

Abstract
Objectives
Obesity is a significant risk factor for many liver diseases, including hepatocellular carcinoma (HCC). Leptin has been identified as a central mediator of factors that regulate energy intake and expenditure, including appetite, metabolism and fat storage. The role of leptin in the initiation, development and progression of HCC remains poorly understood. The aims of this study were to determine the effect(s) of leptin on HCC cell proliferation and to identify potential signalling mechanism(s) by which leptin exerts these effects.
Methods
Rat H4IIE HCC cells and H4IIE-derived HCC tumours were analysed for leptin receptor (LR) expression. H4IIE cells were treated with leptin (0–100 ng/ml) in the absence or presence of pharmacological inhibitors of p42/p44 mitogen-activated protein kinase (MAPK) (PD98059), p38-MAPK (SB202190) or Janus kinase-signal transducers and activators of transcription (JAK-STAT) (AG490; 10 µM) signalling. Cell proliferation was determined and signal pathway activity analysed.
Results
Immunohistochemistry identified increased LR expression in HCC in human tissue. Leptin did not significantly affect H4IIE cell numbers in serum-depleted (0.1% [v/v] foetal bovine serum [FBS]) medium. However, leptin significantly inhibited serum-stimulated (1.0% [v/v] FBS) H4IIE proliferation. Immunoblot analysis demonstrated that leptin significantly activated p42/p44-MAPK, p38-MAPK and STAT3 signalling in a time-dependent manner. Pretreatment of H4IIE cells with SB202190 abrogated leptin-dependent inhibition of H4IIE proliferation, an effect not observed in cells pretreated with PD98059 or AG490.
Conclusions
Leptin inhibits HCC cell growth in vitro via a p38-MAPK-dependent signalling pathway. Identifying similar effects on tumour growth in vivo may provide an attractive therapeutic target for slowing HCC progression.
Keywords: hepatocellular carcinoma, obesity, leptin, p38-MAPK, AG490
Introduction
Hepatocellular carcinoma (HCC) is the most rapidly increasing type of cancer diagnosed in the USA and represents a major health burden globally. Common risk factors for HCC development include chronic alcohol consumption, viral hepatitis infection, aflatoxin exposure and obesity.1–3 Obesity is of particular concern in developed nations because of its rapidly increasing incidence.4 Epidemiological data suggest a strong correlation between obesity and risk for HCC development; men with a body mass index (BMI) > 35 are subject to a 4.52-fold increase in relative risk for HCC.4 Furthermore, risk factors for HCC are noted to act synergistically. For example, patients with hepatitis C virus (HCV) infection who also abuse alcohol have been reported to have an approximately 50-fold increased risk for developing HCC.5 Thus, efforts to reduce obesity and to better understand the mechanism(s) by which obesity contributes to HCC development and/or progression are of clear clinical significance.
One mechanism by which obesity is proposed to increase risk for HCC development is described in the ‘two-hit’ theory. In this model, obesity leads to a fatty liver, steatosis and non-alcoholic steatohepatitis (NASH). Underlying NASH predisposes the liver to development of fibrosis, which can, in turn, progress to cirrhosis, the leading risk factor for subsequent HCC development.6–9 As with liver disease, obesity does not develop acutely. Dysregulation of leptin signalling has been identified as a central factor during the development of diet-induced obesity. Leptin is a 16-kDa pleiotropic hormone involved in a wide range of physiological functions, including thermogenesis, bone formation, pubertal development, angiogenesis and regulation of satiety.10–13 Under normal conditions, leptin exerts a cachectic effect whereby it acts on the brain to suppress appetite and limit food intake.14,15 However, in the setting of obesity the body becomes resistant to the effects of leptin, resulting in paradoxical weight gain in the setting of high levels of circulating leptin.16 Leptin signalling occurs via binding to a specific leptin receptor (LR), a member of the interleukin-6 receptor family of class 1 cytokine receptors. To date, six LR isoforms have been reported, each of which contains an extracellular binding domain. However, only the long form LR (LRb) also possesses the intracellular domains necessary for signal transduction.17 Following ligand binding/LRb activation, several intracellular signalling cascades can be activated, including the Janus-activated kinase/signal transducers and activators of transcription (JAK/STAT), p38-mitogen/(stress)-activated protein kinase (p38-MAPK/SAPK) and p42/p44-MAPK extracellular signal-regulated kinase (ERK) pathways.18,19
Accumulating evidence suggests a potential role for leptin–LR signalling in the development and/or progression of liver diseases, including HCC.20–22 In the setting of HCC, leptin is considered to be important in regulating fat storage and during tumour angiogenesis.12,22,23 Similarly, a rat model of LR deficiency suggests a link between leptin signalling and neovascularization in the progression of NASH to advanced liver injury and tumour formation.24 These experimental data are further supported by pathological evaluation of human samples in which LR expression correlates with increased intratumour microvascularity in HCC.25,26 Whereas these data support a role for leptin during tumour neoangiogenesis, other studies also report that leptin can act as a mitogen in human HCC cells in vitro.27,28 Despite these data, our understanding of the mechanisms by which leptin–LR regulates these events remains poorly defined.
In this study, we analysed the effect of leptin on intracellular signalling and proliferation using a rat HCC cell line in vitro. Our findings demonstrate that leptin inhibits serum-stimulated proliferation in the rat H4IIE HCC cell line via a p38-MAPK-dependent signalling pathway in vitro. Identifying the mechanism(s) by which leptin signalling acts to alter tumour cell proliferation is critical to our understanding of the role of obesity in HCC pathology and provides potential targets for future therapeutic intervention.
