMC
The Mg2+ titration was performed by recording the 1H-15N HSQC spectra as a function of increasing Mg2+ concentration, ranging 0 to 100 mM, in 100 mM KCl at 50°C for the DD mutant. The NMR signals of the amide protons of G96 (EF-hand III) and G132 (EF-hand IV) during the titration are shown in Figure 4(A). Two glycine residues exist at position 4 in each of the binding sequences and are good probes for Mg2+-binding to these sequences.
The amide chemical shifts of G96 and G132 were plotted as a function of the Mg2+ concentration [Fig. 4(B)]. The microscopic Mg2+-binding constants (KIII and KIV) for EF-hands III and IV were calculated using the G96 and G132 data, respectively. EF-hands III and IV bind to two Ca2+ ions with positive cooperativity. In contrast, the Mg2+-induced chemical shift changes agreed with the theoretical curve based on the model in which two Mg2+ ions bind to the protein independently. The binding manner of Mg2+ to the protein was not cooperative. The calculated microscopic Mg2+ dissociation constants were KIII = 29.4 mM and KIV = 2.67 mM. Roughly speaking, Mg2+ binds to the EF-hands IV and III sequentially, since the binding constants extensively differ. In the presence of 100 mM Mg2+, the population of the two Mg2+ form is significantly predominant [Fig. 4(C)].
To separate the effects of the binding of the first and second Mg2+ ions to the protein, the chemical shift differences between 0 and 10 mM Mg2+ and those between 10 mM Mg2+ and 100 mM Mg2+ were plotted in Figure 4(D). The changes up to 10 mM Mg2+ mainly reflect the effects of binding to EF-hand IV, and those beyond 10 mM Mg2+ mainly reflect the binding to EF-hand III. Upon Mg2+ binding to EF-hand IV, large chemical shift changes were observed throughout the EF-hand IV binding sequence. The chemical shifts in the EF-hand III binding sequence were predominantly changed upon Mg2+ binding to EF-hand III. In all other regions, larger chemical shift changes were observed between 0 and 10 mM Mg2+. Even the signals of the F-G linker changed upon Mg2+binding to EF-hand IV, suggesting that either helix F or helix G moved. These results indicated that the first Mg2+ binding event to EF-hand IV has a major impact on the structural change.
Solution structure of the Mg2+-bound form of CaMC
We determined the solution structure of the Mg2+-bound form of CaMC, using the DD mutant. Over 90% of the sequence-specific resonance assignments for the polypeptide backbone and the side chains of the Mg2+-bound form of the DD mutant were obtained, through analyses of the 2D and 3D heteronuclear NMR spectra recorded for the uniformly 13C, 15N-labeled protein. The 3D structure of the Mg2+-bound form of the DD mutant was calculated using 840 restraints derived from 1,159 NOEs from 3D NOESY spectra, supplemented with 84 dihedral angles (φ, ψ, and χ1 angles) and 23 hydrogen bonding restraints. Automatic NOE assignments and structure calculations were performed using CYANA 2.2.29–31 Over 98% of the distance restraints derived from the NOEs were assigned, and the R.M.S.D.s of the backbone atoms of the folded part of the calculated structures were ∼0.5 Å. The structural statistics are summarized in Supporting Information Table S3, and the ensemble of 20 structures of the Mg2+-bound DD mutant is shown in Figure 5(A).
Figure 5.
