

Abstract
How do we quantify patterns (such as responses to local selection) sampled across multiple populations within a single species? Key to this question is the extent to which populations within species represent statistically independent data points in our analysis. Comparative analyses across species and higher taxa have long recognized the need to control for the non-independence of species data that arises through patterns of shared common ancestry among them (phylogenetic non-independence), as have quantitative genetic studies of individuals linked by a pedigree. Analyses across populations lacking pedigree information fall in the middle, and not only have to deal with shared common ancestry, but also the impact of exchange of migrants between populations (gene flow). As a result, phenotypes measured in one population are influenced by processes acting on others, and may not be a good guide to either the strength or direction of local selection. Although many studies examine patterns across populations within species, few consider such non-independence. Here, we discuss the sources of non-independence in comparative analysis, and show why the phylogeny-based approaches widely used in cross-species analyses are unlikely to be useful in analyses across populations within species. We outline the approaches (intraspecific contrasts, generalized least squares, generalized linear mixed models and autoregression) that have been used in this context, and explain their specific assumptions. We highlight the power of ‘mixed models’ in many contexts where problems of non-independence arise, and show that these allow incorporation of both shared common ancestry and gene flow. We suggest what can be done when ideal solutions are inaccessible, highlight the need for incorporation of a wider range of population models in intraspecific comparative methods and call for simulation studies of the error rates associated with alternative approaches.
Keywords: intraspecific, comparative method, contrasts, generalized least squares, autoregression
1. Introduction
Comparative analysis across taxa ranging from individuals through populations to species has been central to the study of evolution. Key to such analyses is the incorporation of the non-independence (autocorrelation) of taxon values resulting from genetic relationships between them. The need to deal with multiple causes of non-independence in biological data has long been recognized [1], and in evolutionary genetics, methodologies are well developed for analyses at both extremes of the taxonomic scale—among individuals linked by a pedigree, and among species (or higher taxa) linked by a phylogeny. Far less consensus exists for analyses addressing patterns across populations within species, many of which assume populations to be statistically independent entities. Here we review sources of non-independence in comparative analysis in general, and highlight the specific challenges associated with examining patterns across populations within species. Given the scope of this special issue, the scenarios we discuss are drawn from the field of community genetics; however, the issues raised apply more broadly to use of population data in studies of local adaptation [2].
Community genetics examines the impact of genetic diversity in one species on associated community structure and ecosystem processes [3–5]. The community genetics paradigm has developed primarily (but not exclusively, e.g. [6]) from studies on plants and their associated animal communities, encompassing issues ranging in scale from patterns among individuals within populations to those among populations within species [5,7–10]). One common question concerns the extent to which individual or population genetic diversity in keystone species predicts associated community species richness and abundance [11–13]. Alternatively, one might wish to know how key phenotypic traits of one trophic level (such as levels of defensive plant compounds, or budburst phenology) influence associated communities [14,15]. While some aspects of a community extended phenotype (sensu [7])—such as sessile herbivores—can be sampled for individual host plants, other community components are only meaningful at the population level (for example, measures of between-plant synchrony in budburst phenology), or can only be sampled using methods that sum over plant stands or populations. Examples of the latter include estimates of insect diversity sampled using malaise traps or moth traps among discrete stands of a single tree species. How, then, should patterns across populations be analysed?
Many community genetic studies use statistical approaches developed in work on local adaptation and quantitative genetics [2,16–18], such as analysis of phenotypic patterns (for example, in herbivore communities) across sets of full- or half-sib plants grown in a common garden [12,15,19–21]. Pursuing the insect–plant example, an alternative (often included alongside the first approach) is to examine patterns in communities sampled from natural populations whose pedigree relationships are unknown [11,22]. Multiple populations are sampled (in situ, or under common garden conditions) to provide degrees of freedom for hypothesis testing.
The key question in this second approach is how we regard the information from each population. Many studies treat populations as statistically independent replicates (if using population means) or incorporate population as a fixed effect in analyses using individual/clone data, with each population contributing equal weight [2,10–13,18,22–25]. However, two processes operating at the population level (figures 1 and 2)—exchange of migrants (gene flow) and shared common ancestry (phylogenetic non-independence)—mean that sampled population phenotypes (and hence associated community-extended phenotypes) are often not statistically independent. Instead, the phenotypes measured in any one population are related to, and influenced by, those in others [26,27]. Revealing the true relationship between variables across a set of related taxa requires that such non-independence is accounted for statistically.
Figure 1.

Sources of non-independence in population data. (a) Diagrammatic representation of the history of population splits and gene flow linking populations within species. This history results in three genetic contributions to measured population phenotypes, shown diagrammatically for a set of four populations: (i) contributions owing to shared common ancestry (represented by the colours of internal branches in the population tree), (ii) evolution specific to each population owing to selection and drift (represented by colour changes along terminal branches), and (iii) impacts of gene flow (exchange of migrants or gametes) between populations (indicated by arrows, for simplicity shown only for population 1). (b) Gene flow brings into a recipient population a subset of the genetic variation in source populations. Three source populations (1–3) contribute migrants to a recipient population (4). Imagine recipient population 4 has a higher value for a trait (distribution x in the frequency distribution diagram at right) under selection/drift equilibrium than the source populations (which, for simplicity, all share distribution y). Migration into population 4 followed by interbreeding displaces the trait value distribution for this population downwards to a new equilibrium (distribution z). The impact of gene flow is greatest when, relative to a recipient population, source populations have very different equilibrium trait distributions and contribute large numbers of migrants. Under such circumstances, the phenotypes measured in any population may be a poor guide to the selective forces acting on it. Migration effects must be accounted for before local selective effects can be estimated. (c) Population models assumed by different analytical approaches discussed in the text. Assumption of population independence implies no impact of either gene flow or history. This occurs when there is no gene flow and populations are either entirely unrelated (i) or influenced only by population-specific processes (ii), as might happen when selection acting on populations is so rapid and strong that ancestral states can be ignored. Analyses that incorporate only population history (iii) assume no gene flow, while analyses that incorporate only gene flow (iv) assume no population similarity through common ancestry.
Figure 2.

Consequences of phylogenetic non-independence for inferring relationships between variables across populations. Consider four populations, with mean values for two variables (independent variable x and dependent response variable y) as shown at top right. Forgetting gene flow for the moment, if these populations are equally unrelated phylogenetically (a), data for them can be considered independent, and the relationship across all four populations is a positive correlation (b). However, imagine that populations 1–2 and 3–4 comprise two pairs of closely related populations (c). The high trait values shared by both 1 and 2 (and the low values shared by both 3 and 4) are likely not to be independent, but to reflect low divergence within each pair from a common ancestor with high and low trait values, respectively. Now the relationship between x and y is negative within each population pair (black lines in (d)), but positive when analysed across the ancestors of each population pair (red line). Each of these three relationships is phylogenetically independent. A different pattern of relationships among the same set of populations can generate diametrically opposing relationships between x and y, as shown in (e). Now the relationship within each species pair is positive (black fitted lines in (f), right), while the relationship across the ancestors of the two species pairs is negative. These issues pertain whether the populations are sampled in the wild or grown in a common garden or provenance trial.
Generally, variance in any population phenotypic trait that we measure comprises components owing to (i) population-specific effects (including local selection and genetic drift), (ii) patterns of shared common ancestry among populations, (iii) gene flow between populations, and (iv) sampling error. The question is how to estimate (i) while controlling for (ii), (iii) and (iv). This requires more than adding population as a factor in our analysis because population history and gene flow influence expected patterns of covariance among traits. Put simply, we expect populations that are more closely related or exchange higher numbers of migrants to have more similar trait values before we even consider any population-specific effects [16,26,28]. Analyses that fail to incorporate such links between populations face two problems: first, where effects of these processes are severe, measured phenotypes can be a poor guide to the processes at work in each population, increasing type 1 (false positive) and type 2 (false negative) error rates. Second, treating sets of linked populations as if they are independent overestimates the number of degrees of freedom available for hypothesis testing, inflating false positive error rates. To estimate the magnitude of population-specific processes (such as responses to local selective forces), and so examine correlations between these and any other variable, we must transform observed phenotypes to control for contributions associated with shared common ancestry, gene flow and (where appropriate) sampling error. The magnitude of these effects must be expected to vary widely across studies and taxa, reflecting variation in the age of populations, rates of migration between them and the strength of local selection. It will not always be safe to assume that these impacts are negligible.
The issues involved are best understood at two ends of a sampling spectrum. At the individual level, well-established quantitative genetic methods control for shared common ancestry by incorporating pedigree information for the individuals involved [29–33]. At the other extreme, analyses across species and higher taxa assume no gene flow but routinely control for phylogenetic non-independence using a range of comparative methods incorporating phylogenetic or taxonomic level information [28,33,34]. Caught in the middle, analyses across populations within species vary widely in the extent to which they consider phylogenetic non-independence and gene flow, and most ignore both. Within-species analyses are perhaps more challenging than either pedigree-based analysis of individuals or phylogeny-based analysis across higher taxa, primarily because (as we will explain below), population histories can be hard to estimate, and we need to consider gene flow—a problem that can usually be ignored in analyses of higher taxa.
In the following sections, we first survey methods that have been developed to incorporate phylogenetic non-independence in cross-species analyses (§2), and then (§3) discuss the extent to which these can be extended to cross-population analyses. We summarize a method developed by one of us [26] to incorporate effects of gene flow without population history, and (with thanks to Jarrod Hadfield) outline how both effects can be incorporated in the mixed model framework developed in quantitative genetics [33].