Materials and methods
Assurances
Institutional review board approval was obtained to collect and analyse tissue samples from the pathology archives of patients who had undergone resection for HCC at Carolinas Medical Center, Charlotte, NC, USA.
Reagents
Minimum essential medium (MEM) and heat-inactivated foetal bovine serum (FBS) were purchased from Invitrogen, Inc. (Carlsbad, CA, USA). AG490, SB202190 and PD98059 were purchased from Sigma-Aldrich Corp. (Saint Louis, MO, USA). Antibodies against β-actin, ERK 1, STAT3 and LR were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Antibodies against p38-MAPK, phospho-p38-MAPK (pp38-MAPK) and phospho-STAT3 (pSTAT3) were purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA). An antibody specific against phospho-ERK 1/2 antibody was purchased from Millipore Corp. (Billerica, MA, USA). Recombinant rat leptin (rr-leptin) was purchased from BioVision, Inc. (Mountain View, CA, USA).
Tissue specimens
Paraffin-embedded specimens from 10 patients undergoing hepatic resection at Carolinas Medical Center were obtained from pathology archives. An independent pathologist confirmed diagnosis, and HCC and adjacent non-tumour liver (NTL) were marked. Representative tissue sections were used for subsequent immunohistochemical study.
Cell culture and treatments
The rat H4IIE hepatoma cell line was obtained from the American Type Culture Collection (ATCC) (Manassas, VA, USA) and maintained in MEM supplemented with 10% (v/v) FBS as previously reported.29 When cells were 70–80% confluent, culture medium was replaced with low serum (0.1% v/v FBS) for 24 h prior to experimentation. To determine the effect of leptin on intracellular signalling, cells were treated with 100 ng/ml leptin or vehicle (dimethyl sulphoxide [DMSO] 0.1% [v/v]) for up to 24 h. In a parallel series, cells were pretreated for 1 h with one of AG490 (STAT3 inhibitor, 10 µM), PD098059 (p42/p44 ERK inhibitor, 10 µM), SB202190 (p38-MAPK inhibitor, 100 nm) or vehicle (0.1% [v/v] DMSO) prior to leptin addition.
To determine the effect of leptin on cell proliferation, H4IIE cells were treated with 100 ng/ml leptin for 1 h prior to the addition of either 0.1% (v/v) FBS (low-serum medium [LSM]) or 1.0% (v/v) FBS. Culture medium was subsequently aspirated and replaced with fresh leptin-containing culture medium (0.1% or 1.0% [v/v] FBS accordingly) every 24 h. To evaluate the role of intracellular signal pathway inhibitors on the effect of leptin on HCC cell proliferation, AG490 (10 µM), PD098059 (10 µM), SB202190 (100 nm) or vehicle (0.1% [v/v] DMSO) were added 1 h prior to the addition of leptin and replaced daily along with culture medium and leptin. Cell numbers and viability at 24-h intervals were established using a Countess Automated Cell Counter (Invitrogen, Inc.).
Western blot analysis
Following treatment, cells were washed with ice-cold phosphate-buffered saline (PBS) and whole-cell lysates prepared using radioimmunoprecipitation assay buffer (1.0% [v/v] NP-40, 0.5% [v/v] deoxycholate, 0.1% [w/v] sodium dodecyl sulphate [SDS], 0.5 mM phenylmethylsulphonyl fluoride, 0.05 mM Na3V04, 2 µg/ml aprotinin in PBS). Protein concentrations were determined using a 660-nm Protein Assay (Thermo Fisher Scientific, Inc., Rockford, IL, USA) according to the manufacturer's specifications. Western blot analysis and detection were performed as previously reported.30 Primary antibodies were diluted 1:1000 with the exception of anti-LR (1:500) in 5.0% (v/v) non-fat dry milk diluted in Tris-Tween 20 buffered saline (NFDM-TTBS) and incubated overnight at 4 °C. Secondary antibodies were diluted 1:5000 in 5.0% NFDM-TTBS for 1 h at room temperature.30
Immunohistochemistry
Formalin-fixed, paraffin-embedded tissue was sectioned (4 µM), dried onto glass slides and processed for immunohistochemistry as previously reported.31 Tissue was probed using the anti-LR antibody (1:50 dilution, overnight at 4°C). Immunodetection was performed using an avidin-biotin complex system as per the manufacturer's instructions (Santa Cruz Biotechnology, Inc.). Slides were counterstained with Mayer's haematoxylin, dehydrated through graded alcohols and mounted with a coverslip. Slides were scored by two blinded, independent investigators and the average of these scores used for subsequent statistical analysis.
Statistical analysis
In vitro experiments were performed a minimum of three times. Data are expressed as mean ± standard error of the mean (SEM). Statistical analysis was performed using one-way anova with Dunnett's post-test. A P-value of < 0.05 was considered significant.
Results
Leptin receptor expression in HCC
Immunohistochemistry was performed to detect LR expression in human HCC specimens and adjacent NTL (Fig. 1A). There was significantly higher staining for LR in the tumour mass compared with NTL (2.26 ± 0.27 and 0.08 ± 0.06, respectively, n = 10; P < 0.001) (Fig. 1B). We next performed Western blot analysis for LR expression in whole-cell lysates prepared from cultured H4IIE cells. These data demonstrate two major bands at 90 kDa and 120 kDa corresponding to the long and short forms of the LR (Fig. 1C) and as previously reported by others.27
Figure 1.
