

Abstract
Excitement with the publication of the human genome has served as catalyst for scientists to uncover the functions of specific genes. The main avenues for understanding gene function have been in behavioral genetics on one end and on the other end, molecular mouse models. Attempts to bridge these approaches have used brain imaging to conveniently link anatomical abnormalities seen in knockout/transgenic mouse models and abnormal patterns of brain activity seen in humans. Although a convenient approach, this article provides examples of challenges for imaging genetics, its application to developmental questions, and promises for future directions. Attempts to link genes, brain, and behavior using behavioral genetics, imaging genetics, and mouse models of behavior are described. Each of these approaches alone, provide limited information on gene function in complex human behavior, but together, they are forming bridges between animal models and human psychiatric disorders. Hum Brain Mapp, 2010. © 2010 Wiley‐Liss, Inc.
Keywords: imaging, genetics, development
INTRODUCTION
The publication of the human genome has set the stage for new opportunities to begin to understand gene function. The two main avenues taken to accomplish this feat include human behavioral or psychiatric genetics, where genes are linked to complex behaviors or clinical diagnosis and mouse molecular genetics, where genes can be linked to alterations in cell physiology and gene expression. The difficulty in reconciling these two avenues of research has been referred to as a “translational bottleneck” [Hyman and Fenton, 2003]. As information accumulates on the human and mouse sides of the bottleneck, there is growing demand for, and hence much opportunity, for expertise in levels of analysis, that lie between purely behavioral and purely cell biological levels of analysis. Attempts to bridge these behavioral and molecular approaches have used brain imaging to conveniently link anatomical abnormalities seen in knockout/transgenic mouse models and abnormal patterns of brain activity seen in certain patient populations. Although a convenient and rapidly growing approach (see Fig. 1), this article provides examples of challenges for imaging genetics, its application to developmental questions, and promises for future directions.
Figure 1.
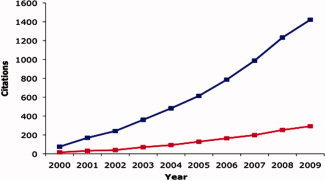
Cumulative citations for neuroimaging genetic papers (in blue) and developmental neuroimaging genetic papers (in red) over the past decade.
Technological advances in brain imaging techniques such as blood oxygenation level‐dependent functional magnetic resonance imaging (fMRI), have allowed researchers to noninvasively assay brain function within discrete brain circuits supporting specific cognitive and emotional processes. Genetic influences on behavior are mediated by the impact of their molecular and cellular effects on brain development and function. As such, genetic imagers have argued that the functional effects of candidate variants on brain structure and function may be more readily measurable than the cognitive and emotional processes supported by these substrates, and thus, functional polymorphisms in genes weakly related to behaviors or psychiatric syndromes may be more strongly related to the function of specific neural systems [Casey and Durston, 2006; Fossella and Casey, 2006]. Currently, 25,000 genes have been identified in the human and mouse genomes. Each of these genes can contain up to 100 polymorphic sites, some of which can impact the expression of those genes in a region‐dependent manner. Thus, in an individual with a brain that can be measured at the 1–3 mm3 voxel level and whose dynamic networks are capable of an infinite variety of computational states, a one‐to‐one statistical mapping of genetic variants to brain function, is challenging, but may be a convenient initial step.
The challenge of linking genes, brain, and behavior becomes even more daunting when we consider gene function in a dynamically changing system, such as the developing brain. In the excitement to use the human genome project to uncover the functions of specific genes, researchers often ignore a fundamental factor: the gradual process of ontogenetic development. Claims about gene/behavior relations are typically based on a phenotypic end‐state and grounded in the neuropsychological tradition of studying adults whose brains were fully and normally developed until a brain insult later in life. The developing brain, of course, is different in that brain regions and circuitry are not specialized at birth. Many years are required for the specialization of neural networks as a result of the complex interaction with the environment in gene expression and the resulting phenotype. Karmiloff‐Smith [ 2006] has argued that “because ontogenetic development and timing play such important roles in development, a tiny impairment in the start state of the brain of a child with a genetic disorder may affect several brain regions, some more profoundly and others more subtly, giving rise in the phenotypic end‐state to what appears to be a domain‐specific outcome.” This neuroconstructivist perspective is an important one to keep in mind when interpreting simple gene‐behavior associations in typical development and in developmental disorders.
The objective of this article is to provide a framework whereby future genetic studies can be constrained and evaluated for validity and relevance with respect to existing data with a converging methods approach. These methods include the combined use of behavioral and imaging genetics together with molecular mouse models of gene function. The consideration of genotypes as dynamic in different environmental and developmental conditions or contexts, rather than static, is underscored.
Genetic Terms and Definitions
Definition of genetic terms may be useful to a predominantly imaging audience in setting the framework for discussing challenges and potentials of genetic imaging studies, beginning with the coined phrase of imaging genetics (see Table I). Imaging Genetics refers to the use of brain imaging to evaluate phenotypic variation in brain morphometry and physiology as a function of genotypic variation [Hariri et al., 2006]. Phenotypic variation or a phenotype is an observable trait or characteristic such as behavior, development, morphology, or physiology whereas a genotype is the inherited genetic code. Not all individuals with the same genotype behave in the same way because behavior is modified by environmental and developmental contexts. Thus, we consider a phenotype to result from the genotype, environment, and development. As such genetic effects are not static, but rather dynamic in changing environments and developmental periods.
Table I.
Definition of genetic terms
| Terms | Definition |
|---|---|
| Heritability | The extent to which variation in a trait (such as cognitive performance) among members of a population is determined by inherited genetic variation. |
| Genome | The aggregate genetic information of an organism comprised of chromosomal and mitochondrial DNA in eukaryotes. The human genome consists of ∼3.2 billion nucleotides. |
| Gene | The functional unit of DNA. Generally refers to protein‐coding units but can also include non‐protein coding regulatory units. |
| Locus | A specific location in the genome. Can be a specific nucleotide or a larger region such a gene or cytogenetic band. |
| Polymorphism | Heritable trait that occurs in multiple forms in populations. Usually refers to variants in DNA but can refer to phenotypic traits. Traditionally refers to genetic variant that occurs in more than 1% of a population with mutation used for more rare variants. |
| Allele | One form of a polymorphic trait. |
| Genotype | The composition of a particular region of the genome. Generally refers to the combination of the maternal and paternal alleles of a polymorphism. |
| Phenotype | An observable trait that results from the combined effects of genetic and environmental factors. |
| Endophenotype | An endpoint for genetic association or linkage studies that is between the genome and clinically‐defined phenotypes. |
| Haplotype | A series of alleles that occur on the same chromosome. Usually inferred statistically from genotype data at multiple loci on a chromosome but can be determined through molecular cloning. |
| Linkage disequilibrium | A significant deviation from random assortment between the alleles of polymorphisms at two or more loci. |
| SNP | Single nucleotide polymorphism. Results from a point mutation during DNA replication which is almost always biallelic; the most common category of polymorphism. |
| VNTR | Variable number of tandem repeats polymorphism. Also referred to as a length polymorphism. Alleles of a VNTR differ by the number of identical or similar repeat elements. |
| GWAS | Genome wide association study. A genetic mapping study is one in which the entire genome is interrogated for significant association between individual polymorphisms and a target phenotype. Currently multiple genomic technologies allow screening the entire human genome at 500,000–1,000,000 SNP's using a single “chip” for each subject. |
Most genetic studies have been designed, not from a whole‐genome perspective [Fossella and Casey, 2006], but rather from having selected from the genome, a candidate gene. The candidate gene study design is appropriate when substantial amounts of converging evidence suggest that a particular gene contributes to heritable variation in behavior or risk for psychopathology. Heritability is the extent to which variation in a trait among members of a population is determined by inherited genetic variation.
For candidate gene association studies, the individual nucleotides that vary from one individual to another are the most important attribute. These variable sites arise from errors in genome replication, which in humans, are rare, but occurs often enough in our genome of three billion nucleotides, so that approximately 100 base‐pair changes accumulate per generation per genome. Most of these minor base pair changes are lost over long time scales as generations come and go. However, some of these changes continue to be passed on and even spread across populations. These ancient deoxyribonucleic acid (DNA) changes, known as polymorphisms or single nucleotide polymorphisms (SNPs) are presently found in slightly more than 1 out of every 1,000 nucleotides in the genome. These changes then lead to an enormous amount of DNA sequence variability across human populations.
