

Abstract
We demonstrate that polyvalent DNA-functionalized gold nanoparticles (DNA-Au NPs) selectively enhance Ribonuclease H (RNase H) activity, while inhibiting most biologically relevant nucleases. This combination of properties is particularly interesting in the context of gene regulation, since high RNase H activity results in rapid mRNA degradation and general nuclease inhibition results in high biological stability. We investigate the mechanism of selective RNase H activation and find that the high DNA density of DNA-Au NPs is responsible for this unusual behavior. This work adds to our understanding of polyvalent DNA-Au NPs as gene regulation agents, and suggests a new model for selectively controlling protein-nanoparticle interactions.
The relationship between a material's nanostructure and its interactions with biomolecules represents an important area of research. The promise of this research lies in the ability to selectively engage specific molecules in complex biological environments, which could potentially lead to more accurate diagnostics and more potent therapeutics.1 The interactions between oligonucleotides and nucleases, enzymes that degrade nucleic acids, are particularly important because they play a central role in gene detection and regulation.2 Unmodified oligonucleotides are rapidly destroyed by non-specific nucleases in most biological environments, rendering them less effective. Chemists have developed designer nucleic acids to combat this problem by retarding nuclease catalyzed degradation.3 This leads to greater oligonucleotide stability, but can have unintended consequences as some nucleases are necessary for biological applications. For example, messenger RNA (mRNA) expression can be regulated by antisense oligonucleotides through interactions with Ribonuclease H (RNase H), but in this case designer nucleic acids are counterproductive, inhibiting RNase H and leading to ineffective gene knockdown.2 This problem is generally overcome through a compromise in which oligonucleotides containing both designer and traditional nucleic acid regions are used. These chimeric molecules have enhanced stability and some RNase H reactivity, but do not maximize either, leading to the desire to create a single agent that inhibits non-specific nucleases, but also selectively enhances the activity of RNase H.
DNA-functionalized gold nanoparticles (DNA-Au NPs) are efficient agents in many applications, including materials assembly,4 detection,5 and gene regulation.6 These nanoparticles have many attributes that make them ideal for these applications, including distance-dependent optical properties,7 efficient programmable binding,5e,8 rapid cell uptake,9 and little innate immune response,10 but biological stability stemming from the nuclease resistant cloud of concentrated DNA and salt surrounding the nanoparticle is particularly important.11 Despite this general nuclease inhibition, DNA-Au NPs are effective gene regulation agents, acting through the RNase H pathway.6d Although these DNA-Au NPs have been well studied, their combination of potent gene regulation and nuclease inhibition is both practically important and scientifically surprising. As such, we chose to directly investigate RNase H activity on DNA-Au NPs. We report that, in contrast to most nucleases studied to date, RNase H activity is enhanced on the nanoparticle surface, making DNA-Au NPs ideal gene regulation agents, both highly stable in biological environments, and highly active in the presence of target mRNA (Figure 1).
Figure 1.
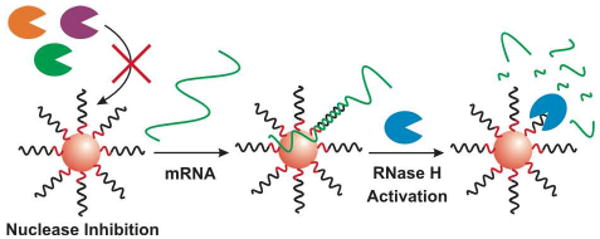
Schematic representation of general nuclease inhibition and RNase H activation by DNA-Au NPs. The Au NP is depicted as a red sphere, functionalized with DNA. The recognition region of the DNA is depicted in black and the A10-thiol spacer, linking the target region to the gold surface is depicted in red. Most nucleases (far left, orange, purple and green) are inhibited by the nanoparticle surface, leading to high stability for DNA-Au NPs. Once target mRNA (green line) binds to the nanoparticle, enhanced RNase H (right side, blue) catalysis at the nanoparticle surface leads to the rapid RNA degradation and efficient gene regulation.
The first step in this investigation was to create a model RNase H substrate containing a fluorophore labeled RNA/DNA heteroduplex. This was accomplished by hybridizing fluorophore labeled RNA to either Dabcyl-DNA (free target) or DNA-Au NPs. Similar substrates were created to compare RNase H to Deoxyribonuclease I (DNase I) and serum nucleases. The number of DNA molecules per nanoparticle was measured using a commercial assay (Invitrogen, Quant-It Oligreen).9 In the intact substrate, the proximity of the fluorophore to the quencher (either the NP or Dabcyl) results in distance-dependent quenching. When the substrate is exposed to the appropriate nuclease, the nucleic acid is degraded, and the fluorophore is released, which can be monitored in real-time as an increase in solution-associated fluorescence (Figure 2A-B).
Figure 2.