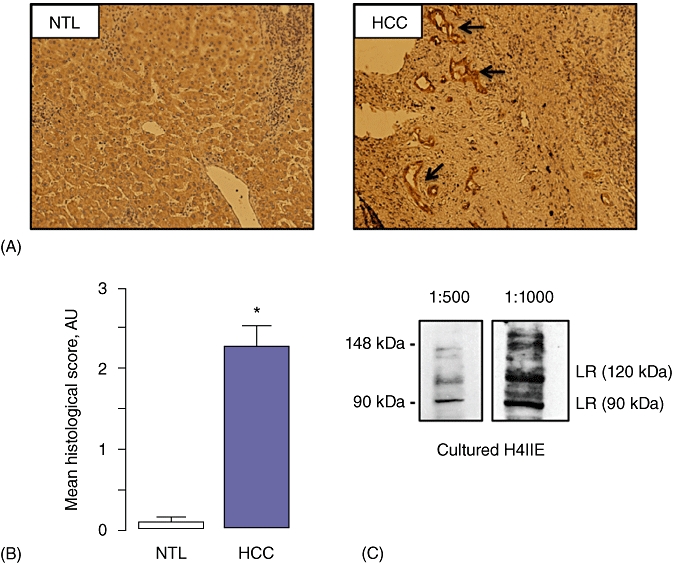
Leptin receptor expression in human and animal models of hepatocellular carcinoma (HCC). (A) Representative immunohistochemical micrographs of leptin receptor (LR) staining (arrows) in human non-tumour liver (NTL) and HCC specimens. (B) Cumulative scoring analysis of LR expression in human NTL and HCC specimens expressed as mean histological score (arbitrary units [AU]; values are mean ± standard error of the mean of five separate fields, independently blind-scored by two different investigators, n = 10; *P < 0.001). (C) Representative Western blot analysis of samples prepared from cultured H4IIE cells culture using an antibody specific against LR at 1:500 and 1:1000 dilutions
Leptin inhibits serum-induced H4IIE proliferation
Cell proliferation was measured for H4IIE cells cultured in 0.1% (v/v) FBS culture medium (LSM) or 1.0% (v/v) FBS with or without leptin pretreatment (100 ng/ml, 1 h prior to FBS addition). In cells maintained in LSM, treatment with leptin failed to significantly alter cell numbers at any point in the 4-day experimental period, an effect not significantly different to that measured in untreated cells (Fig. 2) (n = 6 independent experiments performed in duplicate). By contrast, leptin pretreatment significantly delayed 1.0% (v/v) FBS-stimulated cell proliferation up to 72 h post-FBS stimulation (P < 0.05 for leptin + FBS vs. FBS alone, n = 6 independent experiments performed in duplicate) (Fig. 2). However, by 96 h the inhibitory effect of leptin was exhausted and cell proliferation of leptin-pretreated cells did not significantly differ from that of FBS-only treated cells (n = 6 independent experiments performed in duplicate) (Fig. 2).
Figure 2.
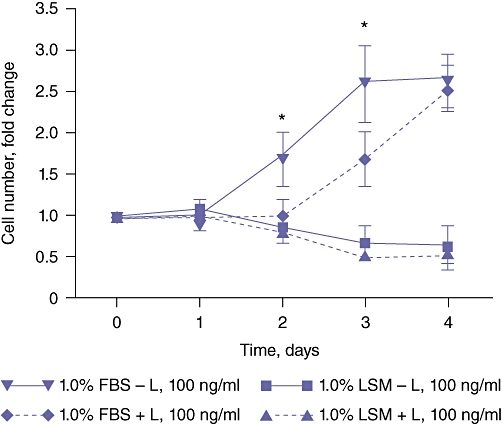
Leptin (L) inhibits serum-stimulated H4IIE cell proliferation in vitro. H4IIE hepatocellular carcinoma cells were cultured in medium containing either 0.1% (v/v) foetal bovine serum (FBS) (LSM) or 1.0% (v/v) FBS in the absence or presence of leptin (L, 100 ng/ml). Number of cells was calculated as a fold change vs. number of cells at day 0, n = 6 separate experiments; *P < 0.05 1.0% (v/v) FBS + L vs. 1.0% (v/v) FBS
Leptin stimulates STAT3, ERK and p38-MAPK activity in H4IIE cells
To gain understanding of the mechanism(s) by which leptin affects H4IIE cell proliferation, we examined the effect of leptin on downstream intracellular signalling cascades. Quiescent H4IIE cells were first treated with leptin (100 ng/ml), after which total and activated STAT3 (STAT3/pSTAT3), p42/p44 ERK-MAPK (ERK 1/pERK 1/2) and p38-MAPK (p38-MAPK/pp38-MAPK) were measured. The results demonstrated that leptin significantly stimulated activation of all three signalling cascades, albeit with different kinetic profiles. STAT3 activation occurred within 20 min and was sustained for the next 2–4 h before decreasing toward baseline activity at 8–24 h (n = 3 independent experiments) (Fig. 3A). Conversely, p42/p44 ERK-MAPK and p38-MAPK remained largely unchanged for the first 1–2 h before increasing over the remainder of the experimental time course (4–24 h, n = 3 independent experiments) (Fig. 3B, C).
Figure 3.
