

Abstract
The TetR family of transcription regulators are diverse proteins capable of sensing and responding to various structurally dissimilar antimicrobial agents. Upon detecting these agents, the regulators allow transcription of an appropriate array of resistance markers to counteract the deleterious compounds. Campylobacter jejuni CmeR is a pleiotropic regulator of multiple proteins, including the membrane-bound multidrug efflux transporter CmeABC. CmeR represses the expression of CmeABC and is induced by bile acids, which are substrates of the CmeABC tripartite pump. The multiligand-binding pocket of CmeR has been shown to be very extensive and consists of several positively charged and multiple aromatic amino acids. Here we describe the crystal structures of CmeR in complexes with the bile acids, taurocholate and cholate. Taurocholate and cholate are structurally related, differing by only the anionic charged group. However, these two ligands bind distinctly in the binding tunnel. Taurocholate spans the novel bile acid binding site adjacent to and without overlapping with the previously determined glycerol-binding site. The anionic aminoethanesulfonate group of taurocholate is neutralized by a charge-dipole interaction. Unlike taurocholate, cholate binds in an anti-parallel orientation but occupies the same bile acid-binding site. Its anionic pentanoate moiety makes a water-mediated hydrogen bond with a cationic residue to neutralize the formal negative charge. These structures underscore the promiscuity of the multifaceted binding pocket of CmeR. The capacity of CmeR to recognize bile acids was confirmed using isothermal titration calorimetry and fluorescence polarization. The results revealed that the regulator binds these acids with dissociation constants in the micromolar region.
Keywords: TetR family, Campylobacter jejuni, transcriptional regulator, multidrug resistance
Introduction
The ability of bacteria to adapt and respond to diverse classes of toxic compounds allows these organisms to survive in a variety of harsh environments. Campylobacter jejuni, the leading bacterial cause of food-borne enteritis in humans, is able to flourish in the intestinal mucosa due to its rapid response to bile acid intrusion.1,2 This Gram-negative enteric pathogen has become increasingly resistant to common antibacterial agents encountered during the course of an infection. The intrinsic and acquired resistance to these diverse classes of toxic chemicals is facilitated through the expression of multidrug resistant (MDR) efflux transporters. The MDR pumps are capable of effectively lowering the intracellular concentration, thus compromising the effectiveness of the antibacterial compounds. Based on the genomic sequence of C. jejuni NCTC 11168, this organism harbors 13 putative MDR transporters that belong to five different classes, including the ATP-binding cassette (ABC) superfamily, the resistance-nodulation-division (RND) family, the multidrug and toxic compound extrusion (MATE) family, the major facilitator (MF) superfamily, and the small multidrug resistance (SMR) family.3,4 Currently, only two RND efflux transporters, CmeABC and CmeDEF, in C. jejuni have been functionally characterized.5–8
Among these five different families of transporters, members of the RND superfamily exhibit the broadest range of substrate specificity and are usually the primary contributor to the intrinsic multidrug resistance associated with Gram-negative organisms.9,10 CmeB, a prototypical RND family transporter, is the major efflux transporter in C. jejuni. This inner membrane efflux pump functions as a tripartite protein complex along with a periplasmic membrane fusion protein, CmeA, and an outer membrane channel, CmeC, to extrude deleterious compounds from the bacterial cell.6 The CmeABC complex recognizes and protects C. jejuni from a diverse set of antibacterial compounds, including commonly used antibiotics, metal ions, and lipophilic compounds.2,6–8,11 In addition, CmeABC plays a major role in conferring resistance to bile acids, which are ubiquitously present in the intestinal tract.2,12 It has been reported that mutant strains of C. jejuni lacking a functional CmeABC are unable to colonize in the intestinal tract of chickens.2 The essential role of CmeABC for the growth of C. jejuni in the intestinal mucosa highlights the importance of this efflux complex to the pathogenicity of the bacterium.
The expression of CmeABC is controlled by the transcriptional regulator CmeR, whose open reading frame is located immediately upstream of the cmeABC operon and is transcribed divergently.13 Transcription of the cmeR gene gives rise to a 210 amino acid protein, which shares N-terminal sequence and structural similarities to members of the TetR family.14,15 CmeR is a two-domain protein with an N-terminal DNA-binding motif and a C-terminal multiligand-binding domain. Experimental evidence suggests that the 16 base pair palindromic inverted repeat (IR) sequence, 5′TGTAATAAATATTACA3′, located between cmeR and cmeABC is the operator site for CmeR binding and transcriptional repression.13 Bile acids induce the expression of cmeABC by inhibiting the binding of CmeR to this operator.12
The crystal structure of CmeR revealed that CmeR exhibits a unique secondary structural feature among all known structures of the TetR family of regulators.16 To date, CmeR is the only regulator in the TetR family that lacks the N-terminal helix-turn-helix DNA-binding motif, in which the recognition helix α3 is replaced by a random coil.16,17 Along with this unique characteristic, a large center-to-center distance (54 Å as measured by the separation between Cα atoms of Y51 and Y51′ from the other subunit) was observed between the two N-termini of the dimer, making it incompatible with the distance between two consecutive major grooves of B-DNA. Each monomer of CmeR consists of nine helices, and numbered with helix α3 being skipped to facilitate comparisons to other members of the TetR family. As a result, the N-terminal domain of CmeR comprises helices α1 and α2 along with this random coil (Fig. 1). The larger C-terminal domain is composed of helices α4-α10, forming a very large hydrophobic tunnel for substrate binding. This tunnel is about 20 Å long with a volume of approximately 1000 Å3, which is distinctly larger than the binding pockets of many other members of the TetR family. Surprisingly, a fortuitous glycerol molecule was found to bind in the binding tunnel of each monomer.16 Residues F99, F103, F137, S138, Y139, V163, C166, T167, and K170 are responsible for forming this glycerol-binding site. The structure also suggests that CmeR binds glycerol in a manner of 1:1 monomer-to-ligand molar ratio. The volume of the ligand-binding tunnel of CmeR is large enough to accommodate a few of the ligand molecules. Additional water molecules fill the portion of the large tunnel that is unoccupied by ligand. Thus, CmeR might be able to bind more than one drug molecule at a time, or possibly accommodate a significantly larger ligand that spans across the entire binding tunnel. This tunnel, possibly consisting of multiple binding sites for different ligands, is rich in aromatic residues and contains four positively charged amino acids (three histidines and one lysine). Based on the structural information and the fact that bile acids induce transcription of cmeABC, we hypothesize that CmeR may utilize these positively charged residues to recognize negatively charged ligands, like bile acids. To elucidate how CmeR recognizes these large anionic ligands, we here report the crystal structures of CmeR in complexes with taurocholate (Tch) and cholate (Chd), respectively.
Figure 1.