Mouse models have been developed to relate changes in gene expression to behavior. One of the most common approaches is to look at relationships between gene dosage and behavior. By breeding knockout mice (−/−) with wild‐type mice (+/+), heterozygote animals (−/+) with one working copy of the gene of interest are produced. Often, such changes in the amount of gene activity lead to variations from the wild type. Knockout mice contain the same, artificially introduced mutation in every cell, abolishing the activity of a preselected gene. The resulting mutant phenotype (e.g., behavior or anatomy) may provide some indication of the gene's role in the mouse, and ultimately, in humans. Knockout mice are produced by a technique called gene targeting that involves the replacement of one gene sequence, with a related sequence that has been modified to contain a mutation.
A transgenic mouse contains additional, artificially‐introduced genetic material in every cell that can lead to a gain of function. The extra genetic material is often described as foreign DNA, but it can come from any source, including another mouse. A knock‐in involves the insertion of a protein coding DNA sequence at a particular locus in an organism's genome. The difference between knock‐in technology and transgenic technology is that a knock‐in involves a gene inserted into a specific locus, and is a “targeted” insertion. Genes are silenced or “knocked down” by short pieces of double‐stranded RNA. Viral vectors can be used to insert interfering RNA into stem cells or neurons to modify the activity of genes in selected tissues.
Challenges for Genetic Studies
With a definition of terms, challenges of and potential solutions for future genetic studies can more easily be discussed. The challenges include imprecise phenotypes, lack of known functionality of genetic variation, underpowered studies, and stratification effects.
Imprecise phenotype
Although the major psychiatric and developmental disorders display a substantial heritable component, very few genetic associations to these phenotypes have proven to be reliable. The majority of allelic associations with neurobehavioral phenotypes are not consistently replicated. For instance, a common variable number of tendem repeats (VNTR) polymorphism in the gene for the serotonin transporter (the serotonin transporter‐linked polymorphic region or 5‐HTTLPR) has been heavily studied and associated with anxious personality traits, amygdala response to emotional stimuli, and susceptibility to depression in the face of environmental stressors. However, a recent metaanalysis of 24 studies (comprised of more than 3,000 subjects) of the association between the serotonin transporter‐linked polymorphic region and anxiety‐related personality traits found “that the effect, if present, is small [Munafo et al., 2005].” A more recent metaanalysis of the role of the 5‐HTTLPR in gene–environment interactions for risk of depression has reported no evidence of association [Risch et al., 2009]. Genetic risk is likely distributed across many allelic variants in psychiatric illness, thus the effect size of any single risk factor for disorders will be small and difficult to reliably identify.
Endophenotypes are heritable, distinct endpoints in biology such as anatomy, biochemistry, and behavior that reflect discrete components of pathophysiologic processes. These refined phenotypes have been proposed as attractive targets for human genetic studies because they are less biologically complex than disease phenotypes and can be more objectively and reliably ascertained than categorical disorders. These attributes suggest that genetic correlation with a specific endophenotype should prove more reliable than associations with disease phenotype, but to date, this is often not the case.
To ensure the utility of endophenotypes, candidate gene studies must be focused on validated endophenotypes that are comprised of basic biological processes, relate more closely to the biology of the candidate gene, and are more precisely measured than categorical disorder phenotypes (see Fig. 2). Specifically, endophenotypes must fulfill several criteria: (1) reflect a biological process that is a component of the more complex disorder phenotype; (2) be more biologically simple than the disorder phenotype to ensure that the effect size of any particular risk factor is relatively large; and (3) the biology of the endophenotype must be understood well enough that it can be related to specific candidate risk factors including genetic, environmental, and developmental ones. These criteria are particularly important for functional imaging genetic studies where the findings are dependent upon validity of the behavioral activation paradigm.
Figure 2.
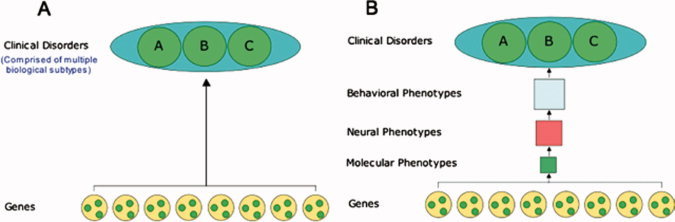
Phenomenological (A) versus biological (B) approach to behavioral genetics [Casey et al., 2009].
Functionality over replication of genetic effects
It is useful when investigating a candidate gene to understand how to classify variable sites according to functionality and frequency. Some variable sites lie within a gene and affect the structure of the protein (e.g., brain derived neurotrophic factor or BDNF). Other sites lie outside the coding sequence, and yet are able to influence the amount of expression of the encoded protein (e.g., 5‐HTTLPR). Still other sites lie inside or outside of a protein‐coding region, and yet, have no effect at all on the encoded protein structure or expression.
Unfortunately, many gene association studies are carried out using such nonfunctional polymorphic sites. A case in point presented by Fossella et al. [ 2006] provides a cautionary example of how simple gene, brain and behavior associations from genetic imaging studies can be over interpreted. They examined a well‐studied genetic polymorphism referred to as the TaqIA restriction fragment length polymorphism (RFLP), which resides downstream from the dopamine D2 receptor (DRD2) gene. A number of electrophysiological and PET imaging studies have been performed on genetic variants of DRD2. Only recently has it been shown that this polymorphism lies 10 kb downstream of the coding region of DRD2 and occurs in Exon 8 of the novel kinase gene, named ankyrin repeat and kinase domain containing 1 (ANKK1) gene, resulting in a glutamate‐to‐lysine (E713K) substitution within the 11th ankyrin repeat. As yet, expression of the ANKK1 gene has not been detected in the developing or adult mammalian brain. Thus, it is surprising, that so many gene association studies have been reported for a polymorphism for this gene. Fossella et al. show an association between this genetic variant and patterns of brain activity during attentional control studies.
Clearly future studies will be needed to understand how a polymorphism that lies downstream of the coding region of gene and in a gene as yet, undetectable in the developing and adult mammalian brain, can impact brain and behavior. One explanation for the existing literature and their finding would be that the TaqIA polymorphism in ANKK1 is in linkage disequilibrium or correlation with upstream polymorphisms in DRD2 and lies within a haplotype block, a series of alleles at multiple loci along a chromosome that spans the overlap between the two genes. This example, however, underscores the caution that should be taken in making simple associations between brain activity and a polymorphism in terms of the gene–behavior association. The need to examine functionality, despite replication, is thus warranted.
Overestimation of genotypic effects
One‐to‐one statistical mappings of genetic variants to brain function or behavior is limited in the context of thousands of genes with up to 100 variable sites within each of these genes in an individual with a brain whose dynamic networks are capable of an infinite variety of computational states. The problems in gaining sufficient power or effect sizes to detect genotypic effects on behavior or on risk for psychopathology would seem insurmountable. Nonetheless, initial studies have suggested as much as 20% of phenotypic variance could be explained by a single genotype. Hariri et al. provide a cautionary note in this regard.
In 2002, Hariri et al. used BOLD fMRI to evaluate the impact of the 5‐HTTLPR on the physiological response of the human amygdala to biologically salient environmental stimuli. In this initial study, healthy adult volunteers from two independent cohorts (n = 14 in each sample, 7 per group) were divided into equal groups based on their 5‐HTTLPR genotype, with the groups matched for age, gender, IQ, and task performance. During BOLD fMRI scanning, the subjects performed a simple perceptual processing task involving the matching of fearful and angry human facial expressions [Hariri et al., 2000]. The fMRI data showed that subjects carrying the low expressing 5‐HTTLPR S allele exhibited significantly increased amygdala activity, as indexed by the BOLD signal, in comparison with subjects homozygous for the L allele [Hariri et al., 2002]. In fact, the difference in amygdala activity between 5‐HTTLPR genotype groups in this study was nearly fivefold, accounting for 20% of the total variance in the amygdala response during this experiment, an effect size greater than any previously reported behavioral associations. This initial finding suggested that the increased anxiety and fearfulness associated with individuals possessing the 5‐HTTLPR S allele may reflect the hyperresponsiveness of their amygdala to relevant environmental stimuli.