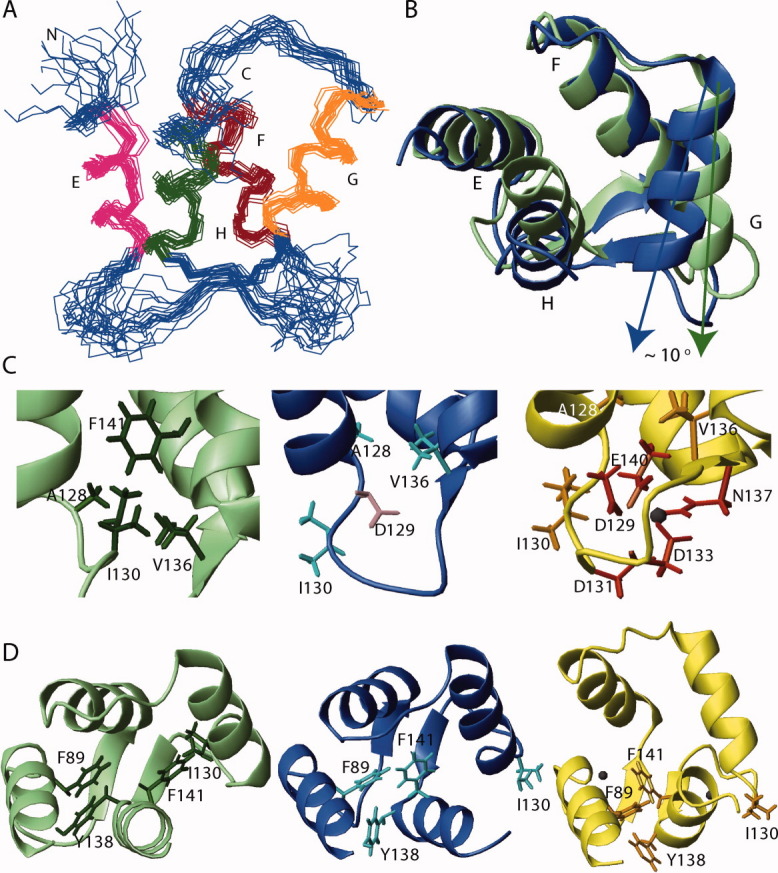
Solution structures of the Mg2+-bound CaMC DD mutant. (A) Ensemble of the backbone heavy atoms of the 20 lowest target functions of the Mg2+-bound form of the CaMC DD mutant. The α-helices are sequentially labeled with letters. Helices E and F form EF-hand III, while G and H form EF-hand IV. (B) Superimposition of the apo form of wild type CaMC (green, PDB ID: 1F71) and the Mg2+-bound form of the CaMC DD mutant (blue). Helices E and F of the apo form are superimposed against the corresponding helices of the Mg2+-bound form. (C) EF-hand IV binding sequence of the apo form of the wild type (left, green), the Mg2+-bound form (center, blue), and the Ca2+-bound form of the wild type (right, yellow, PDB ID: 1J7P) of CaMC. The side chains of A128, I130, V136, and F141 are shown. In the Ca2+-bound form, the side chains of the chelating residues D129, D131, D133, N137, and E140 are shown, and in the Mg2+-bound form, the side chain of the chelating residue D129 is also depicted. The Ca2+ ion is shown in black. (D) Ribbon representation of the apo form of the wild type (left, green), the Mg2+-bound form of the DD mutant (center, blue) and the Ca2+-bound form of the wild type (right, yellow) of CaMC. The side chains of F89, I130, Y138, and F141 are also shown.
The secondary structure elements of the Mg2+-bound form of the DD mutant are essentially identical to those of the apo form and the Ca2+-bound form of the wild type [Fig. 5(B)]. Helices E and F form EF-hand III, and helices G and H form EF-hand IV. The short antiparallel β-sheet connecting EF-hands III and IV is also conserved. The first six residues in the EF-hand III binding sequence and I130-G134 in EF-hand IV were not defined well, due to the lack of medium- and long-range NOEs.
Discussion
Mg2+-induced conformational changes in the EF-hand IV
The Mg2+-induced conformational changes occurred in wide area of EF-hand IV, while the closed conformation was retained. The global fold of the Mg2+-bound form was similar to that of the apo form [Fig. 5(B)]. In particular, the EF-hand III structures were almost identical. For EF-hand III, the backbone R.M.S.D. between the Mg2+-bound and apo forms was 0.85 ± 0.13 Å.