2. Coping with phylogenetic non-independence in cross-species comparative analysis
Comparative analysis across species and higher taxa requires explicit consideration of evolutionary history, because for any group of phylogenetically linked taxa, phenotypic similarity may reflect convergent evolutionary responses to a similar underlying cause, or limited divergence from a shared common ancestor, or both [28,34,35]. Four general approaches (discussed in detail below) have been developed to address phylogenetic non-independence: analysis of phylogenetically independent contrasts (PICs, [34]), generalized least squares (GLS, [35–37]), phylogenetic autoregression [38,39] and phylogenetic mixed models [33,40,41]. The approaches differ in attempting either to incorporate phylogenetic and other effects simultaneously (PIC, GLS, phylogenetic mixed models), or to remove phylogenetic effects and examine patterns in residual trait variation (phylogenetic autoregression). Though the four methods have to some extent evolved in isolation, Lynch [40] and Hadfield & Nakagawa [33] have emphasized the similarity between phylogenetic mixed models (of which GLS can be seen as a restricted case) and the ‘animal model’ developed in quantitative genetics, with phylogeny playing the role of pedigree (§2c,d). Hadfield & Nakagawa [33] have recently shown that existing theoretical and computational algorithmics in quantitative genetics can be usefully brought to bear on comparative questions using existing software.
(a). Analysis of phylogenetically independent contrasts
This approach is based on the ‘radiation principle’—that evolutionary correlations between traits are, in principle, free to evolve anew each time daughter taxa diversify from a shared common ancestor (an internal node in a phylogeny) [34,35,42]. The impact of common ancestry is removed by considering only the variation across the daughter lineages at each internal node in a phylogeny (figure 2), summarized for each trait of interest as a form of weighted mean called a linear contrast ([34,43]; for worked examples, see also [28,44]). To reveal the evolutionary relationship between traits, independent contrasts in one trait are regressed against the corresponding contrasts in another trait for the same nodes. Initially developed for analyses structured using bifurcating phylogenies [34], the linear contrasts approach has been extended to incorporate less resolved phylogenies in a GLS framework [28,33,35,45]. While the number of independent contrasts for n species in a fully bifurcating phylogeny is (n − 1), in GLS approaches using incompletely resolved phylogenies, it is the number of internal nodes in the phylogeny.
Estimation of PICs and their statistical analysis both require assumptions about how evolution happens, (i) to allow the estimation of trait values for shared common ancestors, so that contrasts can be calculated for nodes deeper in the phylogeny, and (ii) to scale contrasts made at different nodes in the phylogeny such that the data meet the equal variance assumption of parametric statistical tests. The first [34] and most widely applied model in analytical software (e.g. Phylip [46], Caic [45], Ape [47]) is the Brownian, which assumes continuous characters to evolve by a random walk, with rates of evolutionary change per unit branch length constant in all branches of the phylogeny, and unaffected by the value of a trait or by other species. Though this assumption might appear restrictive, Brownian motion models can incorporate a wide range of evolutionary scenarios [34,48,49]. Other approaches scale by branch length but do so in a different way (phylogenetic regression [35]), or use weighted regression (GLS) to scale the contributions of individual contrasts [36,37]. Despite its convenience for parametric hypothesis testing, the Brownian motion model of character evolution is not supported for some datasets, and is inappropriate for some evolutionary scenarios [50–52]. Alternative plausible models exist that predict rates of trait change exceeding (e.g. resource-partitioning models) or falling below (e.g. niche-filling models) Brownian predictions [51,52]. Though particularly appropriate to adaptive radiations of species, these alternatives may also apply to trait evolution within species—for example, where populations face a geographical mosaic of contrasting selective pressures [16,53–55]. The extent to which datasets conform to the predictions of a Brownian motion model can be tested [51], and a major strength of generalized linear mixed model approaches discussed below is that they can be applied to non-normally distributed (more generally, non-Gaussian) response variables [33].
PIC methods will only control for the effects of shared common ancestry if the phylogeny used (the ‘working phylogeny’; [35]) actually reflects the true relationships among taxa. This will depend on whether the relationships between sampled taxa are actually tree-like, and, if they are, on whether the phylogeny used has the correct topology (branching pattern; so that the right contrasts are made), and relative branch lengths (so that, in methods using this information, contrasts are scaled appropriately). Computer simulations show that if the working phylogeny is inaccurate, the performance of PIC methods deteriorates significantly [48,56]. This is the major argument against applying this approach to within-species analyses, because population-splitting events can be very hard to reconstruct with any confidence (§3). Although some early PIC analyses used existing taxonomy as a proxy for phylogenetic relationships (e.g. [57–59]), working phylogenies are now usually estimated using sequence data, often from a small number of loci. Reconstructing the working phylogeny from a small sample of gene trees makes the assumption that these accurately represent the species tree. When the periods between the branching events in the true species tree are long in terms of numbers of generations (relative to the effective population size; §3a)—usually true in comparative analyses across species—this assumption is reasonable [60]. Methods have been developed that allow uncertainty in the topology of the working phylogeny to be incorporated into comparative analyses, either by repeating the analysis on a set of trees generated by bootstrapping [61] or by using Bayesian methods [62]. Assuming that the working phylogeny is accurate, calculation of appropriate contrasts will then depend on the fit between real evolutionary processes and the assumptions of the evolutionary model [37,44,51,52,63,64]. Again assuming that the working phylogeny is accurate, it is possible to quantify the extent to which trait values are phylogenetically correlated, and so assess whether phylogenetic non-independence needs to be controlled for [37,64–67]. However, this inference is still only as good as the underlying phylogeny.
(b). Phylogenetic autoregression
Originally developed for analysis of spatial patterns [68,69], autoregression approaches have been widely used in comparative analyses to partition variance in trait values into components attributable to (i) the phylogenetic or spatial relationships among taxa, and (ii) specific, independent evolution in each taxon [38,39,48]. The central tenet of autoregression-based approaches is that only patterns in the specific phenotypic components are free of phylogenetic non-independence (box 1). Though now rarely used in cross-species analyses, spatial and genetic autocorrelation approaches have been used more recently in analyses of within-species character divergence [71] and community genetics [13,23,73].
Box 1. Cheverud's phylogenetic autocorrelation method.
The specific, independent trait value for each species is estimated by subtracting from its total trait value a phylogenetic component owing to shared common ancestry, which is a weighted mean of the values for the same trait in all of the other species sampled. The weight of the contribution of one species i to trait values in another j, wij, is a function of their spatial or genetic similarity, summarized in a relationship matrix W of pairwise values. The pairwise values are modelled as wij = 1/dijα, where dij is the pairwise distance between species i and j, and the exponent α allows scaling of the relative impact of one species on another's phenotype with phylogenetic distance. Where alpha is high, more distant populations have little impact on trait values [39,70]. Pairwise distances can be derived from a user-defined hierarchy (e.g. of spatial groupings or taxonomy) or from empirical, geographical or phylogenetic distances [13,23,70,71]. Specific trait values for each of n species are estimated by fitting an autoregression model of the form:
where (with vectors in bold, lower case, and matrices in bold upper case): x is an n × 1 vector of standardized trait values for n species; ρ is the phylogenetic autocorrelation coefficient; W is an n × n matrix of pairwise phylogenetic similarities between sampled taxa; and e is an n × 1 vector of residuals.
ρWx is taken to represent the phylogenetic component of trait values, while the residuals represent the component that is free of phylogenetic effects, and which may be used to test evolutionary hypotheses. Fitting of the autoregression model requires estimation of ρ and α by maximum likelihood (for example, using the software Compare [72]). Evolutionary correlations between traits can be estimated by fitting separate autoregression models for each trait, and testing the correlation between the specific components of each trait using standard statistical approaches (see [44] for a worked example). See main text for potential drawbacks of this approach.
An attractive aspect of autocorrelation approaches is that the need to control for phylogenetic effects, and the extent to which this is achieved, can be assessed graphically using phylogenetic correlograms [39,48,70,71]. Correlograms show the relationship between the strength of any autocorrelation (usually expressed in terms of Moran's I coefficient) and the pairwise distance between taxa. Moran's I ranges from −1 (maximal negative autocorrelation) to 1 (maximal positive autocorrelation), with an expected value of zero where no association exists. Where there is strong phylogenetic autocorrelation in the original total phenotypic data, Moran's I values at low pairwise distances will be significantly positive, while values at larger distances tend to 0 or negative values. Such a pattern is taken to support fitting of an autoregression model [39]. If phylogenetic and specific effects are effectively partitioned in the model, a correlogram for the specific effects should show no trend in Moran's I with increasing genetic distance.
Autoregression approaches share with PIC methods the assumption that the way in which any two taxa evolve is independent of the relationship between them (i.e. the covariance between specific and phylogenetic components is assumed to be zero). This will not be the case where closely related taxa are exposed to similar habitats and adapt independently to them (parallel evolution). Under such circumstances, resulting similarity in trait values among taxa will be attributed to the phylogenetic component and, so, discarded. Harvey & Pagel [28, p. 134] observed that autoregression approaches ‘assign both parallel evolution and variance due to the interaction of the phylogenetic and specific components solely to the phylogenetic effect’, and for this reason, autoregression approaches have been strongly criticized and are generally avoided in comparative analysis [64,74].
An important assumption of autocorrelation methods is that the relationship matrix W (box 1) provides an appropriate estimate of pairwise taxon relationships; if not, estimates of Wx will be incorrect. Autoregression approaches are, however, less sensitive than PIC methods to errors in identifying the relationships between taxa [44,48,56]. This is important because although contrast/GLS methods can perform better than autocorrelation approaches when the working phylogeny is estimated accurately [48], the performance of contrast-based methods falls substantially when the working phylogeny is significantly wrong [44,48,56]. This is a potential advantage of autocorrelation approaches when the precise relationships linking taxa being compared are either hard to estimate or not tree-like at all [39,70] (see below).
(c). Generalized least squares
What we may call the Cheverud approach [38] to dealing with autocorrelation is not the only way. In the Cheverud approach, the factor causing the autocorrelation is supposedly removed, leaving a set of uncorrelated residuals for analysis. GLS is another method. In familiar regressions and ANOVAs, for example, statistical analyses are based on the premise that our observations are independent observations from the same probability distribution—typically the normal. More technically, we suppose that the variance–covariance matrix of our data is diagonal, with all the covariances between the data equal to zero, as they are independent. But as explained above, taxon data may not be independent. Technically, there may be non-zero covariances. If we have a model to generate these covariances, we may construct a suitable variance–covariance matrix and use it to transform our data. GLS then effectively applies standard regression to these transformed data. This approach has been applied to analyses across species and higher taxa by Pagel [36,37] and Grafen [35], using phylogenetic information and a Brownian motion model of evolution to construct the variance–covariance matrix. Freckleton & Jetz [75] use GLS to study a cross-species model in which both phylogeny and degree of range overlap together determine the variance–covariance structure. GLS has been criticized in a quantitative genetics context because it does not take into account uncertainty in fixed effects [33], and has been largely replaced by more sophisticated generalized linear mixed models, of which GLS can be seen as a restricted case.