A subsequent metaanalysis by Hariri and coworkers [Munafo et al., 2008] showed that the magnitude of the reported associations between amygdala activation and the serotonin transporter gene linked polymorphic region (5‐HTTLPR) was less substantial. Although their analysis provided support for the association of the 5‐HTTLPR polymorphism and amygdala activation, it also suggested that most studies lack sufficient statistical power to make such claims. Increasing the sample sizes of future imaging genetics studies will allow a more accurate characterization of any true effect size and afford adequate power to examine the impact of multiple polymorphisms that likely work in concert to affect gene function and, in turn, bias neural processes mediating emotional related behavior.
Stratification effects
One of the most critical issues in designing candidate gene association studies is a consideration of the frequency of the polymorphic alleles under investigation. Allele frequencies can be extremely rare and vary by population. For example, the frequency of the E4 allele of Apolipoprotein E (APOE) ranges from 5% in Taiwan and Sardinia to 40% in Pygmies [Corbo and Scacchi, 1999; Siest et al., 1995]. It is desirable to choose a polymorphism whose alternate alleles exist at relatively high frequencies within and across populations. Choosing an allele that is present in only 5% of the population creates practical difficulties, from a recruitment perspective, but also can be problematic to interpret. Because allele frequencies can vary by population, it is important to monitor ancestral heritage and population admixture in study samples to avoid spurious associations between a polymorphism and a trait that might simply relate to different prevalence of that polymorphism in different ethnic groups in the sample.
Ethnic heterogeneity is an important concern in genetic association studies where subjects are categorized by phenotype and associations are then identified through statistical differences in genotype distributions. In such a case, phenotypically neutral polymorphisms may appear associated due to different allele frequencies in ethnic groups that are unevenly represented in cases and controls or across a quantitative variable. The problem of population stratification is analogous to that encountered when there is uneven representation of genders across genotype groups in a sample which may result in apparent differences by genotype, that are actually due to the effects of gender. Uneven representation of ethnicities can have the same effect (effects due to ethnicity rather than genotype).
For example, Keen‐Kim et al. [ 2006] recently suggested that the association between a rare non‐coding variant in Slitrk1 with Tourette Syndrome may be due to population stratification effects. The association was reported the year before [Abelson et al., 2005] because it was seen in 2 out of 348 Tourette Syndrome chromosomes (0.6%) but not once in a sample of nearly 5,000 control chromosomes with a significance of P = 0.0056. The following year, Keen‐Kim et al. showed that the prevalence of this mutation was higher in Ashkenazi Jews than other ethnic groups (0.7% in patient cases, 0.8% in parents of cases, and 0.2% in controls). Using a family‐based design they found that when a parent of a child with Tourette Syndrome possessed the risk allele they were almost as likely to pass on the nonrisk allele (two of five transmissions) to their affected child as the risk allele (three of five transmissions) meaning there was no association with Tourette Syndrome in that study. Further, they suggest that the previous report had more Ashkenazi Jews in patient cases than controls. This example demonstrates the potential risks of population stratification and also points to a benefit of family‐based designs in that they automatically control for population stratification.
Stratification in population‐based studies can be identified and adjusted for using genotype information from unlinked ancestry informative markers. Other than family‐based designs, genetic studies where the design employed is to select subjects based on genotype—rather than phenotype—and then test for phenotypic differences, are not sensitive to classic stratification error. Moreover matching genotypic groups on ethnicity, as well as other factors such as gender and age further prevent spurious results. Yet, few genetic imaging studies have matched on ethnic diversity in their samples or had sufficient power to statistically control for such effects.
Similar concerns arise in genetically modified mice. Specifically, it has been shown that there are strain differences in the efficacy of standard animal behavior protocols [Balogh and Wehner, 2003; Bolivar et al., 2001; Brinks et al., 2008; Waddell et al., 2004]. These differences may be due to strain differences in pain sensitivity, learning, or other variables, but genetic background effects may lead to spurious results. Studies that do not account for ethnic variability in humans or genetic background in mice cannot make strong about genotypic effects.
New Directions for Genetic Research
Attempts to bridge behavioral and molecular genetics through brain imaging has provided a convenient link between abnormal patterns of brain activity seen in certain patient populations and anatomical abnormalities seen in knockout/transgenic mouse models. Genetically modified mice provide useful model systems for testing the role of candidate genes in behavior and imaging studies provide neuroanatomical evidence to validate cross‐species translation. The extent to which such genetic manipulations in the mouse and the resulting phenotype can be translated across species, from mouse to human, is beginning to be assessed more directly. A new direction for genetic research is to exploit behavioral paradigms being used with mice, and adapt them for use with humans in the imaging environment. Such an approach provides a direct move toward translating the function of genes in the context of human behavior. In highlighting the promise of imaging genetics, we provide a concrete example of how parallel human and mouse genetic studies can address many of the challenges of genetic studies.
We describe research that focuses on the impact of a polymorphism in the brain‐derived neurotrophic factor (BDNF) gene. BDNF is a member of the neurotrophins a unique family of polypeptide growth factors that influence differentiation and survival of neurons in the developing nervous system. In adults, BDNF is important in regulating synaptic plasticity and connectivity in the brain. Recently, a common single nucleotide polymorphism in the human BDNF gene, resulting in a valine (Met) to methionine (Met) substitution in the prodomain of the peptide (Val66Met), has been shown to lead to memory impairment and risk for psychiatric illness. An understanding of how this naturally occurring polymorphism affects behavior and neuroanatomy is an important first step in linking genetic alterations in the neurotrophin system to definable biological outcomes in humans.
Functionality
BDNF Val66Met is common in human populations with a prevalence of 20–30% [Shimizu et al., 2004] and because it codes for an amino acid substitution has a high likelihood of affecting the biological properties of the BDNF peptide. Indeed, it has been demonstrated that the Met allele leads to reduced activity‐dependent secretion of BDNF from hippocampal neurons in culture [Egan et al., 2003; see Fig. 3A]. This effect is due to a reduced affinity of the variant (Met) BDNF for sortilin an intracellular trafficking molecule [Chen et al., 2005].
Figure 3.
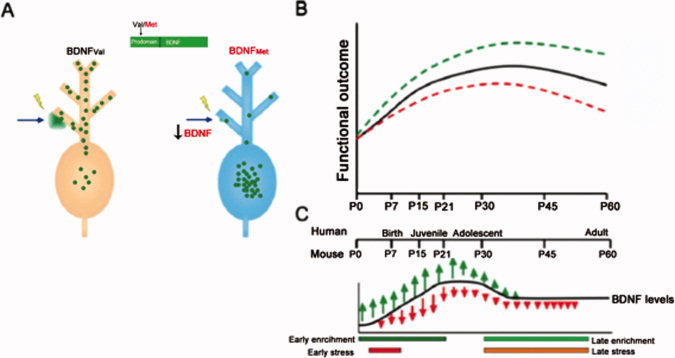
Model of impact of BDNF across development. (A) The genetic variant BDNF Val66Met leads to an amino acid substitution in the BDNF prodomain (Val to Met at position 66) that results in decreased activity‐dependent secretion of BDNF from neurons. Thus, this trafficking defect leads to a decrease in the availability of biologically active BDNF. (B) This model predicts that BDNF levels will have different functional consequences across development. As the variant BDNF (Val66Met) has decreased secretion throughout this period, we anticipate that there will be functional deficits, evident even in childhood, but (C) these deficits will become diminished by adolescence when BDNF levels peak. In addition, BDNF levels will be modulated by environmental stressors. Carriers of the Met allele will have decreased secretion and less neurotrophic support for plasticity and change, whereas Val allele carries will show greater change, including both positive and negative effects on hippocampal structure and function, but potentially greater neurotrophic support for plasticity and resilience once a stressor is removed [Casey et al., 2009].
The BDNFMet allele has been associated with impairment in select forms of learning and memory [Egan et al., 2003] and susceptibility to psychiatric disorders [Neves‐Pereira et al., 2002; Ribases et al., 2003, 2004; Sen et al., 2003; Sklar et al., 2002]. Given the established role of BDNF in promoting learning and memory [Desai et al., 1999; Korte et al., 1995; Patterson et al., 1996], it is likely that impaired BDNF secretion, due to expression of the BDNFMet allele, may have pleiotropic effects (e.g., a single gene impacting multiple phenotypic traits) in a variety of BDNF‐dependent processes.