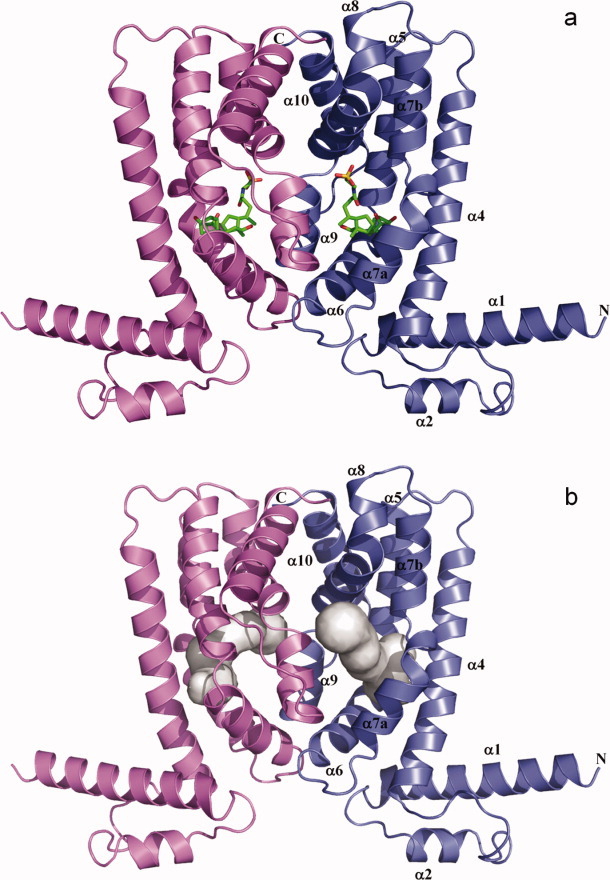
Structure of a CmeR-ligand complex. (a) Ribbon diagram of the taurocholate-bound CmeR homodimer generated by crystallographic symmetry. The taurocholate is shown as a stick model (green, carbon; blue, nitrogen; red, oxygen). (b) The hydrophobic binding tunnel of CmeR. This tunnel was calculated using the program CAVER (http://www.caver.cz), utilizing the position of residue A108 as a starting point. The binding tunnel on each subunit of CmeR is colored gray.
Results
Crystals of the bile acid bound complexes belonged to the space group P21212, with the asymmetric unit being occupied by one CmeR molecule. The symmetry operators were used to identify the dimeric state of CmeR (Fig. 1). These bile acids were found to bind within the ligand-binding tunnel and interact with the regulator using a surprisingly novel binding site. However, the overall conformation of the bile acid bound CmeR structures are very similar to that of the CmeR-glycerol structure.
Structure of the CmeR-Taurocholate complex
The crystal structure of the CmeR-Tch complex (Fig. 1) was refined to 2.20 Å resolution with a final Rwork of 22.2% and Rfree of 28.4% (Table I), revealing that Tch binds within the ligand-binding tunnel in a position adjacent to the previously identified glycerol-binding site. The Tch binding site utilizes a distinct set of amino acids to accommodate the elongated structure of the bile acid, while leaving the glycerol-binding site unoccupied. The simulated annealing electron density omit map of this bound Tch is illustrated in Figure 2.
Table I.
Crystallographic Data and Refinement Statistics
| CmeR-Tch | CmeR-Chd | |
|---|---|---|
| A. Data collection | ||
| Wavelength (Å) | 0.9792 | 0.9792 |
| Space group | P21212 | P21212 |
| Cell constants (Å) | a = 94.0, b = 37.8, c = 57.6 | a = 94.0, b = 37.4, c = 57.8 |
| Resolution (Å) | 2.20 (2.28-2.20) | 2.35 (2.43-2.35) |
| Completeness (%) | 99.8 (99.2) | 98.4 (99.2) |
| Total no. of reflections | 227,441 | 286,493 |
| No. of Unique reflections | 11,011 | 8,984 |
| Redundancy | 4.7 | 3.2 |
| Rmerge (%)a | 4.5 (22.5) | 6.3 (42.1) |
| 〈I/σ(I)〉 | 36.4 (5.0) | 17.9 (2.1) |
| B. Refinement | ||
| Rwork (%) | 22.2 | 20.0 |
| Rfree (%) | 28.4 | 26.3 |
| 〈B〉 | ||
| Overall (Å2) | 45.6 | 43.6 |
| Protein (Å2) | 45.0 | 43.1 |
| Ligand (Å2) | 73.1 | 57.8 |
| Water (Å2) | 43.9 | 40.9 |
| No. of ligands | 1 | 1 |
| No. of waters | 54 | 60 |
| Rms deviations | ||
| Bond angles (°) | 1.0 | 1.3 |
| Bond length (Å) | 0.006 | 0.009 |
| Ramachandran analysis | ||
| Most favored regions (%) | 93.5 | 93.0 |
| Allowed regions (%) | 6.5 | 7.0 |
| Generously allowed regions (%) | 0.0 | 0.0 |
| Disallowed regions (%) | 0.0 | 0.0 |
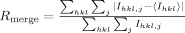 , where Ihkl,j is the jth intensity measurement of reflection hkl and 〈Ihkl〉 is the average intensity from multiple observation.
, where Ihkl,j is the jth intensity measurement of reflection hkl and 〈Ihkl〉 is the average intensity from multiple observation.
Figure 2.
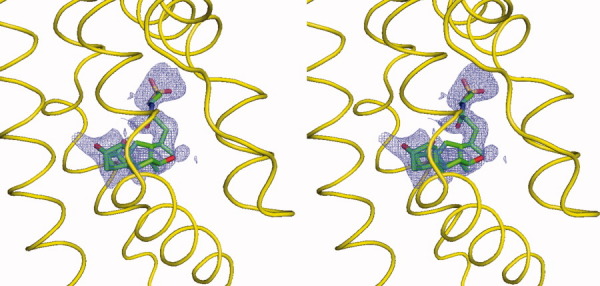
Stereo view of the simulated annealing electron density map of the bound taurocholate. The bound taurocholate in the left subunit of dimeric CmeR (the orientation corresponds to Fig. 1) is shown as a stick model (green, carbon; blue, nitrogen; red, oxygen). The 2Fo-Fc simulated annealing omit map is contoured at 1.2 σ (blue mesh). The electron density for the surrounding protein has been deleted. The surrounding secondary structural elements are shown as yellow ribbons.
The four-ring system of the bound Tch is completely buried in the CmeR binding tunnel, leaving its negatively charged 2-aminoethanesulfonate group in the 5β position oriented at the entry point and exposed to the solvent. This four-ring skeleton, mimicking the steroid backbone, consists of three hydroxyl groups located at the 3α, 7α, and 12α positions. The CmeR-Tch structure demonstrates that the 3α-hydroxyl group in the A ring makes a hydrogen bond with the positively charged residue H72 (Fig. 3). The C ring and the 12α-hydroxyl group of Tch; however, face directly toward helix α8 and subunit interface of the dimer. This orientation facilitates the interaction between the 12α-hydroxyl oxygen and H175′ of the next subunit of CmeR, allowing them to form a second hydrogen bond to anchor the bound Tch. Interestingly, the repressor protein further anchors this bound bile acid molecule through a water-mediated hydrogen bond between K170 of helix α8 and the 7α-hydroxyl group of the B ring to secure the binding.
Figure 3.