Mg2+ binding induced conformational changes in EF-hand IV. Initially, the N-terminal part of the EF-hand IV binding sequence, which directly interacts with Mg2+, was rearranged. In the apo form, the side chain of I130 is buried and interacts with the hydrophobic A128, V136 and F141 residues [Fig. 5(C), left]. In the Mg2+-bound form, D129 occupies this position and forms a hydrogen bond with G134. As a result, I130 was extruded [Fig. 5(C), center]. D129 is an important residue for the coordination to Mg2+ or Ca2+ on the inside of the molecule [Fig. 5(C), center and right]. As previously mentioned, the hydrogen bond formation by G134 was indicated by the amide signal position [Fig. 1(A)]. This was also observed in the Ca2+-induced changes. The rearrangement of the N-terminal part of the EF-hand IV binding sequence shifted the C-terminus of helix G toward the β-sheet. The backbone of A128 was converted into the regular α-helical conformation in the Mg2+-bound form [Fig. 5(C)]. In association with the movement of D129, A128 was also oriented toward the inside of the molecule, resulting in the extension of the α-helix. As compared to the apo form, the averaged φ and ψ angles of A128 changed from −89.3° to −63.9° and from 153.0° to −47.3°, respectively. These changes were also observed in the Ca2+-bound form. Accordingly, helix G was tilted toward the open orientation by approximately 10 degrees, and helix H was also tilted in a similar direction to helix G [Fig. 5(B)]. Consequently, the inter-helical angle between helices G and H in the Mg2+-bound form was close to that in the apo form (Supporting Information Table S4).
Conformational change in the hydrophobic core
The Mg2+-induced changes also occurred in another region, in addition to the postulated Mg2+ position. Around the C-terminal end of the EF-hand IV binding sequence, Y138 and F141 also changed their interaction partners. New aromatic interactions between F89, Y138, and F141 were created in the Mg2+-bound form [Fig. 5(D)]. This may be attributed to the movement of helices G and H. In the apo form, the aromatic ring of Y138 is oriented toward the C-terminus of helix E and the β-sheet.2–4 In the Mg2+-bound form, the ring is away from the β-sheet and faces F89 vertically [Fig. 5(D)]. The up-field ring current shift observed in the δ proton of Y138 can be attributed to the interaction with F89. The orientation of the ring of F141 changed from helix G to helix F, and was close to V108 on helix F and the aromatic ring of F89 on helix E [Fig. 5(D)]. These conformational changes are also observed in the presence of Ca2+. However, the extents of the structural changes are larger in the Ca2+-bound form, and those of the Mg2+-bound form are around the mid points, as seen in Figure 5(D).
In contrast to the effects of Ca2+ binding, the Mg2+-induced structural changes exerted only a minor influence on the inter-helical angles (Supporting Information Table S4). The hydrophobic core, which provides the binding surface for the target molecules, remained buried. The conformation of EF-hand III was minimally affected by Mg2+ binding. The Ca2+-induced movements of Y138 and F141 were more significant, as compared to those induced by Mg2+. Taken together, the structure of the Mg2+-bound form can be considered as an intermediate state of the structural changes caused by Ca2+ binding.
The incorporation of Mg2+ in the binding sequence leads to the formation of ionic interactions between Mg2+ and the negatively charged residues. These ionic interactions contribute to the thermal stability of the Mg2+-bound form of CaMC. The hydrogen bond is an additional stabilizing factor. The extension of the C-terminal end of helix G increases the hydrogen bond network, in addition to the hydrogen bond between G134 and D129.
Mg2+ coordination
The geometry of the Ca2+ coordination can be described as a pentagonal bipyramid, in which six of the seven Ca2+ ligands are provided from the residues at positions 1, 3, 5, 7, and 12 (a bidentate ligand). The remaining Ca2+ ligand is the residue at position 9, via a water molecule. The Ca2+-induced rearrangements of positions 1, 2, and 6 of the EF-hand IV binding sequence were also found in the Mg2+-bound form, suggesting that the accommodation of Mg2+ in this region of the EF-hand IV binding sequence is similar to that of Ca2+, though NMR signal broadening shows dynamic behavior of positions 3 and 5.