Recently, a unique inbred genetic knock‐in mouse strain was developed [Chen et al., 2006] that expresses the variant BDNF allele to recapitulate the specific phenotypic properties of the human polymorphism in vivo. All inbred mouse strains contain a Valine 66 residue in BDNF. The BDNFMet mouse is a transgenic knock‐in of a methionine residue at this position that mimics the human polymorphism. This model is unique in that it is the only animal model that fully recapitulates the established phenotypic effects of a common human polymorphism expressed in the brain. Unlike traditional transgenic mouse models which alter the quantitative expression of targeted genes throughout development or at selected times, this model introduces the single polymorphic amino acid into the murine genome, thereby providing a precise physiologic model of the polymorphic effect of human BDNF Val66Met. Such testable mechanistic approaches cannot be applied to other frequent polymorphisms related to behavior. For example, the 5‐HTTLPR is postulated to be a regulatory polymorphism but its activity has not been consistently identified. Furthermore, the 5‐HTTLPR genetic alteration cannot be fully recapitulated in transgenic mice because the regulatory element that is polymorphic in humans does not exist in nonprimate species [Lesch et al., 1997]. The mouse model of BDNF Val66Met has been validated by studies that have found that animals carrying the Met allele manifested phenotypes (hippocampal size and hippocampal‐dependent learning) that matched differences in humans expressing the BDNFMet allele, as compared to individuals with the Val/Val genotype [Chen et al., 2006].
Constrained model of genotypic effects
We have developed a model with testable hypotheses for how gene‐ or environment‐related alterations in BDNF levels will have a significant impact on behavioral and neuroanatomic changes that vary with age. Such an approach can move the field away from simplistic notions of risk alleles, recognizing that an allele may be a risk factor during one period of development and a protective factor during another. Because the variant BDNFMet allele shows decreased regulated secretion, we predict that there will be functional deficits or biases in BDNF dependent forms of learning when physiologic levels of BDNF are low (Fig. 3B,C). However, when BDNF levels peak [e.g., during adolescence, Katoh‐Semba et al., 1997] this trafficking deficit may yield only minor differences in these measures in stable enviornments. During periods of increased physiologic expression of BDNF, the lower secretion conferred by the BDNFMet allele may actually be protective and lead to risk for individuals in adolescence without this allele [e.g., BDNFVal/Val in substance abuse; see meta‐analysis by Gratacos et al., 2007].
This model also encompasses nongenetic factors. Early environmental risk factors including physiological or psychological stress result in decreased neurotrophic support to certain BDNF‐rich regions like the hippocampus [Smith et al., 1995]. The additional deficit in neurotrophic support in carriers of the Met allele may result in increased vulnerability to stress, and thus put them at greater risk for psychiatric disorders (e.g., anxiety, depression, and schizophrenia) that have been associated with stress. During other developmental windows when BDNF levels are high, carriers of the Val allele may be at greater risk for other psychiatric disorders given that stress can increase BDNF in the amygdala and ventral striatum, areas implicated in bipolar disorder [e.g., Geller et al., 2004] and substance abuse [Liu, 2005; Matsushita et al., 2004]. Thus, it is important to consider changes in the level of BDNF across development and the opposing effects that stress has on BDNF levels in brain regions that support very different forms of learning.
Promise for development
This model distinguishes itself by underscoring the importance of development in examination of genetic effects on behavior. First, BDNF is a molecule that is essential for developmental processes including, neuronal plasticity [Barde et al., 1987; Bramham and Messaoudi, 2005; Leibrock et al., 1989; Liao et al., 2007; Lu, 2003; Rattiner et al., 2005; Thoenen, 1995; Tongiorgi et al., 2006; Yamamoto and Hanamura, 2005]; regulation of both short‐term synaptic function and long‐term activity‐dependent synaptic consolidation [Barco et al., 2005; Black, 1999; Katz and Shatz, 1996; Lohof et al., 1993; Lu and Chow, 1999; McAllister et al., 1999; Patterson et al., 1996; Poo, 2001; Thoenen, 1995]; potentiation of synaptic transmission [Kang and Schuman, 1995; Levine et al., 1995; Lohof et al., 1993]; modulation of long‐term potentiation (LTP) in vitro and in vivo [Korte et al., 1995; Messaoudi et al., 2002; Patterson et al., 1996]; and induction of morphological changes in dendritic spines [Gomes et al., 2006; McAllister et al., 1995]. Thus, BDNF has a role in (1) synaptic plasticity; (2) inducing changes in synaptic morphology; and (3) mediating cell survival and cell proliferation during development. These functions serve to underscore the importance of considering BDNF in any neurodevelopmental disorder of learning.
Second, BDNF availability changes across development (Fig. 1B,C). Although these changes have been shown to differ by region [Hofer et al., 1990; Katoh‐Semba et al., 1997; Maisonpierre et al., 1990; Webster et al., 2006], rodent studies suggest that changes in BDNF levels across development approximate an inverted U‐shape function [Ivanova and Beyer, 2001; Silhol et al., 2005]. In humans, BDNF mRNA levels in cortical regions increase approximately one‐third from infancy to adulthood. They are relatively low during infancy and childhood, peak during young adulthood, and are maintained at a constant level throughout adulthood. The increase in BDNF at this critical time in human development may have important implications for the etiology and treatment of the severe mental disorders that tend to present during this time [Webster et al., 2002]. The BDNF Val66Met mouse model is able to recapitulate this regional and temporal complexity as the single nucleotide polymorphism occurs in the protein coding sequence and leaves the regulatory elements of the gene unaffected, thus maintaining the normal regional and temporal expression of this gene.
Precision of (endo)phenotype
Genetically influenced forms of learning that lie at the core of neurodevelopmental disorders include those that capture the difficulties some individuals have in: (1) recognizing signals of safety or danger (cued learning); and (2) learning to adjust behavior when actual associations no longer exist (extinction). Unlike disease states, the tasks that examine these types of learning can be assessed equivalently in typically and atypically developing humans and mice. Although most studies have emphasized the role of BDNF in learning and memory processes supported by the hippocampus, high levels of BDNF mRNA and protein are expressed in the amygdala [Conner et al., 1997; Yan et al., 1997] suggesting another important potential site for BNDF‐mediated plasticity. In studies focusing on the hippocampus, BDNF has been shown to facilitate long term potentiation (LTP) at hippocampal CA1 synapses [Figurov et al., 1996; Korte et al., 1995; Patterson et al., 1996] and BDNF mRNA levels have been found to increase following induction of LTP [Barco et al., 2005; Bramham et al., 1996; Castren et al., 1993; Pang and Lu, 2004; Patterson et al., 1992, 1996; Radecki et al., 2005; Zakharenko et al., 2003]. The activity‐dependent secretion of BDNF enhances the molecular mechanisms of synaptic restructuring needed to support LTP. We have shown [Chen et al., 2005] that the Val66Met mutation in the BDNF gene leads to a decrease in this regulated secretion of BDNF, suggesting that carriers of this allele would have compromised BDNF‐dependent synaptic modulation. In humans, Val/Met individuals have repeatedly been shown to have a smaller hippocampal volume relative to individuals who are homozygous for the Val allele (Val/Val) [Bueller et al., 2006; Pezawas et al., 2004; Szeszko et al., 2005].
In this context, we provide data that focus on simple measures that reflect adaptation to environmental change/stress (e.g., fear conditioning) and that appear to lie at the very core of a number of clinical disorders [Charney and Manji, 2004; Duman et al., 1997; Nestler et al., 2002; Pine, 2007]. Importantly, these measures can be tested across species and throughout development and have known underlying biological substrates. Using such measures across development and under varying degrees of stress, will ultimately allow us to examine vulnerability and protection of each BDNF allele (Val and Met), in an attempt to understand gene X environment interactions across development. Mouse and human data are presented to illustrate this multilevel approach for understanding gene function. The objective of this study was to test if the Val66Met genotype could impact extinction learning in our mouse model, and if such findings could be generalized to human populations. For preliminary gene X development and gene X environment findings see Casey et al., [ 2009].