(d). Generalized linear mixed models and the phylogenetic mixed model
Mixed models were first developed in quantitative genetics, and applied extensively to analysis of data for individuals linked through a pedigree—an analysis now known as the animal model [76]. This approach was first applied to phylogenetic comparative analysis by Lynch [40], and has been extended within a more inclusive generalized linear mixed model framework by Hadfield & Nakagawa [33]. The essence of what is now called the phylogenetic mixed model is the partitioning of variation in the data into various components. Suppose we denote the average value of a character of interest in taxon i as zi. We model our data as follows:
where m is the expected phenotype at the root of the phylogeny, a is the phylogenetic contribution and e is a residual. Such models are referred to as mixed models because, statistically, they mix both fixed effects (m) and random effects (a, e). They are widely used in many sciences where measurements are made on clusters of related (i.e. non-independent) statistical units (another application is in analyses involving multiple observations on the same individual). For the purposes of intuition, we may suppose that m was the state of the character at the origin of the radiation of the phylogeny we are studying. Our data, the zi, will be non-independent and this will be revealed in the phylogenetic component, a, which will result in more closely related species having more similar values of z. As in other comparative approaches, the ai are assumed to evolve along the phylogeny via Brownian motion, resulting in dependencies in the phylogenetic effects of species that have shared a common ancestor. The rate at which the phylogenetic effects evolve is proportional to the standard deviation of the phylogenetic effects and is estimated from the data. As the standard deviation of the phylogenetic effects increases relative to the standard deviation of the residuals, the dependency between the phenotypes of taxa increases. When the standard deviation of the residuals, is zero, this method then becomes equivalent to PICs. Substitute ‘pedigree’ for ‘phylogeny’ and ‘breeding value’ for ‘phylogenetic effect’ in the above and you have the quantitative genetics animal model [76]. This simplified view captures the essentials and makes clear the essential similarity between phylogenetic and quantitative genetic models first noted by Lynch [40] and recently emphasized by Hadfield & Nakagawa [33]. Phylogenetic or pedigree-based mixed models can both be fitted using existing software, including ASReml [77], BUGS [78] and the MCMCglmm R library [33,79], allowing estimation of the separate contributions of the components of the model to our data. Phylogenetic, taxonomic or pedigree information are incorporated in a covariance matrix, representing the amount of evolutionary history two taxa have shared. As with other comparative approaches, phylogenetic mixed models have usually assumed trait values to be normally distributed (but see [80]). A major strength of the mixed model approach, noted by Hadfield & Nakagawa [33], is the use of link functions to allow analysis of non-normally distributed (more generally, non-Gaussian) data, and hence application to non-Brownian motion models of evolution (§2a).
3. Analysis of patterns across populations within species
Populations within species are to some extent equivalent to species within higher taxonomic levels, in that their mean trait values include a component inherited from ancestral populations, and components owing to local population effects of selection and genetic drift. However, because populations within species can exchange migrants, their phenotypes may also contain additional components resulting from the combination of history, drift, selection and gene flow acting on neighbouring populations [26,81]. As with phylogenetic non-independence, gene flow means that phenotypes sampled in a given population will often not be a good guide to the direction and strength of selection acting on that population; instead, migration smoothes phenotypic variation among sets of connected populations [26] (figure 1). Further, migration means that (as with phylogenetic non-independence) trait values for linked populations cannot be considered statistically independent. Our problem is that the trait values from linked populations are ‘intertwined’, making them totally unsuitable in their current form for any kind of principled inference. For both consequences of migration just mentioned, the more migration there is, the worse it gets.
Analysis of patterns across populations thus requires incorporation of both phylogenetic and gene flow effects. Population trait values will otherwise only be statistically independent where either (i) selection is so rapid and strong that sampled phenotypes are unaffected by either ancestry or gene flow, or (ii) all populations are equally descended from the same common ancestor (i.e. they are branch tips in a star-shaped phylogeny) and there is no migration between them [48,70,82] (figure 1). Analyses that do not incorporate these potential causes of non-independence (and many do not) assume the above. Studies incorporating these issues in analyses across populations have found significant genetic and spatial non-independence [16,27,70,71]. In the following sections, we consider first phylogenetic non-independence (§3a) and then gene flow (§3b), before considering specific approaches to dealing with these issues (§3c–f). In contrast to the well-established methodologies available for cross-species analysis, it is early days for within-species analysis, and from promising beginnings much remains to be done.
(a). Incorporating phylogenetic non-independence in within-species analyses
It is tempting to use analysis of PICs for within-species comparisons, based on a within-species phylogeny constructed from sequence data for an appropriately polymorphic marker (such as the mitochondrial cytochrome oxidase c and cytochrome b genes often used in DNA barcoding in animals). This temptation should be resisted [26]. Analysis of PICs is only appropriate where the relationship between populations is both tree-like (often it will not be! See below.) and can be inferred with reasonable accuracy. If there is extensive gene flow between populations, the genetic relationship between them will not be tree-like, but reticulate, and no appropriate working phylogeny will exist [26,70]. We might be tempted to use the same data to generate a relationship matrix among populations for use in a phylogenetic mixed model, matrix correlation analysis (for a cross-species example, see [67]) or autocorrelation analysis. Here again the key issue is whether such markers accurately capture the expected covariance in trait values among populations. Mitochondrial sequence data, though easy to generate and often useful in resolving population relationships, in arthropods and nematodes have coalescent histories that can be strongly influenced by selective sweeps imposed by maternally inherited symbionts, such as Wolbachia [83,84]. As we explain below, sequence data will often overestimate the age of splits between populations, leading to an inappropriate expectation of covariance in the data.
The relationships between populations inferred from molecular data are sensitive to two processes: the way in which genetic variation present in a parental population separates out between its daughter populations (sorting of ancestral polymorphism), and the independent accumulation of new mutations in each daughter population. As a general rule, the genetic diversity in very recently separated populations will be dominated by sorting of ancestral polymorphism [60,85]. Since ancestral polymorphism by definition evolves before the separation of daughter taxa, the variation it shows provides no information on their relationships. Instead, the ancestral sequences we sample are determined by the dynamics of the coalescent process at each locus (for diagrammatic illustrations of conflicts between gene trees and species trees, see [60,85,86]). As a result, relationships between populations based entirely on ancestral polymorphism are very likely to differ widely among loci, and to differ substantially in both topology and branch lengths from the true underlying population tree. The use of gene trees in comparative analyses within species risks applying a false working phylogeny if using PICs, and applying a false covariance/weighting matrix in any other method. Bad news all ’round.
As the timescale over which populations diverge increases, so patterns owing to sorting of ancestral polymorphism are augmented by the accumulation of lineage-specific mutation, increasing the concordance between gene trees and the underlying species tree [60,87,88]. As long as some lineage-specific signal exists (given also the randomness of mutation and the finiteness of the regions of the genome examined), the underlying population tree can be estimated in principle using coalescent-based approaches that combine information from many loci [89–96]. As genomics data accumulate for model systems in local adaptation and community genetics [97], this approach will become increasingly accessible. However, the effort required to get a useful answer will be beyond most comparative analyses. Confident inference of the topology and branch lengths linking a recent series of branching events in species with large effective population sizes can require data for many loci even for a small number of populations [96,98–100]. Accurate resolution of relationships among many local (and so recently diverged) populations using genetic data may be impossible.
Within species, phylogeny-based methods are only likely to be appropriate where populations are resolvable as independently evolving lineages, i.e. (i) population splits are well-enough separated in time to allow adequate resolution of the population tree, and (ii) migration between populations is minimal. These conditions will only apply when the units we are calling populations are separate enough to effectively constitute incipient species. For multicellular organisms, this is demonstrably the case for populations in long-separated glacial refugia or other habitat islands [101–105]. For very rapidly evolving organisms with low dispersal ability occupying very specific habitat patches, the same may apply over much smaller spatial and temporal scales. In most intraspecific analyses, however, phylogeny-based methods will be inappropriate.
(b). Analysis of patterns across populations linked by migration
As described above, migration influences the component of variance in a trait that we can ascribe to population-specific effects, and hence the statistical independence of population data. Under specific models of population structure, migration can also result in spurious cross-population patterns in phenotypes, suggesting an underlying cause such as clinal selection where none beyond migration and drift exists [26]. This means that even though phylogenetic relationships between populations can be estimated in the face of some gene flow [95,106], applying phylogeny-based methods (such as PICs) is inappropriate if gene flow significantly influences population phenotypes. We might ask what evidence there is for significant gene flow between a set of phylogenetically linked populations, before deciding whether to incorporate its effects in an analysis. The topology of the population tree and migration rates between populations can be estimated simultaneously using multilocus coalescent approaches [91,107,108], recently extended to multiple populations ([95]; http://genfaculty.rutgers.edu/hey/software). However, many loci are required to estimate splitting times and migration rates with enough accuracy to structure comparative analyses.