The binding of Ca2+ or Mg2+ induced the shift of the nitrogen resonance of E139 (Supporting Information Table S5), which is one of the largest chemical shift changes. One explanation for the nitrogen resonance shift is the rearrangement of the main-chain structure, but the φ and ψ backbone dihedral angles of E139 are minimally affected by Ca2+ or Mg2+ binding, since E139 is within a helical structure. Another possible reason is hydrogen bond formation. The disruption of the hydrogen bond affects the nitrogen resonance and induces an up-field shift. The crystal structure of the Ca2+-bound form of CaM revealed that no atom is involved in a hydrogen bond with the main-chain amide of E139.1 In the ensemble of NMR structures of the apo form, the side chain of N137 is located in the vicinity of the E139 main-chain amide, and could potentially form a hydrogen bond.2–4 Although the crystal structure of the apo form of CaM has not been solved, the corresponding hydrogen bond exists in the crystal structure of the Ca2+-free form of the EF-hand protein, aequorin.32 Accordingly, a hydrogen bond may exist between the main-chain amide of E139 and the side-chain oxygen of N137 in the apo form of CaM. The hydrogen bond disappears upon Ca2+ or Mg2+ binding. N137, at position 9 in the binding sequence, uses its side chain to coordinate to Ca2+ via a water molecule. As in Ca2+ binding, Mg2+ binding may involve N137. The disruption of the hydrogen bond to helix H may be related to the neighboring conformational changes, as described above.
Additional features in the wild type Mg2+-bound form
In the structural determination of the Mg2+-bound form of the DD mutant, the structural changes were observed only in EF-hand IV, while in the wild type, the Mg2+-induced broadening ranged from the β-sheet to the core, including EF-hand III. The resonances of the hydrophobic residues at position 8 (I100 and V136) in the binding sequences of EF-hands III and IV were severely broadened in the wild type. The residue at position 8 is located at the center of the β-strand, and links the paired EF-hands structurally. The exchange broadening was also observed in the residues of the hydrophobic core, in the vicinity of the β-sheet region, and the residues between helices E and F (Fig. 6). One explanation for the broadening of the Mg2+-bound form of the wild type is the multiple conformations in the β-sheet region and the surrounding region, including the hydrophobic core. Fewer conformational exchanges in the β-sheet region occurred in the DD mutant. Replacing glutamate with aspartate at position 12 places the carboxylate group farther away from the bound metal, decreasing the population coordinated with the residue at position 12. The DD mutation may have excluded such low-population structures. Mg2+ binding only to EF-hand IV can lead to the simultaneous movement of both EF-hands III and IV, since a low Mg2+ concentration produces overall broadening. This implies that a critical mechanism underlying the cooperativity of the two EF-hands is reflected in the Mg2+-induced broadening.
Figure 6.
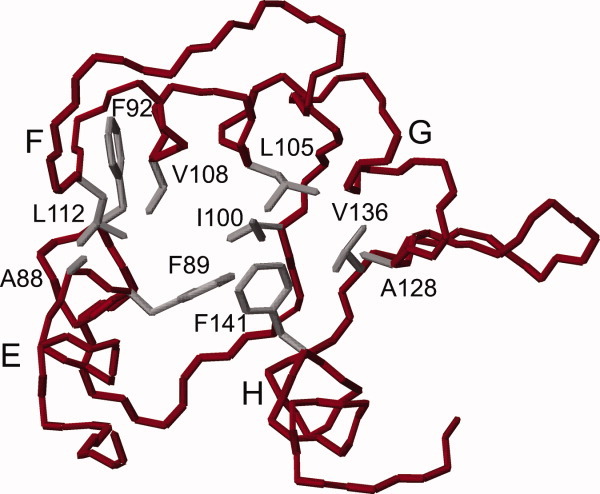
Backbone trace of the Mg2+-bound form of the CaMC DD mutant. The hydrophobic residues with side-chains that undergo exchange broadening in the presence of Mg2+ in the wild type are colored grey.