Cross species validation of genetic results
We examined the impact of the variant BDNF on classic fear conditioning and extinction paradigms [Soliman et al., 2010]. Approximately 70 mice and 70 humans were tested. The mice include 17 BDNFVal/Val, 33 BDNFVal/Met, and 18 BDNFMet/Met. The human sample included 36 Met allele carriers (31 BDNFVal/Met and 5 BDNFMet/Met) and 36 nonMet allele carriers group‐matched on age, gender, and ethnic background. To avoid spurious allelic associations, we balanced demographic factors, including age, gender, and ethnicity across genotype categories [Soliman et al., 2010; Supporting Information Table S1]. We also performed ethnicity‐specific analyses and found that the effect of the Met allele on extinction and conditioning, as measured by change in SCR with time, was not driven by any single ethnic group (extinction: F(3,64) = 0.32, P < 0.81) or (conditioning: F(3,64) = 0.69, P < 0.56]. Fear conditioning consisted of pairing a neutral cue with an unconditioned aversive stimulus until the cue itself took on properties of the unconditioned stimulus (US) of an impending aversive event. The extinction procedure consisted of repeated presentations of the cue (i.e., conditioned stimulus or CS) alone.
There were no effects of BDNF genotype on fear conditioning in mice or humans as measured by freezing behavior to the conditioned stimulus in the mice (F(2,65) = 1.58, P < 0.22) and by skin conductance response in humans to the cue predicting the aversive stimulus relative to a neutral cue (F(1,70) = 0.67, P < 0.42). However, both the mice and humans showed slower extinction in Met allele carriers than in nonMet allele carriers as shown in Figure 4A,B below. Moreover, human functional magnetic resonance imaging data provide neuroanatomical validation of the cross‐species translation. Specifically, we find alterations in frontoamygdala circuitry, known to support fear conditioning and extinction in previous rodent [LeDoux, 2000; Milad and Quirk, 2002; Myers and Davis, 2002; Quirk et al., 2003] and human [Delgado et al., 2008; Gottfried and Dolan 2004; Kalisch et al., 2006; LaBar et al., 1998; Phelps et al., 2004; Schiller et al., 2008] studies, as a function of BDNF genotype. Met allele carriers show less ventromedial prefrontal cortical (vmPFC) activity during extinction relative to nonMet allele carriers (Fig. 4C), but greater amygdala activity relative to nonMet allele carriers (Fig. 4D). These findings suggest that cortical regions essential for extinction in animals and humans [Milad and Quirk, 2002; Milad et al., 2007; Phelps et al., 2004] are less responsive in Met allele carriers during extinction. Morover, amygdala recruitment, which should show diminished activity during the extinction [Phelps et al., 2004] remains elevated in Met allele carriers.
Figure 4.
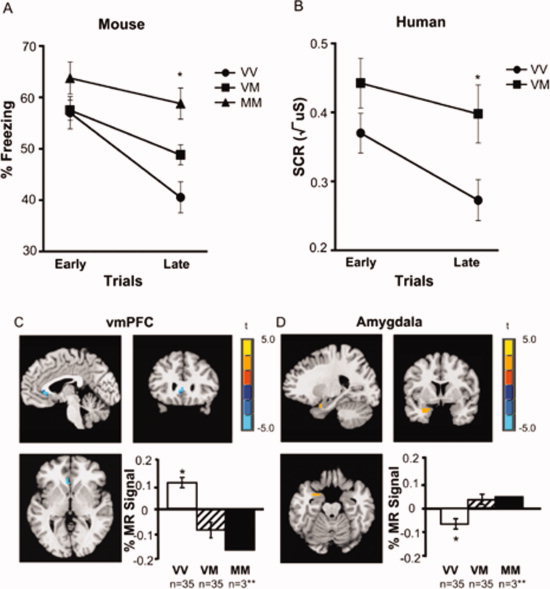
Altered behavior and neural circuitry underlying extinction in mice and humans with BDNF Val66Met. Impaired extinction in Met allele carriers (Val/Met and Met/Met) as a function of time in 68 mice (A) and 72 humans (B) as indexed by percent time freezing in mice and skin conductance response (SCR) in humans to the conditioned stimulus when it was no longer paired with the aversive stimulus. (C) Brain activity as indexed by percent change in MR signal during extinction in the ventromedial prefrontal cortex (vmPFC) by genotype (xyz = −4, 24, 3), with Met allele carriers having significantly less activity than Val/Val homozygotes [VM < VV = blue], image threshold P < 0.05, corrected. (D) Genotypic differences in left amygdala activity during extinction (xyz = −25, 2, −20) in 70 humans, with Met allele carriers having significantly greater activity than Val/Val homozygotes [VM > VV = orange], image threshold P < 0.05, corrected. *P < 0.05. **MM were included in the analysis with VM, but plotted separately to see dose response. All results are presented as a mean ± SEM. VV = Val/Val; VM = Val/Met; MM = Met/Met [Soliman et al., 2010].
The mouse model provides the ability to directly test for dose‐dependent effects of the Met allele (Fig. 4A) that is problematic in human studies given the rarity of humans homozygous for the Met allele. Moreover, the mouse model provides the ability to test the effects of the Met allele in both a controlled genetic and environmental background not feasible in humans. Thus, any behavioral differences observed in the mouse can be reliably assigned to the effects of the Val66Met polymorphism. Together with the human behavioral findings, these data provide confidence of the effects of BDNF Val66Met across species. With the added human imaging data, the effects are shown to be biologically valid as they directly map onto known circuits involved in fear conditioning and extinction.
The findings are exciting as they provide an example of bridging human behavioral and imaging genetics with a molecular mouse model. Each of these approaches alone, provide limited information on gene function in complex human behavior, but together, they are forming bridges between animal models and human psychiatric disorders. Specifically, the findings of impaired extinction in mice and humans with the BDNF Met allele, suggests a role in anxiety disorders showing impaired learning of cues that signal safety versus threat, and in the efficacy of treatments that rely on extinction mechanisms such as exposure therapy.
Mouse strain effects
As noted, previous reports have reported strain differences in the efficacy of standard fear conditioning protocols and the rate of extinction Balogh and Wehner, 2003; Bolivar et al., 2001; Brinks et al., 2008; Waddell et al., 2004]. These differences may be due to strain differences in pain sensitivity, learning, or other variables. The results above are based on data collected from C57BL/6J mice. To directly compare the contribution of the Val66Met polymorphism to fear extinction across genetic backgrounds, we used identical fear conditioning and extinction paradigms in a sample of Val66Met Swiss Webster mice. Fear extinction occurred more rapidly in Swiss Webster mice compared to C57BL/6J mice. Despite these differences, the Swiss Webster homozygous BDNF Met allele mice, like the C57BL/6J Val66Met mice, showed slower extinction to the condition stimulus to than wildtype (Val/Val) mice [see Soliman et al., 2010; Supporting Information].
Future genetic approaches
Although the majority of imaging genetic imaging studies have focused on candidate genes, comparisons of gene expression between individuals or experimental groups has been greatly facilitated in the past decade by the introduction of genome‐wide microarrays or “gene‐chips.” A genome‐wide association study is defined as any study of genetic variation across the entire human genome that is designed to identify genetic associations with observable traits or the presence or absence of a disease or condition. Whole genome information, when combined with clinical and other phenotype data, offers the potential for increased understanding of basic biological processes and those affecting human health, improvement in the prediction of disease and treatment, and ultimately the realization of the promise of personalized medicine.
A potential use for combining imaging genetics with genome‐wide microarray methods is to use a biological signature in much the same way that behavioral signatures such as social responsiveness have been used in genetic research on autism [Duvall et al., 2007]. A neural signature specific to a disorder such as functional coupling between ventromedial prefrontal cortex and amygdala may serve to identify risk genes involved in distinct disorders but with a common pathway in frontolimbic circuitry. Numerous cross‐site imaging initiatives are underway to begin to provide sufficiently large samples of scans needed to pursue this approach.
SUMMARY
With the continued excitement of the publication of the human genome, scientists will no doubt continue to uncover the functions of specific genes. These discoveries will be augmented by connecting major avenues of genetic research across disciplines, using different approaches that bridge animal models with human behavior and evolving imaging methods with genetic technologies. These approaches will provide a more unified understanding of neural mechanisms involved in human behavior and its disruption in psychopathologies. Such an approach may open up new avenues for therapeutic intervention for clinical populations at the pharmacological, genetic and behavioral levels and identify windows of development that may be most optimal to treatment.