One way forward, explored by Felsenstein [26] and discussed in §3c and appendix A, is to consider an explicit population genetic model in which population phenotypes are only influenced by gene flow and local population effects, ignoring population history. An alternative is to use pairwise measures of genetic similarity between populations to sum the contributions of shared common ancestry and gene flow, generating a population relationship matrix for use in phylogenetic autocorrelation or (as the variance–covariance matrix) in a generalized linear mixed model. All of these approaches will be undermined if the population relationship or gene flow matrix is an inaccurate representation of the influences of populations on each others' phenotypes. As explained above, generating appropriate data to construct useful relationship matrices remains a challenge in any system that is not the specific focus of population genetic study. All methods may also fail to discriminate between effects of gene flow and selection where the predicted effects of these processes are confounded, as might be the case where populations facing similar selective pressures are also nearest neighbours experiencing the highest rates of gene flow [26,70,71,109]. The potential for such confounding can be examined post hoc by using matrix correlation analysis to assess the independence of spatial and genetic patterns in phenotypic data across populations (for examples of this approach, see [70,71]). Where population relationships are known in advance, this problem may be avoidable by sampling an appropriate set of populations for which predicted patterns based on relatedness and selection are not the same [26,110].
(c). An intraspecific contrasts method
Felsenstein [26] developed an intraspecific comparative method that generates among-population contrasts controlling for the effects of gene flow between populations. We discuss this approach at some length in appendix A as it makes crystal clear how migration entangles population phenotypes, rendering naive cross-population statistical analyses invalid, and how (under specific conditions) this problem may be fixed.
The intraspecific contrasts approach assumes a network of populations at mutation-drift equilibrium, meaning that in each the measured phenotype represents the local selective optimum distorted only by immigration and genetic drift. The latter effects are incorporated using an explicit underlying model of population structure and character evolution (appendix A). In the example discussed by Felsenstein [26], populations have a constant population size, with equal numbers of migrant individuals exchanged between any pair of populations (requiring smaller populations to contribute a larger proportion of their number as migrants). Each population is selected towards a constant local optimum phenotype and trait heritability is assumed to be constant. The method requires the existence of genetic data that allow the generation of a matrix M containing the pairwise migration rates (mij between populations i and j) scaled by effective population size (i.e. the product nimij). This is possible for multilocus data in a coalescent framework using Migrate [111–113] for a range of population structures assuming constant population size, or for sequence data using IMa2 [95]. Given these assumptions, this intraspecific contrasts approach allows explicit transformation of population phenotypes to values that are independent of migration and reflect the outcome of selection acting on that population (i.e. under the assumptions of the model used, they are locally optimal phenotypes). These corrected phenotypes can be used to reveal correlations between population phenotypes and population-specific explanatory variables. Transformed phenotypic values can be used to fit a model of the form pi = const. + bzi, where pi is the transformed optimal phenotype in population i and zi is a potentially explanatory variable, such as temperature, recorded for that population. Numerous hypotheses can be explored now that the disentangled, transformed data are in place and amenable to statistical analysis. For example, we could test the hypothesis that b = 0, and hence that population-transformed phenotypes are independent of temperature.
As with any comparative method, applying this method requires (i) an appreciation of the extent to which violation of the underlying assumptions compromise the conclusions (and it is very early days as far as addressing this question is concerned) and (ii) generation of an appropriate pairwise migration matrix, M. Departures from the assumption that population phenotypes are in selection/migration/drift equilibrium can influence the strength of selection inferred in this model. For example, if population means differed historically and are now gradually converging owing to gene flow (as we might expect if there is a strong phylogeographic component to population relationships), then assumption of migration/drift/selection equilibrium will overestimate the role of current selection in maintaining the observed phenotypic difference. Similarly, if population phenotypes were historically similar but are now diverging, then the impact of current selection will be underestimated.
Recent work shows that incorporation of information on within-taxon trait variation (rather than simply using a taxon mean) can have important consequences for the scaling of PICs in PIC analyses [43,114,115]. Within-population variation could be incorporated in a similar way as an extension of the within-species contrast method. Other extensions of this method are being developed to take into account sampling variation when there are finite samples from populations, rather than exact knowledge of the population means. The method can also be extended to multiple characters, with additive genetic covariance of characters inferred. In this context, quantitative genetic experiments can connect directly to the method. One particularly troublesome effect for all methods will be direct environmental effects on phenotypes (environmental plasticity), which can mimic genetic differentiation. However, even in that case, one can in principle separate the effects of natural selection from direct effects on the phenotype.
(d). Spatial and genetic autoregression approaches to within-species analysis
Early autoregression studies recognized the approach to be as applicable at the population level as it is to analyses across species [38,39]. Autocorrelation-based analyses of within-species data thus have the advantage of readily available software (e.g. Compare [72]). As with cross-species analyses (and with the same caveats over the limitations of autoregression as before), specific components can be generated for traits individually, and then the relationship between traits examined. Intraspecific analyses have used relationship matrices based on mitochondrial sequence data [70] or a combination of mitochondrial sequence and nuclear allele frequencies [70], and used phylogenetic autocorrelograms to show first that there is a signature of genetic autocorrelation in the data considered, and that fitting of an autoregression model removes this signature in the specific (independent) components of trait values. An alternative is to incorporate a genetic similarity matrix in partial Mantel tests [116] of relationships with other variables (e.g. [21], using multilocus AFLP data for cottonwood poplars). One response to the challenge of obtaining pairwise genetic distances is to control instead for spatial autocorrelation in the data [13,23,73,110]. This will control for genetic non-independence if nearby populations are generally more closely related. However, this model would not be appropriate if populations arise by the sorting of existing intraspecific genetic diversity among microhabitats, for example. If this alternative is the case, neighbouring populations in different habitats may be genetically very divergent.
(e). Generalized least-squares approaches to within-species analysis
Hansen et al. [16] used a GLS model to examine local adaptation in the flower morphology of two Dalechampia species competing for the services of overlapping sets of pollinators. The approach they use is also an early example of a special case of the intraspecific contrasts method [26]. Their hypothesis is that where these species occur together, competition will lead to adaptive divergence in floral morphology. It thus matters whether shifts in phenotype in locations where the two species are sympatric are independently evolved, or derived from widespread dispersal of particular phenotypes in each species (so representing a single divergence event rather than multiple independent events). Their model assumes covariance in trait values between two populations to decay exponentially with their spatial separation, and under this assumption repeated character displacement in the two Dalechampia species was supported. The algebraic machinery employed by Felsenstein [26] (appendix A) also provides the covariance matrix needed for GLS and generalized linear mixed model approaches (below).
(f). Generalized linear mixed models
Notwithstanding the difficulty of accurately estimating both rates of gene flow and branching patterns among populations, the generalized linear mixed models recently discussed in detail by Hadfield & Nakagawa [33] can incorporate both migration and population history. Using the terminology already introduced above and in box 1 and §2d,
where m is the expected phenotype at the root of the phylogeny, a is the phylogenetic contribution, b is the contribution owing to gene flow and e is a residual. This form of model can be fitted using available software, in ASReml [77] and the MCMCglmm R library [78]. The R script for fitting this combined history and gene flow model in MCMCglmm is provided in appendix A. As discussed in §2a,d, a major strength of the mixed model approach is its ability to incorporate non-normally distributed data [33]. However, the reticulate nature of between-population migration may limit development of efficient computational methods, as has been done for pedigree and phylogenetic matrices (see discussion by Hadfield & Nakagawa [33]).
4. Conclusions, and the future of within-species comparative analysis
The need to explore the consequences of non-independence in comparative data, and to control for it if we find it, is clear. The challenges of coping with such non-independence underline the value of using established pedigree-based approaches wherever possible. But what should those working on non-pedigreed populations do? Population genomic data provide the best hope of estimating the parameters required to control for non-independence statistically. Given that generation of population genomic data is in its infancy in almost all systems, an alternative is to use multilocus datasets for whatever neutral genetic markers are available. Use as many informative loci as possible and select those in which likely impacts of recombination and selection are minimized. For reasons described above, under some plausible scenarios, genetic data will be a more reliable guide to population relationships than spatial surrogates. If population trees and migration rates can be estimated with reasonable accuracy, incorporate both into the analysis using generalized linear mixed models to minimize impacts of non-independence.
More work is needed on the impacts of alternative population models (and hence variance–covariance structures) in intraspecific comparative analysis. Examples include the metapopulation models that have received considerable attention in a coalescent framework [117,118], and models incorporating the alternating periods of reproductive isolation and between-population dispersal experienced by many taxa as a result of global climate cycles [119]. Where methods vary in their performance, we should choose the method whose assumptions best match the properties of our data [51,64,120]. Simulation studies have proved very helpful in revealing relative type 1 and type 2 error rates of alternative interspecific comparative methods under different scenarios (levels of phylogenetic information, evolutionary models and levels of within-species sampling) [43,45,48,56,63,65,114,115,121,122]. One outcome of such studies is the realization that even where alternative approaches have similar error rates, the estimates they return for the correlations between variables can be rather different [122]. The field of within-species comparative analysis would benefit enormously from the development of a similar body of critical simulation analysis.
Acknowledgements
G.N.S. is supported by NERC grants NE/H000038/1 and NE/E014453/1 and J.F. was supported by NIH grant GM071639, by NIH grant DEB-0814322 (PI: Mary Kuhner) and, for the period 2009–2010, by Genome Sciences Department ‘life support’ funds. We thank Jarrod Hadfield, David Shuker, Richard Preziosi and Jenny Rowntree for their detailed and helpful feedback, Joe Bailey, Gina Wimp, Tom Whitham, James Nicholls and Konrad Lohse for their useful comments and critical reading of earlier versions and James Cook and Karsten Schönrogge for discussions leading to the writing of this paper.
Appendix A. An intraspecific contrasts method
We elaborate on the within-species contrast method [26] for several reasons. First, it illustrates how between-population genetic exchange entangles the features in which we are interested and, so, emphasizes the nature of the problem of within-species non-independence. Second, the matrix algebra used to disentangle, statistically, the populations will be unfamiliar to many readers, but is widely applied and useful in many contexts. Finally, issues that arise in the context of this method will arise for all within-species comparative methods.
The basic recursion for the evolution of a phenotype, x, over time, t, in a network of n populations is
with (as in box 1), bold face denoting vectors (lower case) and matrices (upper case). x contains the observed phenotype in each population; p contains the optimum phenotype pi for each population; σ is a scalar describing the incremental per generation movement of a population phenotype, xi, towards its local optimum pi. It is the result of both the heritability and the strength of selection. M contains the pairwise migration rates between populations; e is a vector containing terms describing the impact of genetic drift on each population.