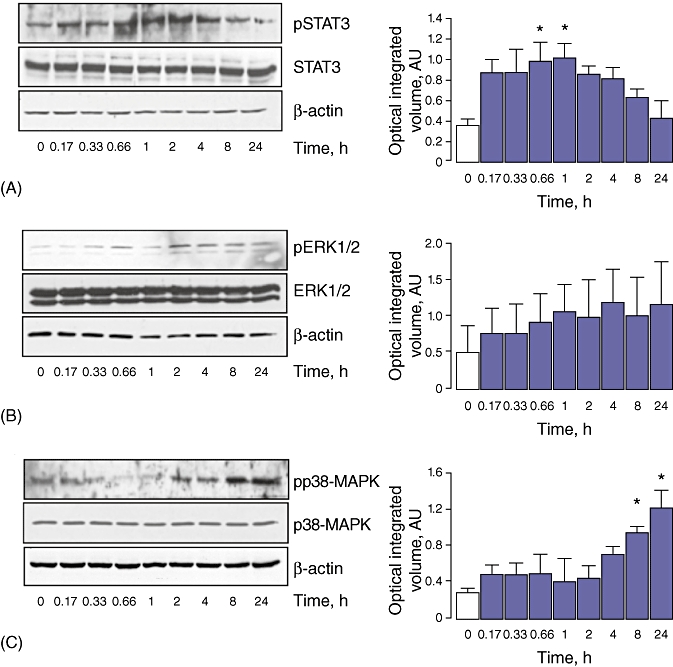
Leptin stimulates STAT3, extracellular signal-regulated kinase (ERK) and p38-MAPK activation in H4IIE cells in vitro. H4IIE hepatocellular carcinoma cells were cultured in medium containing 0.1% (v/v) foetal bovine serum (FBS) and treated in with leptin (100 ng/ml) for 0–24 h. Whole-cell extracts were analysed by Western blot using antibodies against (A) STAT3/pSTAT3, (B) pERK 1/2/ERK 1 or (C) p38-MAPK/pp38-MAPK. In all experiments membranes were stripped and probed with an antibody against β-actin as a housekeeping protein. Bands were analysed by optical integrated volume in arbitrary units (AU) and data expressed as a ratio of activated : total protein. n = 3 separate experiments; *P < 0.05 vs. untreated cells
To confirm the specificity of pharmacological inhibitors, we performed a parallel series of experiments in which cells were pretreated (1 h) with a selective inhibitor of JAK-STAT (AG490, 10 µM), p42/p44 ERK-MAPK (PD98059, 10 µM) or p38-MAPK (SB202190, 100 nm) signalling prior to leptin treatment. These data demonstrated inhibition of the respective signalling pathways using time-points previously established for leptin-dependent activation (Fig. 4A–C).
Figure 4.
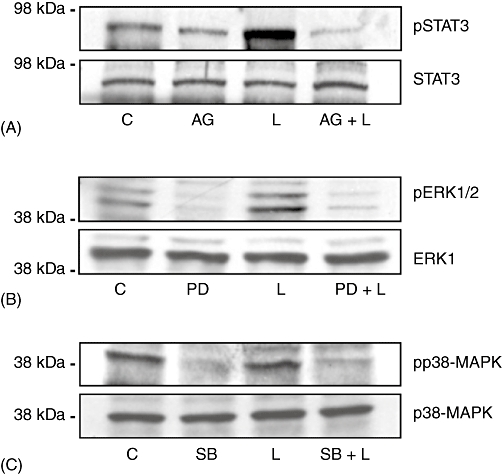
Pharmacological inhibitors alter leptin-dependent signal transduction in H4IIE cells in vitro. H4IIE hepatocellular carcinoma cells were cultured in medium containing 0.1% (v/v) foetal bovine serum (FBS) and treated with one of AG490 (AG; STAT3 inhibitor, 10 µM), PD098059 (PD; pERK1/2 inhibitor, 10 µM), SB202190 (SB; p38-MAPK inhibitor, 100 nm), leptin (L; 100 ng/ml) or inhibitor (1 h) followed by leptin (100 ng/ml). Whole-cell extracts were analysed by Western blot using antibodies against (A) STAT3/pSTAT3, (B) pERK 1/2/ERK 1 or (C) p38-MAPK/pp38-MAPK. C, control
Leptin inhibition of H4IIE proliferation is p38-MAPK-dependent
To determine the role of specific signalling pathways in leptin-mediated inhibition of H4IIE proliferation, we next pretreated quiescent H4IIE cells with AG490 (10 µM), PD98059 (10 µM) or SB202190 (100 nm) followed by leptin (100 ng/ml, 1 h). Cells were then stimulated with 1.0% FBS (v/v) and sequential cell counts performed at 24 h, 48 h and 72 h. As previously, FBS alone stimulated cell proliferation and this effect was significantly inhibited by leptin pretreatment (P < 0.05 for 1.0% [v/v] FBS vs. LSM; P < 0.05 for leptin + FBS vs. FBS; n = 4 independent experiments) (Fig. 5A). Pretreatment of cells with AG490 (a JAK-STAT inhibitor) abrogated FBS-stimulated cell proliferation, in both the absence and presence of leptin (n = 4 independent experiments) (Fig. 4). Similarly, PD98059 significantly inhibited FBS-stimulated proliferation compared with FBS alone, and this effect was not significantly affected by leptin pretreatment (n = 4 independent experiments) (Fig. 5B). Conversely, inhibition of p38-MAPK (SB202190) did not significantly affect FBS only-stimulated proliferation over the first 48 h (Fig. 4). However, pretreatment with SB202190 abrogated the inhibitory effect of leptin on FBS-dependent proliferation to a level not significantly different from that in cells treated with SB202190 and FBS (n = 4 independent experiments) (Fig. 5C). Pretreatment of cells with drug vehicle (0.1% [v/v] DMSO) did not significantly affect FBS-stimulated proliferation or leptin + FBS-stimulated proliferation at any of the time-points assayed (data not shown).