REFERENCES
- Abelson JF, Kwan KY, O'Roak BJ, Baek DY, Stillman AA, Morgan TM, Mathews CA, Pauls DL, Rasin MR, Gunel M, et al. ( 2005): Sequence variants in SLITRK1 are associated with Tourette's syndrome. Science 310: 317–320. [DOI] [PubMed] [Google Scholar]
- Balogh SA, Wehner JM ( 2003): Inbred mouse strain differences in the establishment of long‐term fear memory. Behav Brain Res 140: 97–106. [DOI] [PubMed] [Google Scholar]
- Barco A, Patterson S, Alarcon JM, Gromova P, Mata‐Roig M, Morozov A, Kandel ER ( 2005): Gene expression profiling of facilitated L‐LTP in VP16‐CREB mice reveals that BDNF is critical for the maintenance of LTP and its synaptic capture. Neuron 48: 123–137. [DOI] [PubMed] [Google Scholar]
- Barde YA, Davies AM, Johnson JE, Lindsay RM, Thoenen H ( 1987): Brain derived neurotrophic factor. Prog Brain Res 71: 185–189. [DOI] [PubMed] [Google Scholar]
- Black IB ( 1999): Trophic regulation of synaptic plasticity. J Neurobiol 41: 108–118. [PubMed] [Google Scholar]
- Bolivar VJ, Pooler O, Flaherty L ( 2001): Inbred strain variation in contextual and cued fear conditioning behavior. Mamm Genome 12: 651–656. [DOI] [PubMed] [Google Scholar]
- Bramham CR, Messaoudi E ( 2005): BDNF function in adult synaptic plasticity: The synaptic consolidation hypothesis. Prog Neurobiol 76: 99–125. [DOI] [PubMed] [Google Scholar]
- Bramham CR, Southard T, Sarvey JM, Herkenham M, Brady LS ( 1996): Unilateral LTP triggers bilateral increases in hippocampal neurotrophin and trk receptor mRNA expression in behaving rats: Evidence for interhemispheric communication. J Comp Neurol 368: 371–382. [DOI] [PubMed] [Google Scholar]
- Brinks V, de Kloet ER, Oitzl MS ( 2008): Strain specific fear behaviour and glucocorticoid response to aversive events: Modelling PTSD in mice. Prog Brain Res 167: 257–261. [DOI] [PubMed] [Google Scholar]
- Bueller JA, Aftab M, Sen S, Gomez‐Hassan D, Burmeister M, Zubieta JK ( 2006): BDNF Val66Met allele is associated with reduced hippocampal volume in healthy subjects. Biol Psychiatry 59: 812–815. [DOI] [PubMed] [Google Scholar]
- Casey BJ, Durston S ( 2006): From behavior to cognition to the brain and back: What have we learned from functional imaging studies of attention deficit hyperactivity disorder? Am J Psychiatry 163: 957–960. [DOI] [PubMed] [Google Scholar]
- Casey BJ, Glatt CE, Tottenham N, Soliman F, Bath K, Amso D, Altemus M, Pattwell S, Jones R, Levita L, et al. ( 2009): Brain‐derived neurotrophic factor as a model system for examining gene by environment interactions across development. Neuroscience 164: 108–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castren E, Pitkanen M, Sirvio J, Parsadanian A, Lindholm D, Thoenen H, Riekkinen PJ ( 1993): The induction of LTP increases BDNF and NGF mRNA but decreases NT‐3 mRNA in the dentate gyrus. Neuroreport 4: 895–898. [DOI] [PubMed] [Google Scholar]
- Charney DS, Manji HK ( 2004): Life stress, genes, and depression: Multiple pathways lead to increased risk and new opportunities for intervention. Sci STKE 2004:re5. [DOI] [PubMed] [Google Scholar]
- Chen ZY, Ieraci A, Teng H, Dall H, Meng CX, Herrera DG, Nykjaer A, Hempstead BL, Lee FS ( 2005): Sortilin controls intracellular sorting of brain‐derived neurotrophic factor to the regulated secretory pathway. J Neurosci 25: 6156–6166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZY, Jing D, Bath KG, Ieraci A, Khan T, Siao CJ, Herrera DG, Toth M, Yang C, McEwen BS, et al. ( 2006): Genetic variant BDNF (Val66Met) polymorphism alters anxiety‐related behavior. Science 314: 140–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conner JM, Lauterborn JC, Yan Q, Gall CM, Varon S ( 1997): Distribution of brain‐derived neurotrophic factor (BDNF) protein and mRNA in the normal adult rat CNS: Evidence for anterograde axonal transport. J Neurosci 17: 2295–2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbo RM, Scacchi R ( 1999): Apolipoprotein E (APOE) allele distribution in the world. Is APOE*4 a “thrifty” allele? Ann Hum Genet 63 ( Part 4): 301–310. [DOI] [PubMed] [Google Scholar]
- Delgado MR, Nearing KI, Ledoux JE, Phelps EA ( 2008): Neural circuitry underlying the regulation of conditioned fear and its relation to extinction. Neuron 59: 829–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai NS, Rutherford LC, Turrigiano GG ( 1999): BDNF regulates the intrinsic excitability of cortical neurons. Learn Mem 6: 284–291. [PMC free article] [PubMed] [Google Scholar]
- Duman RS, Heninger GR, Nestler EJ ( 1997): A molecular and cellular theory of depression. Arch Gen Psychiatry 54: 597–606. [DOI] [PubMed] [Google Scholar]
- Duvall JA, Lu A, Cantor RM, Todd RD, Constantino JN, Geschwind DH ( 2007): A quantitative trait locus analysis of social responsiveness in multiplex autism families. Am J Psychiatry 164: 656–662. [DOI] [PubMed] [Google Scholar]
- Egan MF, Kojima M, Callicott JH, Goldberg TE, Kolachana BS, Bertolino A, Zaitsev E, Gold B, Goldman D, Dean M, et al. ( 2003): The BDNF val66met polymorphism affects activity‐dependent secretion of BDNF and human memory and hippocampal function. Cell 112: 257–269. [DOI] [PubMed] [Google Scholar]
- Figurov A, Pozzo‐Miller LD, Olafsson P, Wang T, Lu B ( 1996): Regulation of synaptic responses to high‐frequency stimulation and LTP by neurotrophins in the hippocampus. Nature 381: 706–709. [DOI] [PubMed] [Google Scholar]
- Fossella JA, Casey BJ ( 2006): Genes, brain, and behavior: Bridging disciplines. Cogn Affect Behav Neurosci 6: 1–8. [DOI] [PubMed] [Google Scholar]
- Fossella J, Green AE, Fan J ( 2006): Evaluation of a structural polymorphism in the ankyrin repeat and kinase domain containing 1 (ANKK1) gene and the activation of executive attention networks. Cogn Affect Behav Neurosci 6: 71–78. [DOI] [PubMed] [Google Scholar]
- Geller B, Badner JA, Tillman R, Christian SL, Bolhofner K, Cook EH Jr ( 2004): Linkage disequilibrium of the brain‐derived neurotrophic factor Val66Met polymorphism in children with a prepubertal and early adolescent bipolar disorder phenotype. Am J Psychiatry 161: 1698–1700. [DOI] [PubMed] [Google Scholar]
- Gomes RA, Hampton C, El‐Sabeawy F, Sabo SL, McAllister AK ( 2006): The dynamic distribution of TrkB receptors before, during, and after synapse formation between cortical neurons. J Neurosci 26: 11487–11500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottfried JA, Dolan RJ ( 2004): Human orbitofrontal cortex mediates extinction learning while accessing conditioned representations of value. Nat Neurosci 7: 1144–1152. [DOI] [PubMed] [Google Scholar]
- Gratacos M, Gonzalez JR, Mercader JM, de Cid R, Urretavizcaya M, Estivill X ( 2007): Brain‐derived neurotrophic factor Val66Met and psychiatric disorders: Meta‐analysis of case‐control studies confirm association to substance‐related disorders, eating disorders, and schizophrenia. Biol Psychiatry 61: 911–922. [DOI] [PubMed] [Google Scholar]
- Hariri AR, Bookheimer SY, Mazziotta JC ( 2000): Modulating emotional responses: Effects of a neocortical network on the limbic system. Neuroreport 11: 43–48. [DOI] [PubMed] [Google Scholar]