In summary, the Mg2+-induced conformational change mainly occurs on the helix G and helix H sides of the binding sequence of EF-hand IV, and involves the rearrangement of the hydrophobic interactions. The features of the Mg2+-induced conformational changes of CaMC are included in the Ca2+-induced conformational changes. The structure of the Mg2+-bound form can be regarded as an intermediate state of the structural changes caused by Ca2+ binding. Therefore, the Ca2+-induced conformational changes of CaMC can be divided into two categories, those specific to Ca2+ and those common to Ca2+ and Mg2+.
Materials and Methods
Protein expression and purification
For the recombinant expression and purification of CaMC (residues 78-148), the cDNA encoding this region of Xenopus CaM was cloned into the pET11a vector (Novagen). The N-terminal methionine was removed naturally. All of the CaM mutants were generated by site-directed mutagenesis, using a QuikChange kit (Stratagene). All unlabeled proteins were expressed in Escherichia coli Rosetta2(DE3)pLysS, grown in LB medium at 37 °C. The uniformly 15N or 15N, 13C-labeled proteins were expressed in M9 minimal medium, supplemented with either 15NH4Cl or 15NH4Cl and [13C6]glucose as the sole nitrogen and carbon sources, respectively. The cells were disrupted by sonication, and after clarification by centrifugation, the supernatant was applied to a HiTrap Q FF or HP column (GE Healthcare), in 20 mM Tris-HCl (pH 7.5) containing 5 mM CaCl2, which was eluted with a NaCl gradient. Fractions containing the protein were applied to a HiTrap Phenyl FF or HP column (GE Healthcare) equilibrated with 20 mM Tris-HCl (pH 7.5), containing 5 mM CaCl2 and 2.0M (NH4)2SO4. The column was washed with a buffer containing 20 mM Tris-HCl (pH 7.5), 5 mM EDTA and 2.0M (NH4)2SO4. The protein was eluted with a (NH4)2SO4 gradient. For dialysis and further sample preparation, Chelex 100-treated 10 mM MES (pH 6.5) buffer containing 100 mM KCl was used. The fraction containing the protein was dialyzed against 10 mM MES (pH 6.5) buffer, containing 100 mM KCl. The protein concentration was determined using a molar absorption coefficient of ɛ = 2980 M−1 cm−1.
Hydrophobic interaction chromatography
Hydrophobic interaction chromatography was performed using a 1 mL RESOURCE PHE column (GE Healthcare). The column was equilibrated with an appropriate equilibration buffer, containing 2M (NH4)2SO4 in 20 mM Tris-HCl (pH 7.8). For the apo and Ca2+-loading cases, 5 mM EDTA and 5 mM CaCl2 were included in the equilibration buffer, respectively. For the Mg2+-loading, either 10 mM or 100 mM MgCl2 was included in the equilibration buffer. In all experiments, 0.16 mg of the protein was loaded on the column. All experiments were performed at 10°C. Elution was done by decreasing the ammonium sulfate concentration to 0M in 20 mM Tris-HCl, pH 7.8. The protein eluted under each condition was spectrophotometrically monitored.
NMR measurements and spectral assignments
NMR spectra were measured at 50°C on a Bruker AVANCE600 spectrometer, equipped with pulsed field gradients and triple resonance probes. The samples used in the magnesium titrations initially contained 1 mM of 15N-labeled protein, in a buffer consisting of 10 mM MES and 100 mM KCl in 90% H2O/10% D2O, pH 6.5. Small aliquots of a 100 mM (1 mM to 10 mM) or 1M (10 mM to 100 mM) MgCl2 solution were added to the protein solution, and two-dimensional 1H-15N HSQC spectra were recorded.