Figure 5.
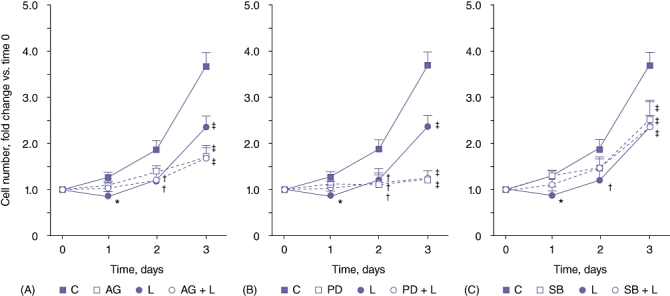
Inhibition of p38-MAPK signalling abrogates leptin-mediated inhibition of H4IIE cell proliferation in vitro. H4IIE hepatocellular carcinoma cells were cultured in medium containing one of 0.1% (v/v) foetal bovine serum (FBS) (low-serum medium [LSM]), LSM + leptin (L; 100 ng/ml), LSM + inhibitor (AG490 [AG]; STAT3 inhibitor, 10 µM), PD098059 (PD; pERK1/2 inhibitor, 10 µM), SB202190 (SB; p38-MAPK inhibitor, 100 nm) or inhibitor + leptin. Number of cells was calculated as fold change vs. number of cells at day 0. n = 4 separate experiments; *P < 0.05 vs. 24-h C; †P < 0.05 vs. 48-h C; ‡P < 0.05 vs. 72-h C. C, control
Discussion
From a global perspective, the leading risk factor for HCC development remains chronic viral hepatitis infection. Despite a global decline in the incidence of hepatitis B virus (HBV), the incidence of HCC is rapidly increasing in developed nations and the most common risk factor is chronic alcohol abuse. However, obesity is reaching epidemic proportions in the USA and other developed countries.32 Obesity is a risk factor for a variety of cancers, including endometrial and breast cancer in women, and pancreatic cancer in both men and women.4,33 Further, a strong association between obesity and risk for HCC has been identified, particularly in males. Interestingly, these data indicate that obesity acts synergistically with HCV and insulin resistance, yet not with HBV.34,35 Despite these epidemiological data, our mechanistic understanding of how obesity contributes to HCC development and/or progression is limited.
Under normal physiological conditions, leptin regulates appetite by acting on the hypothalamus. However, in the setting of obesity, leptin levels rise in correlation with BMI as a result of central leptin resistance and the suppressive effects of leptin on appetite are lost. This has led to the hypothesis that high levels of leptin in obese patients may contribute to the initiation and/or progression of various cancers. In vitro studies utilizing cholangiocarcinoma,36 breast,37 endometrial38 and HCC27,28 cancer cell lines report that leptin is a mitogen. Conversely, leptin was shown to be growth-inhibitory in pancreatic cancer cells39 and cells derived from a colon carcinoma.40 In the current study, leptin treatment of a rat HCC cell line demonstrated leptin inhibition of serum-stimulated proliferation. This finding contrasts with those of other studies in which leptin promoted human HCC cell line proliferation. In addressing these differences, several distinctions become evident between the studies. Saxena et al.27 utilized the HepG2 human hepatoblastoma cell line, whereas Ramani et al.28 used the HuH7 and HepG2 cell lines. Thus species- and tumour-specific variation may explain the differences in response to leptin in vitro. This distinction may be particularly important when comparing results obtained with serum-depleted media to examine the potential mitogenic effects of leptin. In both the Saxena et al.27 and Ramani et al.28 studies, in which leptin stimulated proliferation, cells were exposed to leptin in serum-depleted culture medium. That is, the cells in these studies were quiescent and leptin was the only exogenous agent added. By contrast, leptin failed to stimulate H4IIE cells to proliferate under the same conditions in our study. However, when H4IIE cells were induced to proliferate with FBS, leptin acted to significantly inhibit serum-stimulated, cycling cell proliferation. These data suggest that the inhibitory effects of leptin may be cell cycle-dependent.
Although our data demonstrate the inhibition of serum-stimulated growth by leptin, it is also of interest to note that, after 96 h, leptin appeared unable to continue inhibiting serum-stimulated growth and cell numbers were approximately equal at this time-point. One possible explanation for this observation is that, by 96 h, the FBS-stimulated cells had reached confluence and no additional proliferation was observed (i.e. the growth curve reached a plateau; data not shown). Although contact inhibition of growth explains why FBS-only stimulated cells do not continue to grow, it does not explain why cells pretreated with leptin start to proliferate again after 72–96 h. One possible explanation for this may refer to the clinical paradox whereby obese patients exhibit elevated circulating leptin levels although leptin normally acts to suppress appetite. Using disease models, it has been demonstrated that continued exposure to high circulating leptin leads to a corresponding downregulation of LR expression. In this setting a positive feedback loop exists whereby increased leptin production occurs as a result of the lack of effect caused by diminished receptor expression. A similar series of events may have occurred in our experimental setting whereby continued exposure to high levels of leptin in the culture medium (leptin was replaced every 24 h along with fresh FBS medium) leads to downregulation or internalization of the LR.