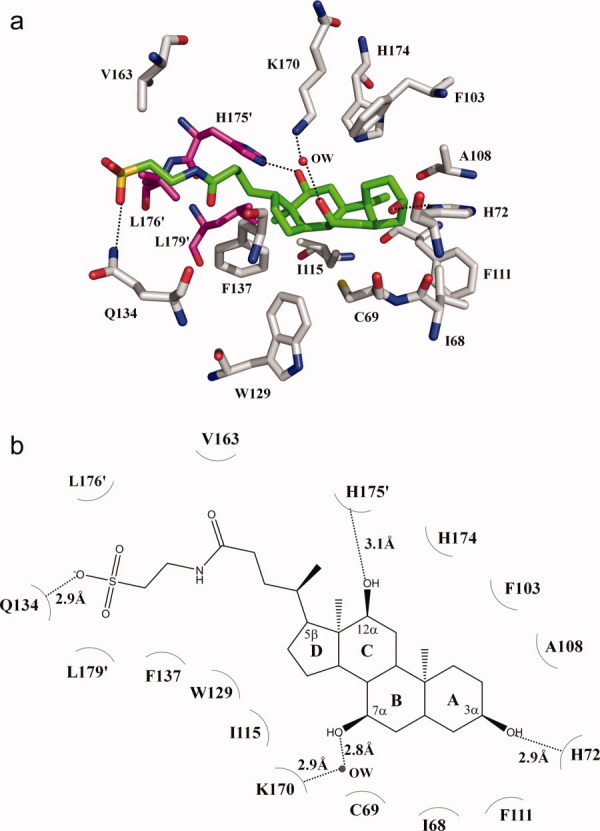
The taurocholate binding site. (a) Amino acid residues within 4.2 Å from the bound taurocholate (green, carbon; blue, nitrogen; red, oxygen; orange, sulfur). The side chains of selected residues are shown as gray sticks (gray, carbon; blue, nitrogen; red, oxygen). Residues from the next subunit of CmeR are shown as magenta sticks (magenta, carbon; blue, nitrogen; red, oxygen). A water molecule (OW) hydrogen-bonded with the bound taurocholate is shown as red sphere. (b) Schematic representation of the CmeR and taurocholate interactions shown in panel a. Dotted lines depict the hydrogen bonds. The hydrogen-bonded distances are also indicated in this figure.
Perhaps, the most striking feature for Tch binding is found at the other end of the molecule which harbors the anionic, conjugated ethanesulfonate tail. Tch is bound in such a way that the long 2-aminoethanesulfonate moiety at the 5β position is extended slightly out of the binding tunnel exposed to the solvent, while still in close enough proximity to interact with residues forming the entrance of the tunnel. Within 5 Å of this negatively charged sulfonate group, there are no positively charged histidines, lysines or arginines available to neutralize the formal negative charge of this sulfonate moiety. Instead, this conjugated acidic tail is engaged to interact with the side chain carbamoyl nitrogen of residue Q134, thus forming an additional hydrogen bond to anchor the bound Tch.
Surprisingly, the large molecule of Tch does not occupy the entire volume of the tunnel. The four-ring backbone and the 2-aminoethanesulfonate tail of the bound Tch are not linear in shape, but rather curve upward and result in a concave conformation. Thus, the end-to-end length of the molecule is significantly shorter than it was expected and only reaches 16.1 Å. In doing so, CmeR is able to accommodate and create a novel bile acid-binding pocket for Tch. This new binding pocket is distinct from the previously determined glycerol-binding site. The bound Tch only spans this Tch-binding site and several solvent molecules are found in the glycerol-binding pocket. Thus, it is very likely that the large ligand-binding tunnel of CmeR could accommodate a bile acid and a glycerol molecule simultaneously. As the binding tunnel of CmeR is mostly hydrophobic in nature, the bound Tch is also found to make extensive hydrophobic contacts with residues forming the wall of this tunnel. It is observed that at least 10 hydrophobic amino acids, including four aromatic residues (F103, F111, W129 and F137), that line the inside wall of the tunnel are involved in Tch binding (Table II).
Table II.
CmeR-ligand Contacts Contacts Within 4.2 Å of Any Ligands are Listed
| Distance (Å) | |||
|---|---|---|---|
| CmeR residues | Taurocholate | Cholate | Glycerol |
| L65 | 3.5 | ||
| I68 | 4.1 | ||
| C69 | 4.0 | ||
| H72 | 2.9 | ||
| F99 | 3.7 | ||
| F103 | 3.2 | 3.7 | 3.4 |
| A108 | 3.5 | 3.0 | |
| F111 | 4.1 | 3.9 | |
| G112 | 3.4 | ||
| I115 | 4.0 | 4.0 | |
| W129 | 3.6 | 3.3 | |
| Q134 | 2.9 | ||
| F137 | 4.0 | 3.6 | 3.8 |
| S138 | 3.1 | ||
| Y139 | 4.0 | 3.3 | |
| C166 | 2.9 | 4.0 | |
| V163 | 3.8 | ||
| K170 | 4.2 | ||
| H174 | 4.0 | ||
| P172′ | 3.8 | ||
| H175′ | 3.1 | 3.0 | |
| L176′ | 4.0 | ||
| L179′ | 4.0 | 4.2 | |
| V163…OW/OW…Gol | 3.0/2.9 | ||
| T167…OW/OW…Gol | 3.2/2.9 | ||
| C69…OW/OW…Tch/Chd | 3.0/2.9 | ||
| K170…OW/OW…Tch/Chd | 2.9/2.8 | 2.9/3.1 | |
| H174…OW/OW…Tch/Chd | 2.7/3.0 | ||
Structure of the CmeR-Cholate complex
The crystal structure of the CmeR-Chd complex was refined to a resolution of 2.35 Å, resulting in Rwork and Rfree of 20.0% and 26.3%, respectively (Table I). This structure revealed that the binding mode for Chd, which differs from Tch by its 5β-cholanoate group, is very distinct. Figure 4 illustrates the simulated annealing electron density omit map of this bound Chd. Surprisingly, the bound Chd was found to orient in an opposite direction when compared with Tch. Thus, the bound Chd and Tch are anti-parallel to each other. For Chd binding, Chd is completely buried within the hydrophobic tunnel in a way that its non-conjugated 5β-cholanoate tail is inserted into the end of the tunnel, leaving its four-ring steroid backbone anchored closer to the entrance. In this manner, the anionic pentanoate moiety of Chd directly interacts with the positively charged H174 of helix α9 through a water-mediated hydrogen bond to anchor this bile acid (Fig. 5). Unlike Tch, in which its anionic ethanesulfoate group is stabilized by charge-dipole interaction, the structure of CmeR-Chd suggests that the negatively charged end of Chd is neutralized by this positively charged histidine residue via a water-mediated hydrogen bond. Important interactions have been found to establish at the 3α, 7α and 12α-hydroxyl groups of the four-ring system. The 3α and 7α-hydroxyl groups contribute two hydrogen bonds with C166 and H175′, respectively, to stabilize the steroid backbone. However, the 12α-hydroxyl moiety participates to form two water-mediated hydrogen bonds with C69 and K170 to further secure the binding. The bound Chd molecule is significantly curved upward and exhibits a boat-like conformation. As a result, the end-to-end length of the molecule is only 11.5 Å. The curved Chd also makes interactions with 11 additional amino acids, including five aromatic residues (F103, F111, W129, F137 and Y139) that create the wall of the tunnel (Table II).