Resonance assignments were accomplished by analyses of multidimensional heteronuclear NMR spectra, 2D 1H-15N HSQC, 2D 1H-13C HSQC, 3D HNCO, HN(CA)CO, C(CO)NH, HNCACB, CBCA(CO)NH, HBHA(CO)NH, H(CCO)NH, HCCH-COSY, HCCH-TOCSY and CCH-TOCSY, acquired on a 1 mM protein sample (uniformly labeled with 13C and 15N), in buffer containing 10 mM MES-d13 (pH 6.5), 100 mM KCl and 100 mM MgCl2 in 90% H2O/10% D2O. All spectra were processed using NMRPipe33 and analyzed using NMRview34 and the integrated KUJIRA module.35
The 13Cα, 13CO secondary shifts were calculated by the table of random coil carbon chemical shifts provided by Wishart and Sykes.36
Structure calculations
NOE restraints were obtained from 3D 15N-edited NOESY and 13C-edited NOESY spectra with mixing times of 160 ms. Backbone φ and ψ torsion angle restraints were derived from an analysis of the Hα, 13Cα, 13Cβ, 13CO and backbone 15N chemical shifts using TALOS,37 including 23 hydrogen bond constraints. The χ1 angle restraints were derived from the HNHB, HN(CO)HB and NOESY spectra. Automated NOE cross-peak assignments and structure calculations with torsion angle dynamics were performed using CYANA 2.2.29–31 A total of 100 structures were independently calculated. The 20 conformers with the lowest final CYANA target functions were used. Figures were generated with MOLMOL. The coordinates for the Mg2+-bound CaMC E104D/E140D mutant have been deposited in the Protein Data Bank (PDB ID: 2EQC).
Acknowledgments
We thank Prof. M. Kainosho and Prof. M. Ikura for providing the Xenopus calmodulin cDNA.
Glossary
Abbrevations
- COSY
correlation spectroscopy
- EDTA
ethylenediaminetetraacetic acid
- HSQC
heteronuclear single quantum correlation
- NMR
nuclear magnetic resonance
- NOE
nuclear Overhauser effect
- NOESY
nuclear Overhauser effect spectroscopy
- RDC
residual dipolar coupling
- R.M.S.D.
root mean square deviation
- TOCSY
total correlation spectroscopy
Supplementary material
References
- 1.Babu YS, Sack JS, Greenhough TJ, Bugg CE, Means AR, Cook WJ. Three-dimensional structure of calmodulin. Nature. 1985;315:37–40. doi: 10.1038/315037a0. [DOI] [PubMed] [Google Scholar]
- 2.Kuboniwa H, Tjandra N, Grzesiek S, Ren H, Klee CB, Bax A. Solution structure of calcium-free calmodulin. Nat Struct Biol. 1995;2:768–776. doi: 10.1038/nsb0995-768. [DOI] [PubMed] [Google Scholar]
- 3.Zhang M, Tanaka T, Ikura M. Calcium-induced conformational transition revealed by the solution structure of apo calmodulin. Nat Struct Biol. 1995;2:759–767. doi: 10.1038/nsb0995-758. [DOI] [PubMed] [Google Scholar]
- 4.Finn BE, Evenäs J, Drakenberg T, Waltho JP, Thulin E, Forsén S. Calcium-induced structural changes and domain autonomy in calmodulin. Nat Struct Biol. 1995;2:777–787. doi: 10.1038/nsb0995-777. [DOI] [PubMed] [Google Scholar]
- 5.Krestinger RH, Nockolds CE. Carp muscle calcium binding protestructure determination and general description. J Biol Chem. 1973;248:3313–3326. [PubMed] [Google Scholar]
- 6.Ikura M, Clore GM, Gronenborn AM, Zhu G, Klee CB, Bax A. Solution structure of a calmodulin-target peptide complex by multidimensional NMR. Science. 1992;256:632–638. doi: 10.1126/science.1585175. [DOI] [PubMed] [Google Scholar]
- 7.Meador WE, Means AR, Quiocho FA. Target enzyme recognition by calmodul2.4 A structure of a calmodulin-peptide complex. Science. 1992;257:1251–1255. doi: 10.1126/science.1519061. [DOI] [PubMed] [Google Scholar]