Although changes in LR expression may be of importance in longterm response to circulating leptin, differences in how leptin signals are transduced within the cell may be equally important. In studies using human HCC cells, and the current study, leptin significantly stimulated STAT3 activation. Conversely, leptin failed to significantly stimulate p42/p44 ERK phosphorylation in H4IIE cells. This finding may be equally important to our understanding of the differences observed in these studies as leptin-dependent proliferation in both HepG2 and HuH7 cells required activation of p42/p44 ERK signalling.27 Indeed, previous reports by our group have demonstrated the central role of p42/p44 ERK in mediating a range of receptor-mediated proliferation in both human and animal models of HCC.41,42
In our experimental model, p42/p44 ERK appears to be less relevant in mediating the effects of leptin, but the opposite is true for p38-MAPK-dependent signalling. That is, pretreatment of H4IIE cells with SB202190 (a p38-MAPK specific inhibitor) reversed the inhibition of proliferation by leptin for up to 48 h. Previous studies report that p38-MAPK exerts growth-inhibitory effects by suppressing cyclin D1 expression.43,44 Saxena et al.27 did not investigate the potential involvement of p38-MAPK signalling in leptin-mediated HepG2 and Huh7 proliferation, and further investigation is required to ascertain whether a role for this pathway exists in these cells.
Canonical leptin-signalling occurs via LR-mediated activation of JAK/STAT. In response to the activation of STAT, upregulation of suppressor of cytokine signalling (SOCS) family members occurs. SOCS3 prevents phosphorylation of JAK2, which, in turn, prevents the further phosphorylation/activation of STAT3.45 Leptin has been demonstrated to upregulate SOCS3 expression, a critical component in establishing central leptin resistance.46 Given that STAT3 phosphorylation in leptin-treated H4IIE cells is only transiently elevated, SOCS3 may play a role in attenuating STAT3 phosphorylation and, in part, explain how leptin fails to stimulate proliferation in H4IIE cells, yet promotes proliferation in HepG2 and HuH7 cells. Inhibition of the JAK/STAT/ERK axis by SOCS3 may enhance the proliferation-inhibiting effects of p38-MAPK on leptin-treated H4IIE cells. SOCS3 has also been suggested as a potential suppressor of HCC tumours as deletion of hepatic SOCS3 promotes diethyl nitrosamine-mediated tumour formation in mice.47 However, the role of SOCS3 remains somewhat ambiguous as examination of HCC sections showed that SOCS3 expression was associated with higher tumour grade and, in a subset of patients with vascular invasion, high SOCS3 expression correlated with decreased survival.48
The data from our study suggest that leptin might exhibit similar antimitogenic effects in HCC cells in vivo, when cells are exposed to a full complement of cytokines and growth factors. However, any discussion of the role of leptin in vivo must consider other factors. Previous studies have reported the requirement of neovascularization in a model of NASH progression to HCC via a vascular endothelial growth factor (VEGF)-dependent mechanism.24 Furthermore, leptin has been reported to promote angiogenesis in vivo in a chick embryo chorioallantoic membrane assay.22 Similarly, increased LR expression has been reported in human HCC, and that LR expression is strongly localized to vascular endothelial cells within the tumour. This has led to speculation that leptin–LR signalling may be a central factor in mediating the neovascularization required for tumour progression in vivo effects that would promote tumorigenesis, particularly if LR expression is downregulated in HCC in the setting of continued high circulating leptin levels (i.e. obesity).
In conclusion, our data demonstrate that leptin inhibits proliferation of H4IIE HCC cells in vitro and this effect is caused, at least in part, by a p38-MAPK-dependent mechanism. These findings, when taken in parallel with those of other studies, typify the diverse nature of HCC and underscore the challenges involved in elucidating common mechanisms in the initiation and progression of HCC in humans.
Conflicts of interest
None declared.
References
- 1.El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132:2557–2576. doi: 10.1053/j.gastro.2007.04.061. [DOI] [PubMed] [Google Scholar]
- 2.Gomaa AI, Khan SA, Toledano MB, Waked I, Taylor-Robinson SD. Hepatocellular carcinoma: epidemiology, risk factors and pathogenesis. World J Gastroenterol. 2008;14:4300–4308. doi: 10.3748/wjg.14.4300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McKillop IH, Schrum LW. Alcohol and liver cancer. Alcohol. 2005;35:195–203. doi: 10.1016/j.alcohol.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 4.Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of US adults. N Engl J Med. 2003;348:1625–1638. doi: 10.1056/NEJMoa021423. [DOI] [PubMed] [Google Scholar]