Figure 4.
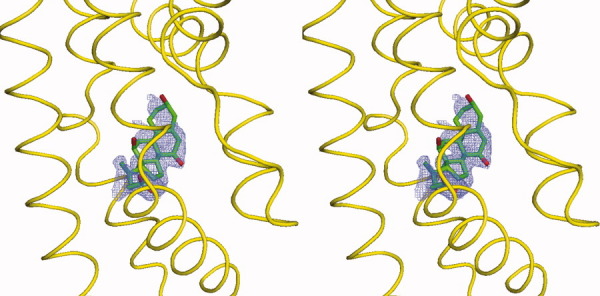
Stereo view of the simulated annealing electron density map of the bound cholate. The bound cholate in the left subunit of dimeric CmeR (the orientation corresponds to Fig. 1) is shown as a stick model (green, carbon; blue, nitrogen; red, oxygen). The 2Fo-Fc simulated annealing omit map is contoured at 1.2 σ (blue mesh). The electron density for the surrounding protein has been deleted. The surrounding secondary structural elements are shown as yellow ribbons.
Figure 5.
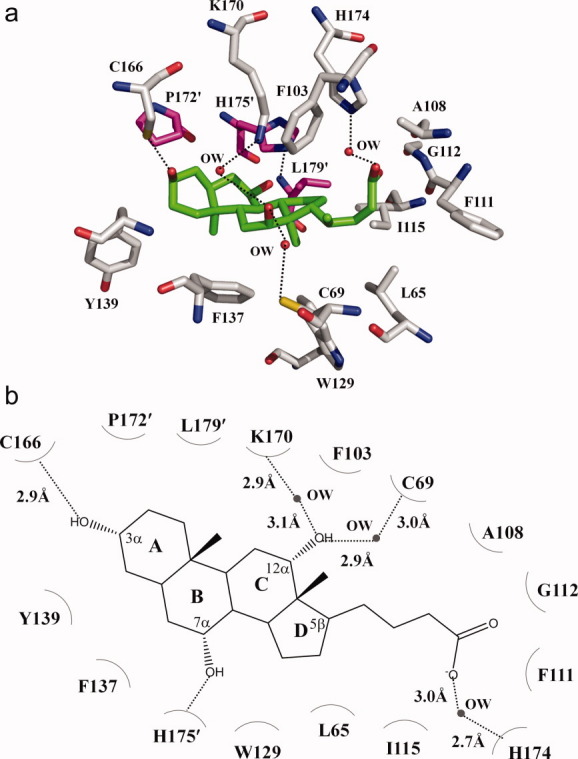
The cholate binding site. (a) Amino acid residues within 4.2 Å from the bound cholate (green, carbon; blue, nitrogen; red, oxygen). The side chains of selected residues are shown as gray sticks (gray, carbon; blue, nitrogen; red, oxygen). Residues from the next subunit of CmeR are shown as magenta sticks (magenta, carbon; blue, nitrogen; red, oxygen). (b) Schematic representation of the CmeR and cholate interactions shown in panel a. Dotted lines depict the hydrogen bonds. The hydrogen-bonded distances are also indicated in this figure.
Overall, Chd and Tch share the same ligand-binding pocket. These bile acids do not span the entire tunnel, but rather bend into a concave structure. In this conformation, these bile acids occupy a novel distinct binding site that is not overlapped with the previously determined glycerol-binding site (Figs. 3 and 5). Based on the structures of CmeR-bile acid complexes, it is observed that the glycerol-binding site in the tunnel remains unoccupied upon bile acid binding. Instead, several solvent molecules are found in this glycerol site. Thus, it is very likely that the large ligand-binding tunnel of CmeR could accommodate a bile acid and a glycerol molecule simultaneously. Tch and Chd are structurally related, differing only by the anionic charged group. Nonetheless, these two bile acids bind distinctly in the binding tunnel. These distinct binding modes indeed underscore the promiscuity of the multifaceted binding pocket of CmeR.
CmeR-bile acid interactions
The binding affinity of each bile acid for the CmeR regulator was determined using isothermal titration calorimetry (ITC), which obtained dissociation constants, KDs, of 1.5 ± 0.1 μM for Tch and 2.5 ± 0.1 μM for Chd. In each case, the titration is characterized by a negative enthalpic contribution, which gives rise to a hyperbolic binding curve (Fig. 6). As expected, the thermodynamic parameters of binding of each bile acid to CmeR are similar, with Tch and Chd displaying enthalpic (ΔH) contributions of −59 ± 1 kcal/mol and -44 ± 4 kcal/mol, respectively. Similar entropic contributions have also been found through these titrations, with ΔS(Tch) = 8 cal mol deg−1 and ΔS(Chd) = 10 cal mol deg−1. The stoichiometries of bile acid binding observed with ITC ranged from 0.9 to 1.0 (bile acid/CmeR monomer).
Figure 6.
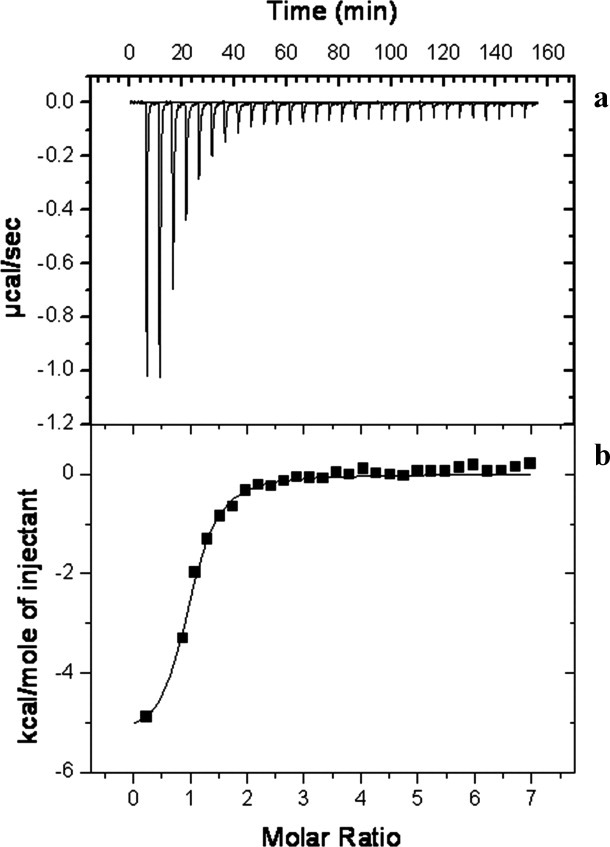
Representative isothermal titration calorimetry for the binding of taurocholate to CmeR. (a) Each peak corresponds to the injection of 10 μL of 0.5 mM Tch in buffer containing 10 mM Na-phosphate (pH 7.2) and 100 mM NaCl into the reaction containing 20 μM CmeR in the same buffer. (b) Cumulative heat of reaction is displayed as a function of the injection number. The solid line is the least-square fit to the experimental data, giving a KD of 1.5 ± 0.1 μM.