- 8.Meador WE, Means AR, Quiocho FA. Modulation of calmodulin plasticity in molecular recognition on the basis of x-ray structures. Science. 1993;262:1718–1721. doi: 10.1126/science.8259515. [DOI] [PubMed] [Google Scholar]
- 9.Chin D, Means AR. Calmodula prototypical calcium sensor. Trends Cell Biol. 2000;10:322–328. doi: 10.1016/s0962-8924(00)01800-6. [DOI] [PubMed] [Google Scholar]
- 10.Ohki S, Ikura M, Zhang M. Identification of Mg2+ binding sites and the role of Mg2+ on target recognition by calmodulin. Biochemistry. 1997;36:4309–4316. doi: 10.1021/bi962759m. [DOI] [PubMed] [Google Scholar]
- 11.Tsai MD, Drakenberg T, Thulin E, Forsén S. Is the binding of magnesium (II) to calmodulin significant? An investigation by magnesium-25 nuclear magnetic resonance. Biochemistry. 1987;26:3635–3643. doi: 10.1021/bi00386a057. [DOI] [PubMed] [Google Scholar]
- 12.Seamon KB. Calcium- and magnesium-dependent conformational states of calmodulin as determined by nuclear magnetic resonance. Biochemistry. 1980;19:207–215. doi: 10.1021/bi00542a031. [DOI] [PubMed] [Google Scholar]
- 13.Drabikowski W, Brzeska H, Venyaminov S. Tryptic fragments of calmodulin. Ca2+- and Mg2+-induced conformational changes. J Biol Chem. 1982;257:11584–11590. [PubMed] [Google Scholar]
- 14.Malmendal A, Evenäs J, Thulin E, Gippert GP, Drakenberg T, Forsén S. When size is important. Accommodation of magnesium in a calcium binding regulatory domain. J Biol Chem. 1998;273:28994–29001. doi: 10.1074/jbc.273.44.28994. [DOI] [PubMed] [Google Scholar]
- 15.Ebel H, Gunther T. Magnesium metabolism: a review. J Clin Chem Clin Biochem. 1980;18:257–270. doi: 10.1515/cclm.1980.18.5.257. [DOI] [PubMed] [Google Scholar]
- 16.Huang H, Ishida H, Vogel HJ. The solution structure of the Mg2+ form of soybean calmodulin isoform 4 reveals unique features of plant calmodulins in resting cells. Protein Sci. 2010;19:475–485. doi: 10.1002/pro.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Masino L, Martin SR, Bayley PM. Ligand binding and thermodynamic stability of a multidomain protein, calmodulin. Protein Sci. 2000;9:1519–1529. doi: 10.1110/ps.9.8.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brzeska H, Venyaminov S, Grabarek Z, Drabikowski W. Comparative studies on thermostability of calmodulin, skeletal muscle troponin C and their tryptic fragments. FEBS Lett. 1983;153:169–173. doi: 10.1016/0014-5793(83)80141-0. [DOI] [PubMed] [Google Scholar]
- 19.Lee SH, Kim JC, Lee MS, Heo WD, Seo HY, Yoon HW, Hong JC, Lee SY, Bahk JD, Hwang I, Cho MJ. Identification of a novel divergent calmodulin isoform from soybean which has differential ability to activate calmodulin-dependent enzyme. J Biol Chem. 1995;270:21806–21812. doi: 10.1074/jbc.270.37.21806. [DOI] [PubMed] [Google Scholar]
- 20.Minowa O, Yagi K. Calcium binding to tryptic fragments of calmodulin. J Biochem. 1984;96:1175–1182. doi: 10.1093/oxfordjournals.jbchem.a134935. [DOI] [PubMed] [Google Scholar]