- Hariri AR, Mattay VS, Tessitore A, Fera F, Smith WG, Weinberger DR ( 2002): Dextroamphetamine modulates the response of the human amygdala. Neuropsychopharmacology 27: 1036–1040. [DOI] [PubMed] [Google Scholar]
- Hariri AR, Drabant EM, Weinberger DR ( 2006): Imaging genetics: Perspectives from studies of genetically driven variation in serotonin function and corticolimbic affective processing. Biol Psychiatry 59: 888–897. [DOI] [PubMed] [Google Scholar]
- Hofer M, Pagliusi SR, Hohn A, Leibrock J, Barde YA ( 1990): Regional distribution of brain‐derived neurotrophic factor mRNA in the adult mouse brain. EMBO J 9: 2459–2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman SE, Fenton WS ( 2003): Medicine: What are the right targets for psychopharmacology? Science 299: 350–351. [DOI] [PubMed] [Google Scholar]
- Ivanova T, Beyer C ( 2001): Pre‐ and postnatal expression of brain‐derived neurotrophic factor mRNA/protein and tyrosine protein kinase receptor B mRNA in the mouse hippocampus. Neurosci Lett 307: 21–24. [DOI] [PubMed] [Google Scholar]
- Kalisch R, Korenfeld E, Stephan KE, Weiskopf N, Seymour B, Dolan RJ ( 2006): Context‐dependent human extinction memory is mediated by a ventromedial prefrontal and hippocampal network. J Neurosci 26: 9503–9511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HJ, Schuman EM ( 1995): Neurotrophin‐induced modulation of synaptic transmission in the adult hippocampus. J Physiol Paris 89: 11–22. [DOI] [PubMed] [Google Scholar]
- Karmiloff‐Smith A ( 2006): The tortuous route from genes to behavior: A neuroconstructivist approach. Cogn Affect Behav Neurosci 6: 9–17. [DOI] [PubMed] [Google Scholar]
- Katoh‐Semba R, Takeuchi IK, Semba R, Kato K ( 1997): Distribution of brain‐derived neurotrophic factor in rats and its changes with development in the brain. J Neurochem 69: 34–42. [DOI] [PubMed] [Google Scholar]
- Katz LC, Shatz CJ ( 1996): Synaptic activity and the construction of cortical circuits. Science 274: 1133–1138. [DOI] [PubMed] [Google Scholar]
- Keen‐Kim D, Mathews CA, Reus VI, Lowe TL, Herrera LD, Budman CL, Gross‐Tsur V, Pulver AE, Bruun RD, Erenberg G, et al. ( 2006): Overrepresentation of rare variants in a specific ethnic group may confuse interpretation of association analyses. Hum Mol Genet 15: 3324–3328. [DOI] [PubMed] [Google Scholar]
- Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T ( 1995): Hippocampal long‐term potentiation is impaired in mice lacking brain‐derived neurotrophic factor. Proc Natl Acad Sci USA 92: 8856–8860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaBar KS, Gatenby JC, Gore JC, LeDoux JE, Phelps EA ( 1998): Human amygdala activation during conditioned fear acquisition and extinction: A mixed‐trial fMRI study. Neuron 20: 937–945. [DOI] [PubMed] [Google Scholar]
- LeDoux JE ( 2000): Emotion circuits in the brain. Annu Rev Neurosci 23: 155–184. [DOI] [PubMed] [Google Scholar]
- Leibrock J, Lottspeich F, Hohn A, Hofer M, Hengerer B, Masiakowski P, Thoenen H, Barde YA ( 1989): Molecular cloning and expression of brain‐derived neurotrophic factor. Nature 341: 149–152. [DOI] [PubMed] [Google Scholar]
- Lesch KP, Meyer J, Glatz K, Flugge G, Hinney A, Hebebrand J, Klauck SM, Poustka A, Poustka F, Bengel D, et al. ( 1997): The 5‐HT transporter gene‐linked polymorphic region (5‐HTTLPR) in evolutionary perspective: Alternative biallelic variation in rhesus monkeys. Rapid communication. J Neural Transm 104: 1259–1266. [DOI] [PubMed] [Google Scholar]
- Levine ES, Dreyfus CF, Black IB, Plummer MR ( 1995): Brain‐derived neurotrophic factor rapidly enhances synaptic transmission in hippocampal neurons via postsynaptic tyrosine kinase receptors. Proc Natl Acad Sci USA 92: 8074–8077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao L, Pilotte J, Xu T, Wong CC, Edelman GM, Vanderklish P, Yates JR III ( 2007): BDNF induces widespread changes in synaptic protein content and up‐regulates components of the translation machinery: An analysis using high‐throughput proteomics. J Proteome Res 6: 1059–1071. [DOI] [PubMed] [Google Scholar]
- Liu QR, Walther D, Drgon T, Polesskaya O, Lesnick TG, Strain KJ, de Andrade M, Bower JH, Maraganore DM, Uhl GR ( 2005): Human brain derived neurotrophic factor (BDNF) genes, splicing patterns, and assessments of associations with substance abuse and Parkinson's Disease. Am J Med Genet B Neuropsychiatr Genet 134B: 93–103. [DOI] [PubMed] [Google Scholar]
- Lohof AM, Ip NY, Poo MM ( 1993): Potentiation of developing neuromuscular synapses by the neurotrophins NT‐3 and BDNF. Nature 363: 350–353. [DOI] [PubMed] [Google Scholar]
- Lu B ( 2003): Pro‐region of neurotrophins: Role in synaptic modulation. Neuron 39: 735–738. [DOI] [PubMed] [Google Scholar]
- Lu B, Chow A ( 1999): Neurotrophins and hippocampal synaptic transmission and plasticity. J Neurosci Res 58: 76–87. [PubMed] [Google Scholar]
- Maisonpierre PC, Belluscio L, Friedman B, Alderson RF, Wiegand SJ, Furth ME, Lindsay RM, Yancopoulos GD ( 1990): NT‐3, BDNF, and NGF in the developing rat nervous system: Parallel as well as reciprocal patterns of expression. Neuron 5: 501–509. [DOI] [PubMed] [Google Scholar]
- Matsushita S, Kimura M, Miyakawa T, Yoshino A, Murayama M, Masaki T, Higuchi S ( 2004): Association study of brain‐derived neurotrophic factor gene polymorphism and alcoholism. Alcohol Clin Exp Res 28: 1609–1612. [DOI] [PubMed] [Google Scholar]
- McAllister AK, Lo DC, Katz LC ( 1995): Neurotrophins regulate dendritic growth in developing visual cortex. Neuron 15: 791–803. [DOI] [PubMed] [Google Scholar]
- McAllister AK, Katz LC, Lo DC ( 1999): Neurotrophins and synaptic plasticity. Annu Rev Neurosci 22: 295–318. [DOI] [PubMed] [Google Scholar]
- Messaoudi E, Ying SW, Kanhema T, Croll SD, Bramham CR ( 2002): Brain‐derived neurotrophic factor triggers transcription‐dependent, late phase long‐term potentiation in vivo. J Neurosci 22: 7453–7461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milad MR, Quirk GJ ( 2002): Neurons in medial prefrontal cortex signal memory for fear extinction. Nature 420: 70–74. [DOI] [PubMed] [Google Scholar]
- Milad MR, Quirk GJ, Pitman RK, Orr SP, Fischl B, Rauch SL ( 2007): A role for the human dorsal anterior cingulate cortex in fear expression. Biol Psychiatry 62: 1191–1194. [DOI] [PubMed] [Google Scholar]
- Munafo MR, Clark T, Flint J ( 2005): Does measurement instrument moderate the association between the serotonin transporter gene and anxiety‐related personality traits? A meta‐analysis. Mol Psychiatry 10: 415–419. [DOI] [PubMed] [Google Scholar]
- Munafo MR, Brown SM, Hariri AR ( 2008): Serotonin transporter (5‐HTTLPR) genotype and amygdala activation: A meta‐analysis. Biol Psychiatry 63: 852–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers KM, Davis M ( 2002): Behavioral and neural analysis of extinction. Neuron 36: 567–584. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Barrot M, DiLeone RJ, Eisch AJ, Gold SJ, Monteggia LM ( 2002): Neurobiology of depression. Neuron 34: 13–25. [DOI] [PubMed] [Google Scholar]