Figure 7(a) illustrates the binding isotherm of CmeR in the presence cholyl-lysyl-fluorescein (Clf, a fluorescein-labeled bile acid) using fluorescence polarization (FP). As presented in the figure, a simple hyperbolic curve was observed for the binding of Clf with a dissociation constant, KD, of 50.2 ± 0.4 μM. A Hill plot of the data yielded a Hill coefficient of 1.07 ± 0.03 [Fig. 7(b)], suggesting a simple drug binding process with a stoichiometry of one CmeR monomer per Clf ligand. These results indeed are in good agreement with the crystal structure that each monomer of CmeR binds one bile acid in the binding tunnel.
Figure 7.
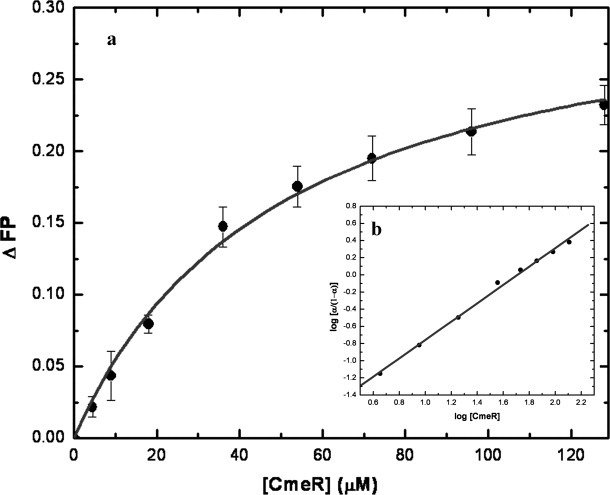
Fluorescence polarization of CmeR with cholyl-lysyl-fluorescein. (a) Binding isotherm of CmeR with cholyl-lysyl-fluorescein, showing a KD of 50.2 ± 0.4 μM, in buffer containing 10 mM Na-phosphate (pH 7.2) and 100 mM NaCl. (b) Hill plot of the data obtained for cholyl-lysyl-fluorescein binding to CmeR. α corresponds to the fraction of bound cholyl-lysyl-fluorescein. The plot gives a slope of 1.07 ± 0.03, indicating a simple binding process with no cooperativity. The interception of the plot provides a KD of 51.1 ± 7.7 μM for the cholyl-lysyl-fluorescein binding.
Discussion
With the rising incidences of MDR strains of bacteria, it has become increasingly important to understand how individual proteins are able to recognize such diverse substrates. The crystal structures of the QacR multidrug binding protein in complex with its respective ligands have provided many insights into the mechanism of multidrug binding,18,19 but these reports have primarily involved positively charged compounds. The CmeR-bile acid complexes reveal how a TetR family protein specifically interacts with negatively charged ligands. To this point, the crystal structure of MarR-salicylate has provided evidence on how regulatory proteins recognize anionic compounds.20 The negatively charged salicylate binds to MarR within a solvent exposed crevice, rather than a large pocket, and interacts with arginines to neutralize its formal charge. The binding crevice lacks the familiar aromatic residues that are critically important in other multidrug binding proteins. It is intriguing that the multidrug binding protein TtgR seems to utilize a different mechanism to recognize negatively charged antibiotics and plant antimicrobials.21 The hydrophobic environment is provided in the ligand binding pocket at the C-terminal regulatory domain. In addition, a positively charged histidine and a polar asparagine are also found to involve in the binding. For CmeR, this regulator seems to share a similar mechanism with TtgR to recognize negatively charged bile acids. Within the multifaceted binding tunnel there are at least seven aromatic residues, five phenylalanines, one tyrosine and one tryptophan, lining the hydrophobic surface to accommodate staking interactions with the ligands. In addition, cationic amino acids are observed to involve in bile acid binding. In fact, within the binding tunnel of CmeR, there are four positively charged residues, including H72, K170, H174 and H175′ (Table II). These residues, which underscore the diversity of the CmeR binding tunnel, probably function to neutralize charges and accommodate the binding of anionic and neutral ligands. This phenomenon is clearly demonstrated in the structures of CmeR-Tch and CmeR-Chd, in which the negatively charged ligands are secured in the binding tunnel by several of these cationic residues. Surprisingly, the two elongated bile acids did not bind in the same orientation inside the tunnel of CmeR, but were actually bound anti-parallel to each other. Chd was bound in an orientation where its A ring was located close to the tunnel opening. However, the bound Tch molecule displayed a contrasting orientation, whereby the corresponding A ring was buried deeply inside the far end of the tunnel. Because of the difference in orientation, the conserved four-ring systems of Tch and Chd were found to bind in different environments. Intriguingly, only two positively charged residues are found to commonly used in the binding of Tch and Chd. Residues K170 and H175′ form important hydrogen bonds to secure the steroid backbones of Tch and Chd. In the case of Tch, CmeR further anchors the steroid backbone of this ligand by using the positively charged H72 residue to form an additional hydrogen bond with the 3α-hydroxyl group. For Chd binding, the regulator chooses H174 to neutralize the anionic charge of the non-conjugated 5β-cholanoate tail of Chd. Tch and Chd are related in chemical structure and have identical charge. Both of these two bile acids are bound by the regulator in the micromolar region. The different binding modes of these two bile acids indeed highlight the promiscuity of the multiligand-binding tunnel of CmeR.
Previously, the crystal structure of CmeR was fortuitously resolved in complex with a glycerol molecule.16 This structure suggested that at least two distinct binding sites existed within the tunnel. Indeed, one of these predicted binding sites was occupied by the bound glycerol. Interestingly, the CmeR-bile acid structures indicated that the large molecules of Chd and Tch did not span both predicted binding sites, but instead took the distinct second site and left the glycerol-binding site unoccupied (Fig. 8). In comparison with the Tch, Chd and glycerol-bound structures, it was found that these three complexes displayed almost an identical structure (with the center-to-centre distance of 54 Å). To bind the operator DNA, this center-to-center distance has to be less than 40 Å because the distance between two consecutive major grooves of B-DNA is 34 Å. Thus, these structures are incompatible with the 16 bp IR operator and should correspond to the induced form of the CmeR regulator. On the basis of these crystal structures, it is possible that CmeR can accommodate a bile acid and a glycerol molecule at the same time (Fig. 8). Such a phenomenon has been previously observed with the crystal structure of QacR simultaneously bound to two ligands, proflavin and ethidium,19 and has been predicted through biochemical analysis to occur in many other proteins, including AcrR,22,23 TtgV24 and MdfA.25 It has also been reported that the QacR repressor can bind a single ligand in multiple positions,26 possibly due to the multifaceted nature of this protein. Thus, there is a chance that the same ligand could interact with these promiscuous multidrug regulators in different orientations within the multifaceted binding pockets.
Figure 8.