- 21.Martin SR, Masion L, Bayley PM. Enhancement by Mg2+ of domain specificity in Ca2+-dependent interactions of calmodulin with target sequences. Protein Sci. 2000;9:2477–2488. doi: 10.1110/ps.9.12.2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Finley NL, Howarth JW, Rosevear PR. Structure of the Mg2+-loaded C-lobe of cardiac troponin C bound to the N-domain of cardiac troponin I: comparison with the Ca2+-loaded structure. Biochemistry. 2004;43:11371–11379. doi: 10.1021/bi049672i. [DOI] [PubMed] [Google Scholar]
- 23.Babini E, Bertini I, Capozzi F, Chirivino E, Luchinat C. A structural and dynamic characterization of the EF-Hand Protein CLSP. Structure. 2006;14:1029–1038. doi: 10.1016/j.str.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 24.Ikura M, Minowa O, Yazawa M, Yagi K, Hikichi K. Sequence-specific assignments of downfield-shifted amide proton resonances of calmodulin. Use of two-dimensional NMR analysis of its tryptic fragments. FEBS Lett. 1987;219:17–21. [Google Scholar]
- 25.da Silva AC, Kendrick-Jones J, Reinach FC. Determinants of ion specificity on EF-hands sites. Conversion of the Ca2+/Mg2+ site of smooth muscle myosin regulatory light chain into a Ca(2+)-specific site. J Biol Chem. 1995;270:6773–6778. doi: 10.1074/jbc.270.12.6773. [DOI] [PubMed] [Google Scholar]
- 26.Blumenschein TM, Reinach FC. Analysis of affinity and specificity in an EF-hand site using double mutant cycles. Biochemistry. 2000;39:3603–3610. doi: 10.1021/bi9924718. [DOI] [PubMed] [Google Scholar]
- 27.Cates MS, Berry MB, Ho EL, Li Q, Potter JD, Phillips GN., Jr. Metal-ion affinity and specificity in EF-hand proteins: coordination geometry and domain plasticity in parvalbumin. Structure. 1999;7:1269–1278. doi: 10.1016/s0969-2126(00)80060-x. [DOI] [PubMed] [Google Scholar]
- 28.Harper ET, Rose GD. Helix stop signals in proteins and peptides: the capping box. Biochemistry. 1993;32:7605–7609. doi: 10.1021/bi00081a001. [DOI] [PubMed] [Google Scholar]
- 29.Güntert P. Automated NMR structure calculation with CYANA. Methods Mol Biol. 2004;278:353–378. doi: 10.1385/1-59259-809-9:353. [DOI] [PubMed] [Google Scholar]
- 30.Güntert P, Mumenthaler C, Wüthrich K. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J Mol Biol. 1997;273:283–298. doi: 10.1006/jmbi.1997.1284. [DOI] [PubMed] [Google Scholar]
- 31.Hermann T, Güntert P, Wüthrich K. Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J Mol Biol. 2002;319:209–227. doi: 10.1016/s0022-2836(02)00241-3. [DOI] [PubMed] [Google Scholar]
- 32.Head JF, Inouye S, Teranishi K, Shimomura O. The crystal structure of the photoprotein aequorin at 2.3 A resolution. Nature. 2000;405:372–376. doi: 10.1038/35012659. [DOI] [PubMed] [Google Scholar]
- 33.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 34.Johnson BA, Blevins RA. NMRView: a computer program for the visualization and analysis of NMR data. J Biomol NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 35.Kobayashi N, Iwahara J, Koshiba S, Tomizawa T, Tochio N, Güntert P, Kigawa T, Yokoyama S. KUJIRA, a package of integrated modules for systematic and interactive analysis of NMR data directed to high-throughput NMR structure studies. J Biomol NMR. 2007;39:31–52. doi: 10.1007/s10858-007-9175-5. [DOI] [PubMed] [Google Scholar]
- 36.Wishart DS, Sykes BD. The 13C chemical-shift index: a simple method for the identification of protein secondary structure using 13C chemical-shift data. J Biomol NMR. 1994;4:171–180. doi: 10.1007/BF00175245. [DOI] [PubMed] [Google Scholar]
- 37.Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.