Each single population evolves as:
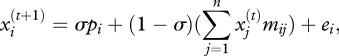 |
where mij is the fraction of newborns in population i that have arrived from population j, and ei is a random variable approximating the effect of genetic drift, drawn from a normal distribution with a mean of 0 and a variance proportional to the inverse of the population size.
The migration matrix M is entangling the evolution of all the data x; the essence of the method is to exploit M and matrix algebra to construct transformations of the data such that the transformed data are disentangled. To do this, we use a result (eigen decomposition, or spectral decomposition) from matrix algebra that is remarkably useful in all sciences, mathematics and statistics and allows us to use the migration matrix M, to obtain uncorrelated variables. The result is
M = C−1ΛC, where C and Λ are matrices generated from M, such that
 |
a diagonal matrix of the eigenvalues of M. C is a matrix of the eigenvectors of M.
We use the matrix C to generate a vector of transformed phenotype values, y, such that
Let us return to our original model in a stripped-down form to let us see clearly the crucial elements: the ‘stripping down’ involves removing the superscripts involving time, t, and looking at the expectation of x, E(x), although we will leave out the ‘E's’ to reduce clutter in this explanatory exercise. This somewhat unusual approach is analogous to the use of pseudo-code when presenting computer algorithms where the emphasis is on exposition, not implementation. Here, matrix algebra is behaving like normal algebra.
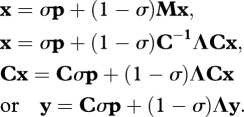 |
Extracting one row of this, and using a as a symbol substitution to simplify visuals:
The crucial thing to note is that by using the migration matrix to provide us with a transformation of our original data, we now have transformed data that are not entangled, but scaled by the single value λi. Obviously, we have swept a lot under the rug to reveal this important core. For example, ‘a’ is a function of the migration matrix and the optimal phenotypes, p.
This is a convenient place to briefly discuss the meaning of the word ‘contrast’ in comparative analysis. ‘Contrasts’ are simply data that have been transformed in such a way that the effects of entangling factors have been removed. The transformation here differs from that used in PICs, but the end result is the same: transformed, disentangled data. In the first case, the entanglement is owing to shared phylogeny, here it is a result of migration.
This model is fitted to some exemplar data in Felsenstein [26], and appropriate software will be made available from http://evolution.gs.washington.edu/programs.html. The explicit underlying model of population structure and character evolution assumed in the application of this approach by Felsenstein [26] is described in the main text. This model can also be fitted in the quantitative genetics package ASReml [77] and using Bayesian approaches, in BUGS [78] and MCMCglmm [33]. With thanks to Jarrod Hadfield, the R scripts for fitting several alternative models in MCMCglmm are as follows. In each, M is the between-population covariance matrix owing to gene flow, equivalent to M above and Msvd is the single value decomposition of M. A generalized linear mixed model (glmm, §3f) incorporating gene flow is fitted using:
Msvd<-svd(M)
Msvd<-Msvd$v%*%(t(Msvd$u)*sqrt(Msvd$d))
testdata$Msvd<-Msvd
M1<-MCMCglmm(x∼1, random=∼idv(Msvd) data=testdata)
To fit a glmm model (M2) that also includes a phylogenetic term, we use:
M2<-MCMCglmm(x∼1, random=∼idv(Msvd)+animal,
pedigree=phylogeny, data=testdata),
where animal is a column in test data that indexes taxa, and phylogeny is a phylogeny stored as a phylo object from the package APE [47].
Felsenstein's model (§3c) is a special case of a simultaneous autoregressive model [123,124], which has been extended to deal with random effects efficiently [125]. With the assumption that the population optimum phenotypes are independent and identically distributed, the Felsenstein model can be fitted using
M3<-MCMCglmm(x∼1+sir(∼M, ∼units), data=testdata)
To fit a phylogenetic component also, we have
M4<-MCMCglmm(x∼1+sir(∼M, ∼units), random=∼animal,
pedigree=phylogeny, data=testdata).
As discussed in Hadfield & Nakagawa [33], Markov Chain Monte Carlo and Gibbs sampling approaches incorporated in BUGS [78] and the MCMCglmm R library [33,79] allow Bayesian approaches to incorporate data for non-Gaussian (including non-normal) trait distributions. See main text, §2a for examples of relevant scenarios involving non-Gaussian trait distributions. The models given above can also be fitted (and perhaps more flexibly) in the R package spdep [126].
Footnotes
One contribution of 13 to a Theme Issue ‘Community genetics: at the crossroads of ecology and evolutionary genetics’.
References
- 1.Ives A. R., Zhu J. 2006. Statistics for correlated data: phylogenies, space, and time. Ecol. Appl. 16, 20–32 10.1890/04-0702 (doi:10.1890/04-0702) [DOI] [PubMed] [Google Scholar]
- 2.Phillimore A. B., Hadfield J. D., Jones O. R., Smithers R. J. 2010. Differences in spawning date between populations of common frog reveal local adaptation. Proc. Natl Acad. Sci. USA 107, 8292–8297 10.1073/pnas.0913792107 (doi:10.1073/pnas.0913792107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Antonovics J. 1992. Toward community genetics. In Plant resistance to herbivores and pathogens: ecology, evolution, and genetics (eds Fritz R. S., Simms E. L.), pp. 426–449 Chicago, IL: University of Chicago Press [Google Scholar]
- 4.Helfield J. M., Naiman R. J. 2001. Effects of salmon-derived nitrogen on riparian forest growth and implications for stream productivity. Ecology 82, 2403–2409 10.1890/0012-9658(2001)082[2403:EOSDNO]2.0.CO;2 (doi:10.1890/0012-9658(2001)082[2403:EOSDNO]2.0.CO;2) [DOI] [Google Scholar]
- 5.Whitham T. G., et al. 2006. A framework for community and ecosystem genetics: from genes to ecosystems. Nat. Rev. Genet. 7, 510–523 10.1038/nrg1877 (doi:10.1038/nrg1877) [DOI] [PubMed] [Google Scholar]
- 6.Bassara R. D., et al. 2010. Local adaptation in Trinidadian guppies alters ecosystem processes. Proc. Natl Acad. Sci. USA 107, 3616–3621 10.1073/pnas.0908023107 (doi:10.1073/pnas.0908023107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Whitham T. G., et al. 2003. Community and ecosystem genetics: a consequence of the extended phenotype. Ecology 84, 559–573 10.1890/0012-9658(2003)084[0559:CAEGAC]2.0.CO;2 (doi:10.1890/0012-9658(2003)084[0559:CAEGAC]2.0.CO;2) [DOI] [Google Scholar]
- 8.Bailey J. K., Wooley S. C., Lindroth R. L., Whitham T. G. 2006. Importance of species interactions to community heritability: a genetic basis to trophic-level interactions. Ecol. Lett. 9, 78–85 10.1111/j.1461-0248.2005.00844.x (doi:10.1111/j.1461-0248.2005.00844.x) [DOI] [PubMed] [Google Scholar]
- 9.Crutsinger G. M., Collins M. D., Fordyce J. A., Gompert Z., Nice C. C., Sanders N. J. 2006. Plant genotypic diversity predicts community structure and governs an ecosystem process. Science 313, 966–968 10.1126/science.1128326 (doi:10.1126/science.1128326) [DOI] [PubMed] [Google Scholar]
- 10.Hughes A. R., Stachowicz J. J. 2009. Ecological impacts of genotypic diversity in the clonal seagrass Zostera marina. Ecology 90, 1412–1419 10.1890/07-2030.1 (doi:10.1890/07-2030.1) [DOI] [PubMed] [Google Scholar]
- 11.Wimp G. M., Young W. P., Woolbright S. A., Martinsen G. D., Keim P., Whitham T. G. 2004. Conserving plant genetic diversity for dependent animal communities. Ecol. Lett. 7, 776–780 10.1111/j.1461-0248.2004.00635.x (doi:10.1111/j.1461-0248.2004.00635.x) [DOI] [Google Scholar]
- 12.Bangert R. K., Turek R. J., Martinsen G. D., Wimp G. M., Bailey J. K., Whitham T. G. 2005. Benefits of conservation of plant genetic diversity to arthropod diversity. Conserv. Biol. 19, 379–390 10.1111/j.1523-1739.2005.00450.x (doi:10.1111/j.1523-1739.2005.00450.x) [DOI] [Google Scholar]
- 13.Bangert R. K., Allan G. J., Turek R. J., Wimp G. M., Meneses N., Martinsen G. D., Whitham T. G. 2006. From genes to geography: a genetic similarity rule for arthropod community structure at multiple geographic scales. Mol. Ecol. 15, 4215–4228 10.1111/j.1365-294X.2006.03092.x (doi:10.1111/j.1365-294X.2006.03092.x) [DOI] [PubMed] [Google Scholar]
- 14.Crawley M. J., Akhteruzzaman M. 1988. Individual variation in the phenology of oak trees and its consequences for herbivorous insects. Funct. Ecol. 2, 409–415 10.2307/2389414 (doi:10.2307/2389414) [DOI] [Google Scholar]