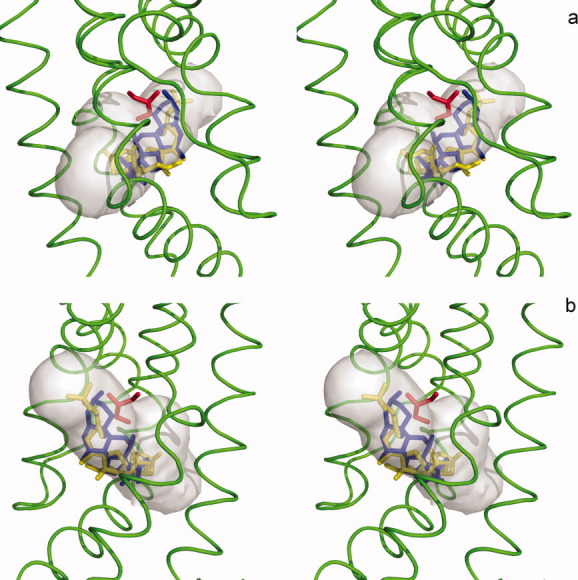
Stereo view of the bile acid and glycerol-binding sites of CmeR. This is a composite figure showing the locations of the bound ligands in the ligand binding tunnel of the (a) left and (b) right subunits of the CmeR dimer. The ligands shown in stick models are taurocholate (yellow), cholate (blue) and glycerol (red). The hydrophobic binding tunnel is colored gray. The surrounding secondary structural elements, based on the structure of the CmeR-glycerol complex, are shown as green ribbons.
The plasticity and promiscuity of the multiligand-binding tunnel of CmeR were further underscored by these CmeR-ligand complex structures. As mentioned previously, glycerol and bile acids have distinct binding sites within the tunnel. In the glycerol-bound structure, the bile acid binding site was unoccupied and filled with water molecules. This empty site was surrounded with several hydrophobic residues, including F111, I115, F137 and Y139. When Tch occupied this bile acid site, which was observed from the CmeR-Tch structure, some of these residues were found to significantly change in position. For example, residues I115, F137 and Y139, which form the wall of the binding tunnel, appeared to shift outward and seemingly participated to expand the internal volume of this binding site, probably accommodating the large size of the bile acid (Fig. 9). Interestingly, residue K170, which was found to form a hydrogen bond with the bound bile acid, reoriented its side chain to accommodate the ligand. Additional movements were also seen through the side chains of H72 and F111. These residues appeared to adjust their orientation to facilitate Tch binding (Fig. 9). It is worth noting that the formal negative charge of Tch was not neutralized by positively charged residues. Instead, electrostatic neutralization was achieved by interaction between the anionic Tch and the positive dipole of the side chain of Q134. Thus, charge-charge electrostatic interaction is not essential for binding negatively charged ligands. Similar drug-regulator interaction has been found in QacR, in which the QacR regulator neutralized one end of the positively charged pentamidine by using carbonyl and side chain oxygen atoms.27 Interestingly, the bound Chd rather employed another mechanism to neutralize its formal negative charge, whereas the anionic pentanoate group was compensated by the formal positive charge of H174.
Figure 9.
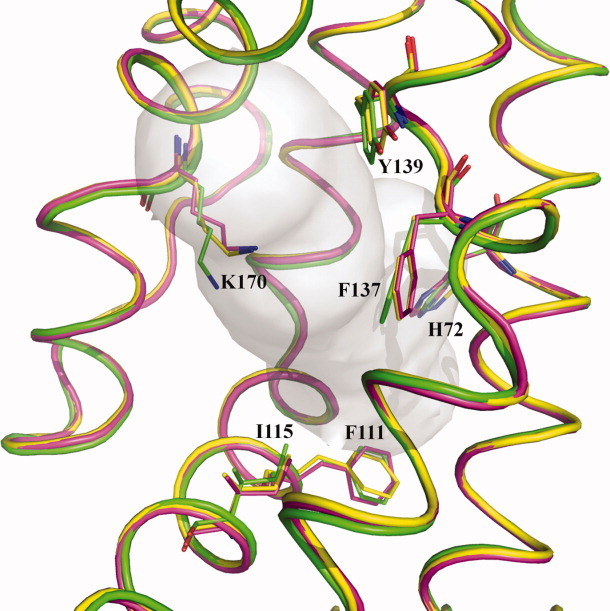
The change in conformation of the binding residues. This is a composite figure showing the conformational change of the side chains H72, F111, I115, F137, Y139, and K170 to accommodate ligand binding. The structural elements of CmeR-glycerol, CmeR-Tch and CmeR-Chd are colored green, yellow and pink, respectively.
In summary, the ability of CmeR to bind two very similar bile acids in quite distinct manners highlighted the plasticity and promiscuity of the ligand-binding tunnel of this regulator. This plasticity is very likely applicable to other multiligand binding proteins, including the AcrR multidrug regulator. Further, neutralization of the negatively charged bile acids can take place using the proximal positively charge residues or the nearby polar groups. The proximal and distinct bile acid and glycerol-binding sites of CmeR highlights the capacity of this regulator, whereby the sizeable hydrophobic tunnel indeed consists of multiple mini-pockets to accommodate diverse ligands.
Materials and Methods
Preparation and crystallization of the CmeR-ligand complexes
Recombinant CmeR containing a 6xHis tag at the N-terminus was overexpressed in Escherichia coli strain JM109 using the pQE30 vector. The cloning, expression and purification procedures have been described previously.16,28 The purified protein was extensively dialyzed against buffer containing 10 mM Na-phosphate pH 7.2 and 100 mM NaCl and concentrated to 10-15 mg/mL. Prior to crystallization trials, Tch or Chd was added to the protein solution at a final concentration of 2 mM and then incubated overnight at 4°C. The stock solution of Tch was prepared by dissolving the sodium salt of Tch (Sigma-Aldrich) in a buffer containing 10 mM Na-phosphate pH 7.2. The Chd solution was made by solubilizing cholate acid (Sigma-Aldrich) in 500 mM NaOH. The resulting solution was then adjusted to a pH of 7.2 with 10 mM Na-phosphate.
Crystals of the 6xHis CmeR were crystallized at room temperature using hanging-drop vapor diffusion as described.16 Briefly, a 4-μL drop containing equal volume of protein solution and reservoir buffer (30% PEG 3350, 0.1 M Tris-HCl pH 8.5 and 0.16 M MgCl2) was equilibrated against 500 μl of reservoir buffer. Crystals of apo-CmeR appeared within two weeks with typical dimensions of 0.2 × 0.2 × 0.2 mm. The CmeR-Tch and CmeR-Chd complex crystals were then prepared by incubating crystals of apo-CmeR in solution containing 30% PEG 3350, 0.1 M Tris-HCl pH 8.5, 0.16 M MgCl2, and 10 mM Tch or Chd for 24 h at 25°C.