- 5.Yuan JM, Govindarajan S, Arakawa K, Yu MC. Synergism of alcohol, diabetes, and viral hepatitis on the risk of hepatocellular carcinoma in blacks and whites in the US. Cancer. 2004;101:1009–1017. doi: 10.1002/cncr.20427. [DOI] [PubMed] [Google Scholar]
- 6.McKillop IH, Moran DM, Jin X, Koniaris LG. Molecular pathogenesis of hepatocellular carcinoma. J Surg Res. 2006;136:125–135. doi: 10.1016/j.jss.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 7.Brunt EM. Non-alcoholic steatohepatitis. Semin Liver Dis. 2004;24:3–20. doi: 10.1055/s-2004-823098. [DOI] [PubMed] [Google Scholar]
- 8.Bugianesi E. Non-alcoholic steatohepatitis and cancer. Clin Liver Dis. 2007;11:191–207. doi: 10.1016/j.cld.2007.02.006. x–xi. [DOI] [PubMed] [Google Scholar]
- 9.Smedile A, Bugianesi E. Steatosis and hepatocellular carcinoma risk. Eur Rev Med Pharmacol Sci. 2005;9:291–293. [PubMed] [Google Scholar]
- 10.Janeckova R. The role of leptin in human physiology and pathophysiology. Physiol Res. 2001;50:443–459. [PubMed] [Google Scholar]
- 11.Shimizu F, Matsuzaki T, Iwasa T, Tanaka N, Minakuchi M, Kuwahara A, et al. Transition of leptin receptor expression during pubertal development in female rat pituitary. Endocr J. 2008;55:191–198. doi: 10.1507/endocrj.k07e-029. [DOI] [PubMed] [Google Scholar]
- 12.Sierra-Honigmann MR, Nath AK, Murakami C, Garcia-Cardena G, Papapetropoulos A, Sessa WC, et al. Biological action of leptin as an angiogenic factor. Science. 1998;281:1683–1686. doi: 10.1126/science.281.5383.1683. [DOI] [PubMed] [Google Scholar]
- 13.Thomas T, Gori F, Khosla S, Jensen MD, Burguera B, Riggs BL. Leptin acts on human marrow stromal cells to enhance differentiation to osteoblasts and to inhibit differentiation to adipocytes. Endocrinology. 1999;140:1630–1638. doi: 10.1210/endo.140.4.6637. [DOI] [PubMed] [Google Scholar]
- 14.Halaas JL, Gajiwala KS, Maffei M, Cohen SL, Chait BT, Rabinowitz D, et al. Weight-reducing effects of the plasma protein encoded by the obese gene. Science. 1995;269:543–546. doi: 10.1126/science.7624777. [DOI] [PubMed] [Google Scholar]
- 15.Pelleymounter MA, Cullen MJ, Baker MB, Hecht R, Winters D, Boone T, et al. Effects of the obese gene product on body weight regulation in ob/ob mice. Science. 1995;269:540–543. doi: 10.1126/science.7624776. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Y, Scarpace PJ. The role of leptin in leptin resistance and obesity. Physiol Behav. 2006;88:249–256. doi: 10.1016/j.physbeh.2006.05.038. [DOI] [PubMed] [Google Scholar]
- 17.Tartaglia LA. The leptin receptor. J Biol Chem. 1997;272:6093–6096. doi: 10.1074/jbc.272.10.6093. [DOI] [PubMed] [Google Scholar]
- 18.Kloek C, Haq AK, Dunn SL, Lavery HJ, Banks AS, Myers MG., Jr Regulation of Jak kinases by intracellular leptin receptor sequences. J Biol Chem. 2002;277:41547–41555. doi: 10.1074/jbc.M205148200. [DOI] [PubMed] [Google Scholar]
- 19.Ahima RS, Osei SY. Leptin signalling. Physiol Behav. 2004;81:223–241. doi: 10.1016/j.physbeh.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 20.Wang SN, Yeh YT, Yang SF, Chai CY, Lee KT. Potential role of leptin expression in hepatocellular carcinoma. J Clin Pathol. 2006;59:930–934. doi: 10.1136/jcp.2005.035477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang YY, Lin SY. Leptin in relation to hepatocellular carcinoma in patients with liver cirrhosis. Horm Res. 2003;60:185–190. doi: 10.1159/000073231. [DOI] [PubMed] [Google Scholar]
- 22.Ribatti D, Belloni AS, Nico B, Di Comite M, Crivellato E, Vacca A. Leptin–leptin receptor are involved in angiogenesis in human hepatocellular carcinoma. Peptides. 2008;29:1596–1602. doi: 10.1016/j.peptides.2008.05.011. [DOI] [PubMed] [Google Scholar]
- 23.Barinaga M. Leptin sparks blood vessel growth. Science. 1998;281:1582. doi: 10.1126/science.281.5383.1582. [DOI] [PubMed] [Google Scholar]
- 24.Kitade M, Yoshiji H, Kojima H, Ikenaka Y, Noguchi R, Kaji K, et al. Leptin-mediated neovascularization is a prerequisite for progression of non-alcoholic steatohepatitis in rats. Hepatology. 2006;44:983–991. doi: 10.1002/hep.21338. [DOI] [PubMed] [Google Scholar]
- 25.Wang SN, Chuang SC, Yeh YT, Yang SF, Chai CY, Chen WT, et al. Potential prognostic value of leptin receptor in hepatocellular carcinoma. J Clin Pathol. 2006;59:1267–1271. doi: 10.1136/jcp.2005.033464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou J, Lei W, Shen L, Luo HS, Shen ZX. Primary study of leptin and human hepatocellular carcinoma in vitro. World J Gastroenterol. 2008;14:2900–2904. doi: 10.3748/wjg.14.2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saxena NK, Sharma D, Ding X, Lin S, Marra F, Merlin D, et al. Concomitant activation of the JAK/STAT, PI3K/AKT, and ERK signalling is involved in leptin-mediated promotion of invasion and migration of hepatocellular carcinoma cells. Cancer Res. 2007;67:2497–2507. doi: 10.1158/0008-5472.CAN-06-3075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ramani K, Yang H, Xia M, Ara AI, Mato JM, Lu SC. Leptin's mitogenic effect in human liver cancer cells requires induction of both methionine adenosyltransferase 2A and 2beta. Hepatology. 2008;47:521–531. doi: 10.1002/hep.22064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moran DM, Koniaris LG, Jablonski EM, Cahill PA, Halberstadt CR, McKillop IH. Microencapsulation of engineered cells to deliver sustained high circulating levels of interleukin-6 to study hepatocellular carcinoma progression. Cell Transplant. 2006;15:785–798. doi: 10.3727/000000006783981477. [DOI] [PubMed] [Google Scholar]