- 15.Andrew R. L., Wallis I. R., Harwood C. E., Foley W. J. 2010. Genetic and environmental contributions to variation and population divergence in a broad-spectrum foliar defence of Eucalyptus tricarpa. Ann. Bot. 105, 707–717 10.1093/aob/mcq034 (doi:10.1093/aob/mcq034) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hansen T. F., Armbruster W. S., Antonsen L. 2000. Comparative analysis of character displacement and spatial adaptations as illustrated by the evolution of Dalechampia blossoms. Am. Nat. 156, S17–S34 10.1086/303413 (doi:10.1086/303413) [DOI] [PubMed] [Google Scholar]
- 17.De la Mata R., Zas R. 2010. Transferring Atlantic maritime pine improved material to a region with marked Mediterranean influence in inland NW Spain: a likelihood-based approach on spatially adjusted field data. Eur. J. Forest Res. 129, 645–658 10.1007/s10342-010-0365-4 (doi:10.1007/s10342-010-0365-4) [DOI] [Google Scholar]
- 18.Torång P., Ehrlén J., Ågren J. 2010. Habitat quality and among-population in reproductive effort and flowering in the perennial herb Primula farinosa. Evol. Ecol. 24, 715–729 10.1007/s10682-009-9327-z (doi:10.1007/s10682-009-9327-z) [DOI] [Google Scholar]
- 19.Lawrence R., Potts B. M., Whitham T. G. 2003. Relative importance of plant ontogeny, host genetic variation, and leaf age for a common herbivore. Ecology 84, 1171–1178 10.1890/0012-9658(2003)084[1171:RIOPOH]2.0.CO;2 (doi:10.1890/0012-9658(2003)084[1171:RIOPOH]2.0.CO;2) [DOI] [Google Scholar]
- 20.Wimp G. M., Martinsen G. D., Floate K. D., Bangert R. K., Whitham T. G. 2005. Plant genetic determinants of arthropod community structure and diversity. Evolution 59, 61–69 [PubMed] [Google Scholar]
- 21.Wimp G. M., Wooley S., Bangert R. K., Young W. P., Martinsen G. D., Keim P., Rehill G. D., Lindroth R. L., Whitham T. G. 2007. Plant genetics predicts intra-annual variation in phytochemistry and arthropod community structure. Mol. Ecol. 16, 5057–5069 10.1111/j.1365-294X.2007.03544.x (doi:10.1111/j.1365-294X.2007.03544.x) [DOI] [PubMed] [Google Scholar]
- 22.Schweitzer J. A., Bailey J. K., Rehill B. J., Martinsen G. D., Hart S. C., Lindroth R. L., Keim P., Whitham T. G. 2004. Genetically based trait in a dominant tree affects ecosystem processes. Ecol. Lett. 7, 127–134 10.1111/j.1461-0248.2003.00562.x (doi:10.1111/j.1461-0248.2003.00562.x) [DOI] [Google Scholar]
- 23.Bangert R. K., Lonsdorf E. V., Wimp G. M., Shuster S. M., Fischer D., Schweitzer J. A., Allan G. J., Bailey J. K., Whitham T. G. 2008. Genetic structure of a foundation species: scaling community phenotypes from the individual to the region. Heredity 100, 121–131 10.1038/sj.hdy.6800914 (doi:10.1038/sj.hdy.6800914) [DOI] [PubMed] [Google Scholar]
- 24.Tovar-Sanchez E., Oyama K. 2006. Effect of hybridization of the Quercus crassifolia × Quercus crassipes complex on the community structure of endophagous insects. Oecologia 147, 702–713 10.1007/s00442-005-0328-5 (doi:10.1007/s00442-005-0328-5) [DOI] [PubMed] [Google Scholar]
- 25.Barbour R. C., O'Reilly-Wapstra J. M., De Little D. W., Jordan G. J., Steane D. A., Humphreys J. R., Bailey J. K., Whitham T. G., Potts B. M. 2009. A geographic mosaic of genetic variation within a foundation tree species and its community-level consequences. Ecology 90, 1762–1772 10.1890/08-0951.1 (doi:10.1890/08-0951.1) [DOI] [PubMed] [Google Scholar]
- 26.Felsenstein J. 2002. Contrasts for a within-species comparative method. In Modern developments in theoretical population genetics (eds Slatkin M., Veuille M.), pp. 118–129 Oxford, UK: Oxford University Press [Google Scholar]
- 27.Ashton K. G. 2004. Comparing phylogenetic signal in intraspecific and interspecific body size datasets. J. Evol. Biol. 17, 1157–1161 10.1111/j.1420-9101.2004.00764.x (doi:10.1111/j.1420-9101.2004.00764.x) [DOI] [PubMed] [Google Scholar]
- 28.Harvey P. H., Pagel M. D. 1991. The comparative method in evolutionary biology. Oxford, UK: Oxford University Press [Google Scholar]
- 29.Falconer D. S. 1981. Introduction to quantitative genetics, 2nd edn. New York, NY: Longman [Google Scholar]
- 30.Kruuk L. E. B. 2004. Estimating genetic parameters in natural populations using the ‘animal model’. Phil. Trans. R. Soc. Lond. B 359, 873–890 10.1098/rstb.2003.1437 (doi:10.1098/rstb.2003.1437) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kruuk L. E. B., Hadfield J. D. 2007. How to separate genetic and environmental causes of similarity between relatives. J. Evol. Biol. 20, 1890–1903 10.1111/j.1420-9101.2007.01377.x (doi:10.1111/j.1420-9101.2007.01377.x) [DOI] [PubMed] [Google Scholar]
- 32.Pemberton J. M. 2008. Wild pedigrees: the way forward. Proc. R. Soc. B 275, 613–621 10.1098/rspb.2007.1531 (doi:10.1098/rspb.2007.1531) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hadfield J. D., Nakagawa S. 2010. General quantitative genetic methods for comparative biology: phylogenies, taxonomies and multitrait models for continuous and categorical characters. J. Evol. Biol. 23, 494–508 10.1111/j.1420-9101.2009.01915.x (doi:10.1111/j.1420-9101.2009.01915.x) [DOI] [PubMed] [Google Scholar]
- 34.Felsenstein J. 1985. Phylogenies and the comparative method. Am. Nat. 125, 1–15 10.1086/284325 (doi:10.1086/284325) [DOI] [Google Scholar]
- 35.Grafen A. 1989. The phylogenetic regression. Phil. Trans. R. Soc. Lond. B 326, 119–157 10.1098/rstb.1989.0106 (doi:10.1098/rstb.1989.0106) [DOI] [PubMed] [Google Scholar]
- 36.Pagel M. D. 1997. Inferring evolutionary processes from phylogenies. Zool. Scripta 26, 331–348 10.1111/j.1463-6409.1997.tb00423.x (doi:10.1111/j.1463-6409.1997.tb00423.x) [DOI] [Google Scholar]
- 37.Pagel M. D. 1999. Inferring the historical patterns of biological evolution. Nature 401, 877–884 10.1038/44766 (doi:10.1038/44766) [DOI] [PubMed] [Google Scholar]
- 38.Cheverud J. M., Dow M. M. 1985. An autocorrelation analysis of genetic variation due to lineal fission in social groups of rhesus macaques. Am. J. Phys. Anthropol. 67, 113–121 10.1002/ajpa.1330670206 (doi:10.1002/ajpa.1330670206) [DOI] [Google Scholar]
- 39.Gittleman J. L., Kot M. 1990. Adaptation: statistics and a null model for estimating phylogenetic effects. Syst. Zool. 39, 227–241 10.2307/2992183 (doi:10.2307/2992183) [DOI] [Google Scholar]
- 40.Lynch M. 1991. Methods for the analysis of comparative data in evolutionary biology. Evolution 45, 1065–1080 10.2307/2409716 (doi:10.2307/2409716) [DOI] [PubMed] [Google Scholar]
- 41.Housworth E. A., Martins E. P., Lynch M. 2004. The phylogenetic mixed model. Am. Nat. 163, 84–96 10.1086/380570 (doi:10.1086/380570) [DOI] [PubMed] [Google Scholar]
- 42.Ridley M. 1983. The explanation of organic diversity. Oxford, UK: Clarendon Press [Google Scholar]
- 43.Felsenstein J. 2008. Comparative methods with sampling error and within-species variation: contrasts revisited and revised. Am. Nat. 171, 713–725 10.1086/587525 (doi:10.1086/587525) [DOI] [PubMed] [Google Scholar]
- 44.Gittleman J. L., Luh H.-K. 1992. On comparing comparative methods. Annu. Rev. Ecol. Syst. 23, 383–404 10.1146/annurev.es.23.110192.002123 (doi:10.1146/annurev.es.23.110192.002123) [DOI] [Google Scholar]
- 45.Purvis A., Rambaut A. 1995. Comparative analysis by independent contrasts (CAIC): an Apple Macintosh application for analysing comparative data. CABIOS 11, 247–251 [DOI] [PubMed] [Google Scholar]
- 46.Felsenstein J. 2005. Phylip (Phylogeny Inference Package), v. 3.6. Distributed by the author Seattle, WA: Department of Genome Sciences, University of Washington [Google Scholar]
- 47.Paradis E., et al. 2009. Ape (Analyses of Phylogenetics and Evolution), R package v. 2.4 See http://ape.mpl.ird.fr/ [DOI] [PubMed]
- 48.Martins E. P. 1996. Phylogenies, spatial autoregression, and the comparative method: a computer simulation study. Evolution 45, 534–557 10.2307/2409910 (doi:10.2307/2409910) [DOI] [PubMed] [Google Scholar]
- 49.Martins E. P. 2000. Adaptation and the comparative method. Trends Ecol. Evol. 15, 296–299 10.1016/S0169-5347(00)01880-2 (doi:10.1016/S0169-5347(00)01880-2) [DOI] [PubMed] [Google Scholar]
- 50.Hansen T. F. 1997. Stabilizing selection and the comparative analysis of adaptation. Evolution 51, 1341–1351 10.2307/2411186 (doi:10.2307/2411186) [DOI] [PubMed] [Google Scholar]
- 51.Freckleton R. P., Harvey P. H. 2006. Detecting non-Brownian trait evolution in adaptive radiations. PloS Biol. 4, 11, e373. 10.1371/journal.pbio.0040373 (doi:10.1371/journal.pbio.0040373) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hansen T. F., Pienaar J., Orzack S. H. 2008. A comparative method for studying adaptation to a randomly evolving environment. Evolution 62, 1965–1977 [DOI] [PubMed] [Google Scholar]
- 53.Kraaijeveld A. R., Godfray H. C. J. 1999. Geographic patterns in the evolution of resistance and virulence in Drosophila and its parasitoids. Am. Nat. 153, S61–S74 10.1086/303212 (doi:10.1086/303212) [DOI] [PubMed] [Google Scholar]