X-ray data collection, processing, and structural refinement
X-ray intensity data were collected at 100 K using beamline-24IDC at the Advanced Photon Source. Crystals of CmeR-Tch and CmeR-Chd were cryoprotected with a solution containing 32% PEG 3350, 0.1 M Tris-HCl pH 8.5, 0.16 M MgCl2 and 10 mM of the corresponding bile acid (Tch or Chd). Diffraction data sets were processed with DENZO and scaled with SCALEPACK.29 Both the CmeR-Tch and CmeR-Chd crystals took the space group of P21212 with unit cell dimensions that were isomorphous to the previously determined CmeR-glycerol complex (Table I).
The structures of the CmeR-Tch and CmeR-Chd complexes were determined using the PHENIX suite of programs for crystallographic computing.30 The initial phases were calculated by molecular replacement as implemented in Phaser31 using the previously determined CmeR-glycerol structure (2QCO) with the bound glycerol and water molecules removed as the starting model. Based on the simulated annealing electron density omit maps, the molecule of the corresponding taurocholate (PDB: tch) or cholate (PDB: chd) was manually added into the binding tunnel. Model building was performed using the program Coot.32 Refinement of both structures was carried out using CNS33 and PHENIX.30 The final model was verified by inspection of the simulated annealing composite omit maps. The 2Fo-Fc simulated annealing electron density omit maps of the bound Tch and Chd are shown in Figures 2 and 4. In the final models of both Tch and Chd bound structures, 100% of the residues are within either the most favored or additional allowed regions of the Ramachandran plot analysis, as defined by the program PROCHECK.34
Isothermal titration calorimetry
We used ITC to examine the binding of Tch or Chd to the purified CmeR regulator. Measurements were performed on a VP-Microcalorimeter (MicroCal, Northampton, MA) at 25°C. Before titration, the protein was thoroughly dialyzed against buffer containing 10 mM Na-phosphate pH 7.2 and 100 mM NaCl. The protein concentration was determined using the Bradford assay. The protein sample was then adjusted to a final concentration of 20 μM. Ligand solution consisting of 0.5 mM Tch or Chd in 10 mM Na-phosphate pH 7.2 and 100 mM NaCl was prepared as the titrant. The protein and ligand samples were degassed before they were loaded into the cell and syringe. Binding experiments were carried out with the protein solution (1.5 mL) in the cell and the ligand as the injectant. Ten microliter injections of the ligand solution were used for data collection.
Injections occurred at intervals of 300 s, and the duration time of each injection was 10 s. Heat transfer (μcal/s) was measured as a function of elapsed time (s). The mean enthalpies measured from injection of the ligand in the buffer were subtracted from raw titration data before data analysis with ORIGIN software (MicroCal). Titration curves were fitted by a nonlinear least squares method to a function for the binding of a ligand to a macromolecule. Nonlinear regression fitting to the binding isotherm provided us with the equilibrium binding constant (KA = 1/KD) and enthalpy of binding (ΔH). Based on the values of KA, the change in free energy (ΔG) and entropy (ΔS) were calculated with the equation: ΔG = - RT lnKA = ΔH - TΔS, where T is 273 K and R is 1.9872 cal/K per mol. Calorimetry trials were also carried out in the absence of CmeR in the same experimental conditions. No change in heat was observed in the injections throughout the experiment.
Fluorescence polarization
FP was used to determine the bile acid binding affinity of CmeR. As Tch and Chd are not fluorescent chemicals, we used Clf (BD Biosciences, San Jose, CA) as a fluorescent ligand to perform this binding assay. The experiment was done using a ligand binding solution containing 10 mM Na-phosphate (pH 7.2), 100 mM NaCl and 1 μM Clf. The protein solution consisting of CmeR in 10 mM Na-phosphate (pH 7.2), 100 mM NaCl and 1 μM Clf was titrated into the ligand binding solution until the polarization (P) was unchanged. As this is a steady-state approach, fluorescence polarization measurement was taken after a 5 min incubation for each corresponding concentration of the protein and bile acid to ensure that the binding has reached equilibrium. All measurements were performed at 25°C using a PerkinElmer LS55 spectrofluorometer equipped with a Hamamatsu R928 photomultiplier. The excitation and emission wavelengths were 480 and 517 nm. Fluorescence polarization signal (in ΔP) was measured at the emission wavelength. Each titration point recorded was an average of 15 measurements. Data were analyzed using the equation, P = {(Pbound – Pfree)[protein]/(KD + [protein])} + Pfree, where P is the polarization measured at a given total protein concentration, Pfree is the initial polarization of free ligand, Pbound is the maximum polarization of specifically bound ligand, and [protein] is the protein concentration. The titration experiments were repeated for three times to obtain the average KD value. Curve fitting was accomplished using the program ORIGIN (OriginLab Corporation, Northampton, MA).
Accession Numbers
Coordinates and structural factors have been deposited in the Protein Data Bank with accession numbers 3HGY (CmeR-taurocholate) and 3HGG (CmeR-cholate).
Acknowledgments
This work is based upon research conducted at the Northeastern Collaborative Access Team beamlines of the Advanced Photon Source, supported by award RR-15301 from the National Center for Research Resources at the National Institutes of Health. Use of the Advanced Photon Source is supported by the U.S. Department of Energy, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357.
Glossary
Abbrevations
- MDR
multidrug resistant
- ABC
ATP-binding cassette
- RND
resistance-nodulation-division
- MATE
multidrug and toxic compound extrusion
- MF
major facilitator
- SMR
small multidrug resistance
- Tch
taurocholate
- Chd
cholate
- ITC
isothermal titration calorimetry
- FP
fluorescence polarization
- Clf
cholyl-lysyl-fluorescein
- IR
inverted repeat
References
- 1.Friedman CR, Neimann J, Wegener HC, Tauxe RV. Epidemiology of Campylobacter jejuni infections in the United States and other industrialized nations. In: Nachamkin I, Blaser MJ, editors. Campylobacter. Washington DC: ASM Press; 2000. pp. 121–138. [Google Scholar]
- 2.Lin J, Sahin O, Michel LO, Zhang Q. Critical role of multidrug efflux pump CmeABC in bile resistance and in vivo colonization of Campylobacter jejuni. Infect Immun. 2003;71:4250–4259. doi: 10.1128/IAI.71.8.4250-4259.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Parkhill J, Wren BW, Mungall K, Ketley JM, Churcher C, Basham D, Chillingworth T, Davies RM, Feltwell T, Holrovd S, Jagels K, Karlvshev AV, Moule S, Pallen MJ, Penn CW, Quail MA, Raiandream MA, Rutherford KM, van Vliet AH, Whitehead S, Barrell BG. The genome sequence of the food-borne pathogen Campylobacter jejuni reveals hypervariable sequences. Nature. 2000;403:665–668. doi: 10.1038/35001088. [DOI] [PubMed] [Google Scholar]