- Neves‐Pereira M, Mundo E, Muglia P, King N, Macciardi F, Kennedy JL ( 2002): The brain‐derived neurotrophic factor gene confers susceptibility to bipolar disorder: Evidence from a family‐based association study. Am J Hum Genet 71: 651–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang PT, Lu B ( 2004): Regulation of late‐phase LTP and long‐term memory in normal and aging hippocampus: Role of secreted proteins tPA and BDNF. Ageing Res Rev 3: 407–430. [DOI] [PubMed] [Google Scholar]
- Patterson SL, Grover LM, Schwartzkroin PA, Bothwell M ( 1992): Neurotrophin expression in rat hippocampal slices: A stimulus paradigm inducing LTP in CA1 evokes increases in BDNF and NT‐3 mRNAs. Neuron 9: 1081–1088. [DOI] [PubMed] [Google Scholar]
- Patterson SL, Abel T, Deuel TA, Martin KC, Rose JC, Kandel ER ( 1996): Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron 16: 1137–1145. [DOI] [PubMed] [Google Scholar]
- Pezawas L, Verchinski BA, Mattay VS, Callicott JH, Kolachana BS, Straub RE, Egan MF, Meyer‐Lindenberg A, Weinberger DR ( 2004): The brain‐derived neurotrophic factor val66met polymorphism and variation in human cortical morphology. J Neurosci 24: 10099–10102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelps EA, Delgado MR, Nearing KI, LeDoux JE ( 2004): Extinction learning in humans: Role of the amygdala and vmPFC. Neuron 43: 897–905. [DOI] [PubMed] [Google Scholar]
- Pine DS ( 2007): A neuroscience perspective on pediatric anxiety disorders. J Child Psychol Psychiatry 48: 631–648. [DOI] [PubMed] [Google Scholar]
- Poo MM ( 2001): Neurotrophins as synaptic modulators. Nat Rev Neurosci 2: 24–32. [DOI] [PubMed] [Google Scholar]
- Quirk GJ, Likhtik E, Pelletier JG, Pare D ( 2003): Stimulation of medial prefrontal cortex decreases the responsiveness of central amygdala output neurons. J Neurosci 23: 8800–8807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radecki DT, Brown LM, Martinez J, Teyler TJ ( 2005): BDNF protects against stress‐induced impairments in spatial learning and memory and LTP. Hippocampus 15: 246–253. [DOI] [PubMed] [Google Scholar]
- Rattiner LM, Davis M, Ressler KJ ( 2005): Brain‐derived neurotrophic factor in amygdala‐dependent learning. Neuroscientist 11: 323–333. [DOI] [PubMed] [Google Scholar]
- Ribases M, Gratacos M, Armengol L, de Cid R, Badia A, Jimenez L, Solano R, Vallejo J, Fernandez F, Estivill X ( 2003): Met66 in the brain‐derived neurotrophic factor (BDNF) precursor is associated with anorexia nervosa restrictive type. Mol Psychiatry 8: 745–751. [DOI] [PubMed] [Google Scholar]
- Ribases M, Gratacos M, Fernandez‐Aranda F, Bellodi L, Boni C, Anderluh M, Cavallini MC, Cellini E, Di Bella D, Erzegovesi S, et al. ( 2004): Association of BDNF with anorexia, bulimia and age of onset of weight loss in six European populations. Hum Mol Genet 13: 1205–1212. [DOI] [PubMed] [Google Scholar]
- Risch N, Herrell R, Lehner T, Liang KY, Eaves L, Hoh J, Griem A, Kovacs M, Ott J, Merikangas KR ( 2009): Interaction between the serotonin transporter gene (5‐HTTLPR), stressful life events, and risk of depression: A meta‐analysis. JAMA 301: 2462–2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller D, Levy I, Niv Y, LeDoux JE, Phelps EA ( 2008): From fear to safety and back: Reversal of fear in the human brain. J Neurosci 28: 11517–11525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen S, Nesse RM, Stoltenberg SF, Li S, Gleiberman L, Chakravarti A, Weder AB, Burmeister M ( 2003): A BDNF coding variant is associated with the NEO personality inventory domain neuroticism, a risk factor for depression. Neuropsychopharmacology 28: 397–401. [DOI] [PubMed] [Google Scholar]
- Shimizu E, Hashimoto K, Iyo M ( 2004): Ethnic difference of the BDNF 196G/A (val66met) polymorphism frequencies: The possibility to explain ethnic mental traits. Am J Med Genet B Neuropsychiatr Genet 126B: 122–123. [DOI] [PubMed] [Google Scholar]
- Siest G, Pillot T, Regis‐Bailly A, Leininger‐Muller B, Steinmetz J, Galteau MM, Visvikis S ( 1995): Apolipoprotein E: An important gene and protein to follow in laboratory medicine. Clin Chem 41( 8, Part 1): 1068–1086. [PubMed] [Google Scholar]
- Silhol M, Bonnichon V, Rage F, Tapia‐Arancibia L ( 2005): Age‐related changes in brain‐derived neurotrophic factor and tyrosine kinase receptor isoforms in the hippocampus and hypothalamus in male rats. Neuroscience 132: 613–624. [DOI] [PubMed] [Google Scholar]
- Sklar P, Gabriel SB, McInnis MG, Bennett P, Lim YM, Tsan G, Schaffner S, Kirov G, Jones I, Owen M, et al. ( 2002): Family‐based association study of 76 candidate genes in bipolar disorder: BDNF is a potential risk locus. Brain‐derived neutrophic factor. Mol Psychiatry 7: 579–593. [DOI] [PubMed] [Google Scholar]
- Smith MA, Makino S, Kvetnansky R, Post RM ( 1995): Stress and glucocorticoids affect the expression of brain‐derived neurotrophic factor and neurotrophin‐3 mRNAs in the hippocampus. J Neurosci 15( 3, Part 1): 1768–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soliman F, Glatt CE, Bath KG, Levita L, Jones RM, Pattwell SS, Jing D, Tottenham N, Amso D, Sommerville L, et al. ( 2010): A genetic variant BDNF polymorphism alters extinction learning in both mouse and human. Science 327: 863–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szeszko PR, Lipsky R, Mentschel C, Robinson D, Gunduz‐Bruce H, Sevy S, Ashtari M, Napolitano B, Bilder RM, Kane JM, et al. ( 2005): Brain‐derived neurotrophic factor val66met polymorphism and volume of the hippocampal formation. Mol Psychiatry 10: 631–636. [DOI] [PubMed] [Google Scholar]
- Thoenen H ( 1995): Neurotrophins and neuronal plasticity. Science 270: 593–598. [DOI] [PubMed] [Google Scholar]
- Tongiorgi E, Domenici L, Simonato M ( 2006): What is the biological significance of BDNF mRNA targeting in the dendrites? Clues from epilepsy and cortical development. Mol Neurobiol 33: 17–32. [DOI] [PubMed] [Google Scholar]
- Waddell J, Dunnett C, Falls WA ( 2004): C57BL/6J and DBA/2J mice differ in extinction and renewal of extinguished conditioned fear. Behav Brain Res 154: 567–576. [DOI] [PubMed] [Google Scholar]
- Webster MJ, Weickert CS, Herman MM, Kleinman JE ( 2002): BDNF mRNA expression during postnatal development, maturation and aging of the human prefrontal cortex. Brain Res Dev Brain Res 139: 139–150. [DOI] [PubMed] [Google Scholar]
- Webster MJ, Herman MM, Kleinman JE, Shannon Weickert C ( 2006): BDNF and trkB mRNA expression in the hippocampus and temporal cortex during the human lifespan. Gene Expr Patterns 6: 941–951. [DOI] [PubMed] [Google Scholar]
- Yamamoto N, Hanamura K ( 2005): Formation of the thalamocortical projection regulated differentially by BDNF‐ and NT‐3‐mediated signaling. Rev Neurosci 16: 223–231. [DOI] [PubMed] [Google Scholar]
- Yan Q, Rosenfeld RD, Matheson CR, Hawkins N, Lopez OT, Bennett L, Welcher AA ( 1997): Expression of brain‐derived neurotrophic factor protein in the adult rat central nervous system. Neuroscience 78: 431–448. [DOI] [PubMed] [Google Scholar]
- Zakharenko SS, Patterson SL, Dragatsis I, Zeitlin SO, Siegelbaum SA, Kandel ER, Morozov A ( 2003): Presynaptic BDNF required for a presynaptic but not postsynaptic component of LTP at hippocampal CA1‐CA3 synapses. Neuron 39: 975–990. [DOI] [PubMed] [Google Scholar]