- 54.Johnson C. R., Boerlijst M. C. 2002. Selection at the level of the community: the importance of spatial structure. Trends Ecol. Evol. 17, 83–90 10.1016/S0169-5347(01)02385-0 (doi:10.1016/S0169-5347(01)02385-0) [DOI] [Google Scholar]
- 55.Toju H. 2009. Natural selection drives the fine-scale divergence of a coevolutionary arms race involving a long-mouthed weevil and its obligate host plant. BMC Evol. Biol. 9, 273. 10.1186/1471-2148-9-273 (doi:10.1186/1471-2148-9-273) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Purvis A., Gittleman J. L., Luh K. 1994. Truth or consequences: effects of phylogenetic accuracy on two comparative methods. J. Theor. Biol. 167, 293–300 10.1006/jtbi.1994.1071 (doi:10.1006/jtbi.1994.1071) [DOI] [Google Scholar]
- 57.Stone G. N., Willmer P. G. 1989. Warm-up rates and body temperatures in bees: the importance of body size, thermal regime and phylogeny. J. Exp. Biol. 147, 303–328 [Google Scholar]
- 58.Stone G. N., Purvis A. 1992. Warm-up rates during arousal from torpor in heterothermic mammals: physiological correlates and a comparison with heterothermic insects. J. Comp. Physiol. B 162, 284–295 10.1007/BF00357536 (doi:10.1007/BF00357536) [DOI] [PubMed] [Google Scholar]
- 59.Stone G. N. 1994. Patterns of evolution of warm-up rates and body temperatures in flight in solitary bees of the genus Anthophora. Funct. Ecol. 8, 324–335 10.2307/2389825 (doi:10.2307/2389825) [DOI] [Google Scholar]
- 60.Edwards S. V. 2009. Is a new and general theory of molecular systematics emerging? Evolution 63, 1–19 10.1111/j.1558-5646.2008.00549.x (doi:10.1111/j.1558-5646.2008.00549.x) [DOI] [PubMed] [Google Scholar]
- 61.Felsenstein J. 1988. Phylogenies and quantitative characters. Annu. Rev. Ecol. Syst. 19, 445–471 10.1146/annurev.es.19.110188.002305 (doi:10.1146/annurev.es.19.110188.002305) [DOI] [Google Scholar]
- 62.Huelsenbeck J. P., Rannala B., Masly J. P. 2000. Accommodating phylogenetic uncertainty in evolutionary studies. Science 288, 2349–2350 10.1126/science.288.5475.2349 (doi:10.1126/science.288.5475.2349) [DOI] [PubMed] [Google Scholar]
- 63.Diaz-Uriate R., Garland T. 1996. Testing hypotheses of correlated evolution using phylogenetically independent contrasts: sensitivity to deviations from Brownian motion. Syst. Biol. 45, 27–47 10.1093/sysbio/45.1.27 (doi:10.1093/sysbio/45.1.27) [DOI] [Google Scholar]
- 64.Freckleton R. P. 2009. The seven deadly sins of comparative analysis. J. Evol. Biol. 22, 1367–1375 10.1111/j.1420-9101.2009.01757.x (doi:10.1111/j.1420-9101.2009.01757.x) [DOI] [PubMed] [Google Scholar]
- 65.Freckleton R. P., Harvey P. H., Pagel M. 2002. Phylogenetic analysis and comparative data: a test and review of evidence. Am. Nat. 160, 712–726 10.1086/343873 (doi:10.1086/343873) [DOI] [PubMed] [Google Scholar]
- 66.Blomberg S. P., Garland T., Ives A. R. 2003. Testing for phylogenetic signal in comparative data: behavioural traits are more labile. Evolution 57, 717–745 [DOI] [PubMed] [Google Scholar]
- 67.Bailey R., Schönrogge K., Cook J. M., Melika G., Csóka G., Thúroczy C., Stone G. N. 2009. Host niches and defensive extended phenotypes structure parasitoid wasp communities. PLoS Biol. 7, e1000179. 10.1371/journal.pbio.1000179 (doi:10.1371/journal.pbio.1000179) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sokal R. R., Oden N. L. 1978. Spatial autocorrelation in biology. I. Methodology. Biol. J. Linn. Soc. 10, 199–228 10.1111/j.1095-8312.1978.tb00013.x (doi:10.1111/j.1095-8312.1978.tb00013.x) [DOI] [Google Scholar]
- 69.Sokal R. R., Oden N. L. 1978. Spatial autocorrelation in biology. 2. Some biological implications and four applications of evolutionary and ecological interest. Biol. J. Linn. Soc. 10, 229–249 10.1111/j.1095-8312.1978.tb00014.x (doi:10.1111/j.1095-8312.1978.tb00014.x) [DOI] [Google Scholar]
- 70.Edwards S. V., Kot M. 1995. Comparative methods at the species level: geographic variation in morphology and group size in grey-crowned babblers (Pomatostomus temporalis). Evolution 49, 1134–1146 10.2307/2410438 (doi:10.2307/2410438) [DOI] [PubMed] [Google Scholar]
- 71.Marko P. B. 2005. An intraspecific comparative analysis of character divergence between sympatric species. Evolution 59, 554–564 [PubMed] [Google Scholar]
- 72.Martins E. P. 2004. COMPARE, version 4.6b. Computer programs for the statistical analysis of comparative data. Bloomington, IN: Department of Biology, Indiana University; Distributed by the author at http://compare.bio.indiana.edu/ [Google Scholar]
- 73.Evans L. M., Allan G. J., Shuster S. M., Woolbright S. A., Whitham T. G. 2008. Tree hybridisation and genotypic variation drive cryptic speciation of a specialist mite herbivore. Evolution 62, 3027–3040 10.1111/j.1558-5646.2008.00497.x (doi:10.1111/j.1558-5646.2008.00497.x) [DOI] [PubMed] [Google Scholar]
- 74.Pagel M. D. 1989. Comparative methods for examining adaptation depend on evolutionary models. Folia Primatol. 53, 203–220 10.1159/000156417 (doi:10.1159/000156417) [DOI] [PubMed] [Google Scholar]
- 75.Freckleton R. P., Jetz W. 2009. Space versus phylogeny: disentangling phylogenetic and spatial signals in comparative data. Proc. R Soc. B 276, 21–30 10.1098/rspb.2008.0905 (doi:10.1098/rspb.2008.0905) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Henderson C. R. 1976. Simple method for computing the inverse of a numerator relationship matrix used in prediction of breeding values. Biometrics 32, 69–83 10.2307/2529339 (doi:10.2307/2529339) [DOI] [Google Scholar]
- 77.Gilmour A. R., Gogel B. J., Cullis B. R., Welham S. J., Thompson R. 2002. ASReml User Guide Release 1.0 www document. See http://www.VSN-Intl.com
- 78.Lunn D. J., Thomas A., Best N., Spiegelhalter D. 2000. WinBUGS—a Bayesian modelling framework: concepts, structure, and extensibility. Stat. Comp. 10, 325–337 10.1023/A:1008929526011 (doi:10.1023/A:1008929526011) [DOI] [Google Scholar]
- 79.Hadfield J. D. 2010. MCMC methods for multi-response generalized linear mixed models: the MCMCglmm R Package. J. Stat. Softw. 33, 1–2220808728 [Google Scholar]
- 80.Felsenstein J. 2005. Using the quantitative genetic threshold model for inferences between and within species. Phil. Trans. R. Soc. B 360, 1427–1434 10.1098/rstb.2005.1669 (doi:10.1098/rstb.2005.1669) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tack A. J. M., Roslin T. 2010. Overrun by the neighbours: landscape context affects strength and sign of local adaptation. Ecology 91, 2253–2260 10.1890/09-0080.1 (doi:10.1890/09-0080.1) [DOI] [PubMed] [Google Scholar]
- 82.Revell L. J., Harmon L. J., Collar D. C. 2008. Phylogenetic signal, evolutionary process, and rate. Syst. Biol. 57, 591–601 10.1080/10635150802302427 (doi:10.1080/10635150802302427) [DOI] [PubMed] [Google Scholar]
- 83.Hurst G. D. D., Jiggins F. M. 2005. Problems with mitochondrial DNA as a marker in population, phylogeographic and phylogenetic studies: the effects of inherited symbionts. Proc. R. Soc. B 272, 1525–1534 10.1098/rspb.2005.3056 (doi:10.1098/rspb.2005.3056) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rokas A., Atkinson R., Brown G., West S. A., Stone G. N. 2001. Understanding patterns of genetic diversity in the oak gallwasp Biorhiza pallida: demographic history or a Wolbachia selective sweep? Heredity 87, 294–305 10.1046/j.1365-2540.2001.00872.x (doi:10.1046/j.1365-2540.2001.00872.x) [DOI] [PubMed] [Google Scholar]
- 85.Degnan J. H., Rosenberg N. A. 2006. Discordance of species trees with their most likely gene trees. PloS Genet. 2, 762–768 10.1371/journal.pgen.0020068 (doi:10.1371/journal.pgen.0020068) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Degnan J. H., Rosenberg N. A. 2009. Gene tree discordance, phylogenetic inference and the multispecies coalescent. Trends Ecol. Evol. 24, 332–340 10.1016/j.tree.2009.01.009 (doi:10.1016/j.tree.2009.01.009) [DOI] [PubMed] [Google Scholar]
- 87.Rosenberg N. 2002. The probability of topological concordance of gene trees and species trees. Theoret. Popul. Biol. 61, 225–247 10.1006/tpbi.2001.1568 (doi:10.1006/tpbi.2001.1568) [DOI] [PubMed] [Google Scholar]
- 88.Maddison W. P., Knowles L. L. 2006. Inferring phylogeny despite incomplete lineage sorting. Syst. Biol. 55, 21–30 10.1080/10635150500354928 (doi:10.1080/10635150500354928) [DOI] [PubMed] [Google Scholar]