- 4.Lin J, Akiba M, Zhang Q. Multidrug efflux systems in Campylobacter. In: Ketley JM, Konkel ME, editors. Campylobacter: Molecular and Cell Biology. Norfolk: Horizon Bioscience; 2005. pp. 205–218. [Google Scholar]
- 5.Akiba M, Lin J, Barton YW, Zhang QJ. Interaction of CmeABC and CmeDEF in conferring antimicrobial resistance and maintaining cell viability in Campylobacter jejuni. J Antimicrob Chemother. 2006;57:52–60. doi: 10.1093/jac/dki419. [DOI] [PubMed] [Google Scholar]
- 6.Lin J, Michel LO, Zhang Q. CmeABC functions as a multidrug efflux system in Campylobacter jejuni. Antimicrob Agents Chemother. 2002;46:2124–2131. doi: 10.1128/AAC.46.7.2124-2131.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pumbwe L, Piddock LJ. Identification and molecular characterization of CmeB, a Campylobacter jejuni multidrug efflux pump. FEMS Microbiol Lett. 2002;206:185–189. doi: 10.1111/j.1574-6968.2002.tb11007.x. [DOI] [PubMed] [Google Scholar]
- 8.Cagliero C, Mouline C, Payot S, Cloeckaert A. Involvement of the CmeABC efflux pump in the macrolide resistance of Campylobacter coli. J Antimicrob Chemother. 2005;56:948–950. doi: 10.1093/jac/dki292. [DOI] [PubMed] [Google Scholar]
- 9.Tseng TT, Gratwick KS, Kollman J, Park D, Nies DH, Goffeau A, Saier MH., Jr The RND permease superfamily: an ancient, ubiquitous and diverse family that includes human disease and development proteins. J Mol Microbiol Biotechnol. 1999;1:107–125. [PubMed] [Google Scholar]
- 10.Schweizer HP. Efflux as a mechanism of resistance to antimicrobials in Pseudomonas aeruginosa and related bacteria: unanswered questions. Genet Mol Res. 2003;2:48–62. [PubMed] [Google Scholar]
- 11.Luo N, Sahin O, Lin J, Michel LO, Zhang Q. In vivo selection of Campylobacter isolates with high levels of fluoroquinolone resistance associated with gyrA mutations and the function of the CmeABC efflux pump. Antimicrob Agents Chemother. 2003;47:390–394. doi: 10.1128/AAC.47.1.390-394.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin J, Cagliero C, Guo B, Barton YW, Maurel MC, Payot S, Zhang Q. Bile salts modulate expression of the CmeABC multidrug efflux pump in Campylobacter jejuni. J Bacteriol. 2005;187:7417–7424. doi: 10.1128/JB.187.21.7417-7424.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin J, Akiba M, Sahin O, Zhang Q. CmeR functions as a transcriptional repressor for the multidrug efflux pump CmeABC in Campylobacter jejuni. Antimicrob Agents Chemother. 2005;49:1067–1075. doi: 10.1128/AAC.49.3.1067-1075.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grkovic S, Brown MH, Skurray RA. Regulation of bacterial drug export systems. Microbiol Mol Biol Rev. 2002;66:671–701. doi: 10.1128/MMBR.66.4.671-701.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ramos JL, Martinez-Bueno M, Molina-Henares AJ, Teran W, Watanabe K, Zhang XD, Gallegos MT, Brennan R, Tobes R. The TetR family of transcriptional repressors. Microbiol Mol Biol Rev. 2005;69:326–356. doi: 10.1128/MMBR.69.2.326-356.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gu R, Su C-C, Shi F, Li M, McDermott G, Zhang Q, Yu EW. Crystal Structure of the transcriptional regulator CmeR from Campylobacter jejuni. J Mol Biol. 2007;372:583–593. doi: 10.1016/j.jmb.2007.06.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Routh MD, Su C-C, Zhang Q, Yu EW. Structures of CmeR and AcrR: insight into the mechanisms of transcriptional repression and multi-drug recognition in the TetR family of regulators. Biochim Biophys Acta. 2009;1794:844–851. doi: 10.1016/j.bbapap.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schumacher MA, Miller MC, Brennan RG. Structural mechanisms of QacR induction and multidrug recognition. Science. 2001;294:2158–2163. doi: 10.1126/science.1066020. [DOI] [PubMed] [Google Scholar]
- 19.Schumacher MA, Miller MC, Brennan RG. Structural mechanism of the simultaneous binding of two drugs to a multidrug-binding protein. EMBO J. 2004;23:2923–2930. doi: 10.1038/sj.emboj.7600288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alekshun MN, Levy SB, Mealy TR, Seaton BA, Head JS. The crystal structure of MarR, a regulator of multiple antibiotic resistance, at 2.3 Å resolution. Nature Struct Biol. 2001;8:710–714. doi: 10.1038/90429. [DOI] [PubMed] [Google Scholar]
- 21.Alguel Y, Meng C, Terán W, Krell T, Ramos JL, Gallegos M-T, Zhang X. Crystal structures of multidrug binding protein TtgR in complex with antibiotics and plant antimicrobials. J Mol Biol. 2007;369:829–840. doi: 10.1016/j.jmb.2007.03.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Su C-C, Rutherford DJ, Yu EW. Characterization of the multidrug efflux regulator AcrR from Escherichia coli. Biochem Biophys Res Commun. 2007;361:85–90. doi: 10.1016/j.bbrc.2007.06.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li M, Gu R, Su C-C, Routh MD, Harris KC, Jewell ES, McDermott G, Yu EW. Crystal structure of the transcriptional regulator AcrR from Escherichia coli. J Mol Biol. 2007;374:591–603. doi: 10.1016/j.jmb.2007.09.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guazzaroni M-E, Krell T, Felipe A, Ruiz R, Meng C, Zhang X, Gallegos M-T, Ramos JL. The multidrug efflux regulator TtgV recognizes a wide range of structurally different effectors in solution and complexed with target DNA. J Biol Chem. 2005;280:20887–20893. doi: 10.1074/jbc.M500783200. [DOI] [PubMed] [Google Scholar]
- 25.Lewinson O, Bibi E. Evidence for simultaneous binding of dissimilar substrates by the Escherichia coli mulitdrug transporter MdfA. Biochemistry. 2001;40:12612–12618. doi: 10.1021/bi011040y. [DOI] [PubMed] [Google Scholar]
- 26.Brooks BE, Piro KM, Brennan RG. Multidrug-binding transcription factor QacR binds the bivalent aromatic diamidines DB75 and DB359 in multiple positions. J Am Chem Soc. 2007;129:8389–8395. doi: 10.1021/ja072576v. [DOI] [PubMed] [Google Scholar]
- 27.Murray DS, Schumacher MA, Brennan RG. Crystal structures of QacR-diamidine complexes reveal additional multidrug-binding modes and a novel mechanism, of drug charge neutralization. J Biol Chem. 2004;279:14365–14371. doi: 10.1074/jbc.M313870200. [DOI] [PubMed] [Google Scholar]
- 28.Su C-C, Shi F, Gu R, Li M, McDermott G, Yu EW, Zhang Q. Preliminary structural studies of the transcriptional regulator CmeR from Camplobacter jejuni. Acta Crystallog F. 2007;63:34–36. doi: 10.1107/S1744309106053127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Otwinowski Z, Minor M. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 30.Afonine PV, Grosse-Kunstleve RW, Adams PD. 2005. The Phenix refinement framework. CCP4 Newsletter 42: Contribution 8.
- 31.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallog. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallog D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 33.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallog D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 34.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallog. 1993;26:283–291. [Google Scholar]