Comparison of RNase H and serum nuclease activity. (A, B) General schematic representations for the fluorescence-based degradation assays using free (A) and nanoparticle bound (B) target. DNA, RNA and nuclease are depicted in black, green and blue respectively. The molecular quenchers (small orange circles) and the excited fluorophores (pink stars) are also shown. (C, D) Progress curves of nucleic acid degradation are shown for nonspecific serum nucleases (C) and RNase H (D). In each plot, the reaction rates of free substrate (blue), and nanoparticle bound substrate (red), were compared. Each curve represents the average from three independent experiments.
The activities of serum nucleases on DNA-Au NPs and free DNA substrates were compared using the materials described above and established protocols (Figure 1).11b Consistent with previous results,11a serum nucleases are strongly inhibited by the nanoparticle conjugate (Figure 2C). RNase H was tested (Figure 2D), and surprisingly, RNase H exhibited 2.5 ± 0.1 times faster degradation on the polyvalent oligonucleotide-functionalized nanoparticle, compared to the same substrate free in solution. This preferential interaction of RNA/DNA-Au NPs with RNase H explains why these nanoparticles are active in RNase H mediated gene regulation, while still preventing degradation by other nucleases.
In order to investigate the surprising activity of RNase H on polyvalent DNA-Au NPs, we looked at the enzyme's salt-tolerance, because DNA-Au NPs inhibit general nucleases through the high local salt concentration around the nanoparticle surface.11b The effects of salt concentration on the degradation of free substrates by RNase H and DNase I, a well-studied model nuclease, were compared. The initial reaction rates for each enzyme were determined and plotted as a function of KCl concentration (Figure 3). RNase H is clearly more active than DNase I in high KCl conditions, for example, RNase H is 20-fold faster than DNase I at 225 mM KCl. Similar results were observed using NaCl (Figure S1). The salt-tolerance of RNase H explains its lack of inhibition by nanoparticles, but it does not fully explain the observed increase in activity (Figure 2D).
Figure 3.
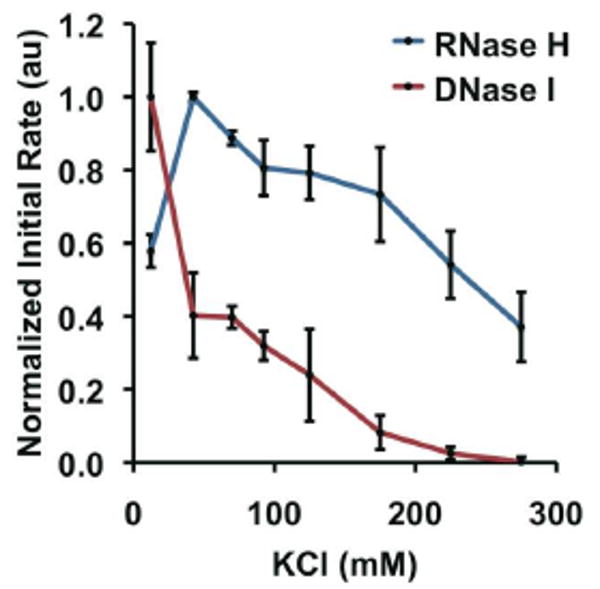
Salt-sensitivity of RNase H (blue) and DNase I (red). Each point represents the normalized initial rate of substrate degradation; the errors bars represent the standard error from three independent experiments.
To further explore nanoparticle-RNase H interactions, we investigated the role of local DNA density, which is much higher on the polyvalent DNA-Au NP surface than with conventional oligonucleotides. The first step was to synthesize nanoparticles with different DNA densities by changing the distance between the Au NP and the DNA. DNA-Au NPs were made with the DNA either bound directly to the nanoparticle surface or bound through a ten unit propyl-phosphodiester spacer (P10) (Figure 4). Without the spacer, the DNA is approximately 2.2 times more dense, due to the radius of curvature of the nanoparticle (calculated based on previous results,12 see Supporting Information). The increase in density leads to an increase in RNase H activity from 0.25 ± 0.01, to 0.46 ± 0.03, to 0.68 ± 0.04 hr-1 for free targets, moderate density NPs, and high density NPs respectively (Figure 4). These results were confirmed with nanoparticles containing different spacer compositions (Figure S2-3). The correlation between high local target concentration and high RNase H activity indicates steric hindrance is not a major issue under the conditions tested.
Figure 4.
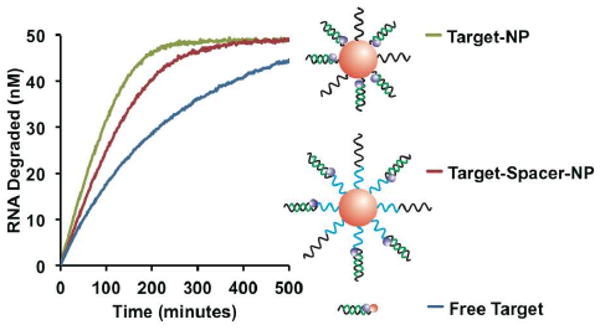
Role of local substrate density on reaction rate. Progress curves for three substrates were compared, free target (blue), target bound to an Au NP through a P10 spacer (red), and the target bound directly to the Au NP surface (green). Schematic diagrams of each substrate are shown with the position of the spacer highlighted in blue. As in previous schemes, DNA and RNA are depicted in black and green respectively. Each curve represents the average from three independent experiments.