- 89.Yang Z. 2002. Likelihood and Bayes estimation of ancestral population sizes in hominoids using data from multiple loci. Genetics 162, 1811–1823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rannala B., Yang Z. 2003. Bayes estimation of species divergence times and ancestral population sizes using DNA sequences from multiple loci. Genetics 164, 1645–1656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hey J., Nielsen R. 2004. Multilocus methods for estimating population sizes, migration rates and divergence time, with applications to the divergence of Drosophila pseudobscura and D. persimilis. Genetics 167, 747–760 10.1534/genetics.103.024182 (doi:10.1534/genetics.103.024182) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hey J., Nielsen R. 2007. Integration within the Felsenstein equation for improved Markov chain Monte Carlo methods in population genetics. Proc. Natl Acad. Sci. USA 104, 2785–2790 10.1073/pnas.0611164104 (doi:10.1073/pnas.0611164104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cornuet J. M., Santos F., Beaumont M. A., Robert C. P., Marin J. M., Balding D. J., Guillemaud T., Estoup A. 2008. Inferring population history with DIY ABC: a user-friendly approach to approximate Bayesian computation. Bioinformatics 24, 2713–2719 10.1093/bioinformatics/btn514 (doi:10.1093/bioinformatics/btn514) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kubatko L. S., Carstens B. C., Knowles L. L. 2009. STEM: species tree estimation using maximum likelihood for gene trees under coalescence. Bioinformatics 25, 971–973 10.1093/bioinformatics/btp079 (doi:10.1093/bioinformatics/btp079) [DOI] [PubMed] [Google Scholar]
- 95.Hey J. 2010. Isolation with migration models for more than two populations. Mol. Biol. Evol. 27, 905–920 10.1093/molbev/msp296 (doi:10.1093/molbev/msp296) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lohse K., Sharanowski B., Stone G. N. 2010. Quantifying the Pleistocene history of the oak gall parasitoid Cecidostiba fungosa using twenty intron loci. Evolution 64, 2664–2681 10.1111/j.1558-5646.2010.01024.x (doi:10.1111/j.1558-5646.2010.01024.x) [DOI] [PubMed] [Google Scholar]
- 97.Whitham T. G., DiFazio S. P., Schweitzer J. A., Shuster S. M., Allan G. J., Bailey J. K., Woolbright S. A. 2008. Extending genomics to natural communities and ecosystems. Science 320, 492–494 10.1126/science.1153918 (doi:10.1126/science.1153918) [DOI] [PubMed] [Google Scholar]
- 98.Jennings W. B., Edwards S. V. 2005. Speciational history of Australian grass finches (Poephila) inferred from thirty gene trees. Evolution 59, 2033–2047 [PubMed] [Google Scholar]
- 99.Patterson N., Richter D. J., Gnerre S., Lander E. S., Reich D. 2006. Genetic evidence for complex speciation of humans and chimpanzees. Nature 441, 1103–1108 10.1038/nature04789 (doi:10.1038/nature04789) [DOI] [PubMed] [Google Scholar]
- 100.Carstens B. C., Knowles L. L. 2007. Estimating species phylogeny from gene-tree probabilities despite incomplete lineage sorting: an example from Melanoplus grasshoppers. Syst. Biol. 56, 400–411 10.1080/10635150701405560 (doi:10.1080/10635150701405560) [DOI] [PubMed] [Google Scholar]
- 101.Das A., Mohanty S., Stephan W. 2004. Inferring the population structure and demography of Drosophila ananassae from multilocus data. Genetics 168, 1975–1985 10.1534/genetics.104.031567 (doi:10.1534/genetics.104.031567) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Knowles L. L., Carstens B. C. 2007. Estimating a geographically explicit model of population divergence. Evolution 61, 477–493 10.1111/j.1558-5646.2007.00043.x (doi:10.1111/j.1558-5646.2007.00043.x) [DOI] [PubMed] [Google Scholar]
- 103.Stone G. N., et al. 2007. The phylogeographic clade trade: tracing the impact of human-mediated dispersal on the colonisation of northern Europe by the oak gallwasp Andricus kollari. Mol. Ecol. 16, 2768–2781 10.1111/j.1365-294X.2007.03348.x (doi:10.1111/j.1365-294X.2007.03348.x) [DOI] [PubMed] [Google Scholar]
- 104.Nicholls J. A., et al. 2010. Concordant phylogeography and cryptic speciation in two Western Palaearctic oak gall parasitoid species complexes. Mol. Ecol. 19, 592–609 10.1111/j.1365-294X.2009.04499.x (doi:10.1111/j.1365-294X.2009.04499.x) [DOI] [PubMed] [Google Scholar]
- 105.Rokas A., Atkinson R. J., Webster L., Stone G. N. 2003. Out of Anatolia: longitudinal gradients in genetic diversity support a Turkish origin for a circum-Mediterranean gallwasp Andricus quercustozae. Mol. Ecol. 12, 2153–2174 10.1046/j.1365-294X.2003.01894.x (doi:10.1046/j.1365-294X.2003.01894.x) [DOI] [PubMed] [Google Scholar]
- 106.Eckert A. J., Carstens B. C. 2008. Does gene flow destroy phylogenetic signal? The performance of three methods for estimating species phylogenies in the presence of gene flow. Mol. Phylogenet. Evol. 49, 832–842 10.1016/j.ympev.2008.09.008 (doi:10.1016/j.ympev.2008.09.008) [DOI] [PubMed] [Google Scholar]
- 107.Becquet C., Przeworski M. 2007. A new approach to estimate parameters of speciation models with application to apes. Genome Res. 17, 1505–1519 10.1101/gr.6409707 (doi:10.1101/gr.6409707) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wilkinson-Herbots H. M. 2008. The distribution of the coalescence time and the number of pairwise nucleotide differences in the ‘isolation with migration’ model. Theor. Popul. Biol. 73, 277–288 10.1016/j.tpb.2007.11.001 (doi:10.1016/j.tpb.2007.11.001) [DOI] [PubMed] [Google Scholar]
- 109.Cheverud J. M., Dow M. M., Leutenegger W. 1985. The quantitative assessment of phylogenetic constraints in comparative analyses: sexual dimorphism in body weight among primates. Evolution 39, 1335–1351 10.2307/2408790 (doi:10.2307/2408790) [DOI] [PubMed] [Google Scholar]
- 110.Walker J. A. 1997. Ecological morphology of lacustrine threespine stickleback Gasterosteus aculeatus L. (Gasterosteidae) body shape. Biol. J. Linn. Soc. 61, 3–50 [Google Scholar]
- 111.Beerli P., Felsenstein J. 1999. Maximum-likelihood estimation of migration rates and effective population numbers in two populations using a coalescent approach. Genetics 152, 763–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Beerli P., Felsenstein J. 2001. Maximum likelihood estimation of a migration matrix and effective population sizes in n subpopulations by using a coalescent approach. Proc. Natl Acad. Sci. USA 98, 4563–4568 10.1073/pnas.081068098 (doi:10.1073/pnas.081068098) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Beerli P., Palczewski M. 2010. Unified framework to evaluate panmixia and migration direction among multiple sampling locations. Genetics 185, 313–326 10.1534/genetics.109.112532 (doi:10.1534/genetics.109.112532) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Harmon L. J., Losos J. B. 2005. The effect of intraspecific sample size on Type 1 and Type 2 error rates in comparative studies. Evolution 59, 2705–2710 [PubMed] [Google Scholar]
- 115.Ives A. R., Midford P. E., Garland T. 2007. Within-species variation and measurement error in phylogenetic comparative methods. Syst. Biol. 56, 252–270 10.1080/10635150701313830 (doi:10.1080/10635150701313830) [DOI] [PubMed] [Google Scholar]
- 116.Legendre P., Legendre L. 1998. Numerical ecology , 2nd edn. Amsterdam, The Netherlands: Elsevier [Google Scholar]
- 117.Wakeley J., Aliacar N. 2001. Gene genealogies in a metapopulation. Genetics 159, 893–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wakeley J. 2004. Metapopulation models for historical inference. Mol. Ecol. 13, 865–875 10.1111/j.1365-294X.2004.02086.x (doi:10.1111/j.1365-294X.2004.02086.x) [DOI] [PubMed] [Google Scholar]
- 119.Jesus F. F., Wilkins J. F., Solferini V. N., Wakeley J. 2006. Expected coalescence times and segregating sites in a model of glacial cycles. Genet. Mol. Res. 5, 466–474 [PubMed] [Google Scholar]
- 120.Freckleton R. P. 2000. Phylogenetic tests of ecological and evolutionary hypotheses: checking for phylogenetic independence. Funct. Ecol. 14, 129–134 10.1046/j.1365-2435.2000.00400.x (doi:10.1046/j.1365-2435.2000.00400.x) [DOI] [Google Scholar]
- 121.Martins E. P., Garland T. 1991. Phylogenetic analyses of the correlated evolution of continuous characters: a simulation study. Evolution 45, 534–557 10.2307/2409910 (doi:10.2307/2409910) [DOI] [PubMed] [Google Scholar]
- 122.Martins E. P., Diniz J. A. F., Housworth E. A. 2002. Adaptive constraints and the phylogenetic comparative method: a computer simulation test. Evolution 56, 1–13 [PubMed] [Google Scholar]
- 123.Cliff A. D., Ord J. K. 1981. Spatial processes: models and applications, 271pp. London UK: Taylor & Francis [Google Scholar]
- 124.Kissling W. D., Carl G. 2008. Spatial autocorrelation and the selection of simultaneous autoregressive models. Global Ecol. Biogeog. 17, 59–71 10.1111/j.1466-8238.2007.00334.x (doi:10.1111/j.1466-8238.2007.00334.x) [DOI] [Google Scholar]
- 125.Gianola D., Sorensen D. 2004. Quantitative genetic models for describing simultaneous and recursive relationships between phenotypes. Genetics 167, 1407–1424 10.1534/genetics.103.025734 (doi:10.1534/genetics.103.025734) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. spdep: Spatial dependence: weighting schemes, statistics and models. R package available at http://cran.r-project.org/web/packages/spdep/index.html . The full description of the package is available at http://cran.r-project.org/web/packages/spdep/spdep.pdf . [Google Scholar]