- 30.Moran DM, Mayes N, Koniaris LG, Cahill PA, McKillop IH. Interleukin-6 inhibits cell proliferation in a rat model of hepatocellular carcinoma. Liver Int. 2005;25:445–457. doi: 10.1111/j.1478-3231.2005.01083.x. [DOI] [PubMed] [Google Scholar]
- 31.Padma S, Smeltz AM, Banks PM, Iannitti DA, McKillop IH. Altered aquaporin 9 expression and localization in human hepatocellular carcinoma. HPB (Oxford) 2009;11:66–74. doi: 10.1111/j.1477-2574.2008.00014.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Flegal KM, Carroll MD, Ogden CL, Johnson CL. Prevalence and trends in obesity among US adults, 1999–2000. JAMA. 2002;288:1723–1727. doi: 10.1001/jama.288.14.1723. [DOI] [PubMed] [Google Scholar]
- 33.Michaud DS, Giovannucci E, Willett WC, Colditz GA, Stampfer MJ, Fuchs CS. Physical activity, obesity, height, and the risk of pancreatic cancer. JAMA. 2001;286:921–929. doi: 10.1001/jama.286.8.921. [DOI] [PubMed] [Google Scholar]
- 34.Moller H, Mellemgaard A, Lindvig K, Olsen JH. Obesity and cancer risk: a Danish record-linkage study. Eur J Cancer. 1994;30A:344–350. doi: 10.1016/0959-8049(94)90254-2. [DOI] [PubMed] [Google Scholar]
- 35.Wolk A, Gridley G, Svensson M, Nyren O, McLaughlin JK, Fraumeni JF, et al. A prospective study of obesity and cancer risk (Sweden) Cancer Causes Control. 2001;12:13–21. doi: 10.1023/a:1008995217664. [DOI] [PubMed] [Google Scholar]
- 36.Fava G, Alpini G, Rychlicki C, Saccomanno S, DeMorrow S, Trozzi L, et al. Leptin enhances cholangiocarcinoma cell growth. Cancer Res. 2008;68:6752–6761. doi: 10.1158/0008-5472.CAN-07-6682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rose DP, Komninou D, Stephenson GD. Obesity, adipocytokines, and insulin resistance in breast cancer. Obes Rev. 2004;5:153–165. doi: 10.1111/j.1467-789X.2004.00142.x. [DOI] [PubMed] [Google Scholar]
- 38.Carino C, Olawaiye AB, Cherfils S, Serikawa T, Lynch MP, Rueda BR, et al. Leptin regulation of proangiogenic molecules in benign and cancerous endometrial cells. Int J Cancer. 2008;123:2782–2790. doi: 10.1002/ijc.23887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Somasundar P, Yu AK, Vona-Davis L, McFadden DW. Differential effects of leptin on cancer in vitro. J Surg Res. 2003;113:50–55. doi: 10.1016/s0022-4804(03)00166-5. [DOI] [PubMed] [Google Scholar]
- 40.Fenton JI, Birmingham JM. Adipokine regulation of colon cancer: adiponectin attenuates interleukin-6-induced colon carcinoma cell proliferation via STAT-3. Mol Carcinog. 2010;49:700–709. doi: 10.1002/mc.20644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McKillop IH, Schmidt CM, Cahill PA, Sitzmann JV. Altered expression of mitogen-activated protein kinases in a rat model of experimental hepatocellular carcinoma. Hepatology. 1997;26:1484–1491. doi: 10.1002/hep.510260615. [DOI] [PubMed] [Google Scholar]
- 42.Schmidt CM, McKillop IH, Cahill PA, Sitzmann JV. Increased MAPK expression and activity in primary human hepatocellular carcinoma. Biochem Biophys Res Commun. 1997;236:54–58. doi: 10.1006/bbrc.1997.6840. [DOI] [PubMed] [Google Scholar]
- 43.Thoms HC, Dunlop MG, Stark LA. p38-mediated inactivation of cyclin D1/cyclin-dependent kinase 4 stimulates nucleolar translocation of RelA and apoptosis in colorectal cancer cells. Cancer Res. 2007;67:1660–1669. doi: 10.1158/0008-5472.CAN-06-1038. [DOI] [PubMed] [Google Scholar]
- 44.Casanovas O, Miro F, Estanyol JM, Itarte E, Agell N, Bachs O. Osmotic stress regulates the stability of cyclin D1 in a p38SAPK2-dependent manner. J Biol Chem. 2000;275:35091–35097. doi: 10.1074/jbc.M006324200. [DOI] [PubMed] [Google Scholar]
- 45.Bjorbaek C, El-Haschimi K, Frantz JD, Flier JS. The role of SOCS-3 in leptin signalling and leptin resistance. J Biol Chem. 1999;274:30059–30065. doi: 10.1074/jbc.274.42.30059. [DOI] [PubMed] [Google Scholar]
- 46.Bjorbaek C, Elmquist JK, Frantz JD, Shoelson SE, Flier JS. Identification of SOCS-3 as a potential mediator of central leptin resistance. Mol Cell. 1998;1:619–625. doi: 10.1016/s1097-2765(00)80062-3. [DOI] [PubMed] [Google Scholar]
- 47.Riehle KJ, Campbell JS, McMahan RS, Johnson MM, Beyer RP, Bammler TK, et al. Regulation of liver regeneration and hepatocarcinogenesis by suppressor of cytokine signalling 3. J Exp Med. 2008;205:91–103. doi: 10.1084/jem.20070820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang SF, Yeh YT, Wang SN, Hung SC, Chen WT, Huang CH, et al. SOCS-3 is associated with vascular invasion and overall survival in hepatocellular carcinoma. Pathology. 2008;40:558–563. doi: 10.1080/00313020802320432. [DOI] [PubMed] [Google Scholar]