Since RNase H is known to associate with single-stranded DNA (ssDNA),13 we hypothesized that the relatively high DNA density increases RNase H activity by binding and localizing the enzyme at the nanoparticle surface, positioning it in close proximity with its target (Figure 5).14 To test this, RNase H affinity for DNA-Au NPs and free target was quantitatively compared by a Michaelis-Menten analysis. The measured Michaelis constant (KM) for the nanoparticle is enhanced by approximately twofold compared to the free duplex, indicating that the rapid reaction rates on the nanoparticle are, at least in part, the result of relatively high affinity between the enzyme and the DNA-Au NP (Figure S4). This is consistent with RNase H localization at the nanoparticle surface due to polyvalent ssDNA interactions, but KM analysis does not directly address the role of ssDNA (Figure 5A). To confirm that RNase H associates with ssDNA-Au NPs even without target RNA, the system was studied by Dynamic Light Scattering (DLS) (Figure 5B). Particle diameter increased 2.5 ± 0.3 nm in the presence of excess RNase H, consistent with RNase H binding.15 Again, this is consistent with the polyvalent ssDNA surface localizing RNase H near its RNA/DNA substrate. To confirm that ssDNA polyvalency is critical for the observed increase in rate, we investigated the effect of excess ssDNA on free duplex degradation (Figure 5C). In this case, ssDNA inhibits, rather than facilitates, the reaction, likely because the free ssDNA is not localized near the DNA/RNA target, so it competes for RNase H binding. Similar experiments were performed in the nanoparticle-bound system to further support this finding (Figure S5). Taken together, these results support the proposed mechanism of enhanced RNase H activity, in which enzyme-target association is facilitated by the localization of both RNase H and its RNA target in close proximity at the nanoparticle surface. This is not observed for other nucleases, most likely because they are inhibited by the high local salt concentration, which is present in high DNA density environments.11b As a result, increasing DNA density can be used to specifically increase RNase H activity while simultaneously decreasing degradation by other nucleases.
Figure 5.

High DNA density localizes RNase H near its target. (A) Proposed intermediate with RNase H and target bound to the NP. (B) DLS measurements of particle diameter at multiple RNase H concentrations. (C) RNase H inhibition by ssDNA when not particle bound. Each point represents the normalized initial rate of substrate degradation. The error bars represent the standard error from three independent experiments. (D, E) Schematic of free (D) and nanoparticle bound (E) substrates. As in previous schemes, DNA, RNA, and RNase H are depicted in black, green and blue respectively.
Polyvalent DNA-Au NPs are highly stable in biological environments, because most nucleases are inhibited by the high local salt concentration around the nanoparticle surface,11 which has made them excellent agents for many applications, including gene regulation.6b However, DNA-Au NPs require a nuclease, RNase H, to engage the gene regulation pathway. This apparent inconsistency has been resolved; unlike other nucleases, RNase H activity is enhanced by the DNA-Au NP (Figure 2). We have identified two biochemical properties that help explain this surprising observation. First, RNase H is a particularly salt-tolerant nuclease, and as such, it is relatively unperturbed by the high salt environment of the nanoparticle (Figure 3); and second, the high DNA density on the nanoparticle surface leads to faster reaction rates, likely due to the high effective substrate concentration around the nanoparticle surface (Figures 4-5). These features make DNA-Au NPs both highly stable in biological environments and highly active in the RNase H-based gene regulation pathway. This work not only accounts for the polyvalent DNA-Au NP's remarkable utility, but also establishes a nanochemistry-based method for selectively activating a specific enzyme, while simultaneously inhibiting closely related enzymes, and represents a significant step towards creating programmable nanoparticle-protein interactions.
Supplementary Material
Acknowledgments
This material is based upon work supported by the DARPA US Army RDECOM Acquisition Center under Award No. W911NF-09-1-0069 and W81XWH-08-1-0766. Additionally, the project was supported by Award Numbers U54CA151880 and U54CA119341 from the National Cancer Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health. A.E.P. and A.H.A. acknowledge the Ryan Fellowship and the K.S.A King Abdullah Scholarship respectively.
Footnotes
Supporting Information Available: Supporting figures, experimental methods, and calculations are available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Niemeyer CM. Angew Chem, Int Ed. 2001;40:4128–4158. doi: 10.1002/1521-3773(20011119)40:22<4128::AID-ANIE4128>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]; (b) Kim J, Grate JW, Wang P. Chem Eng Sci. 2006;61:1017–1026. [Google Scholar]; (c) Rosi NL, Mirkin CA. Chem Rev (Washington, DC, U S) 2005;105:1547–1562. doi: 10.1021/cr030067f. [DOI] [PubMed] [Google Scholar]; (d) Giljohann DA, Mirkin CA. Nature. 2009;462:461–464. doi: 10.1038/nature08605. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Chen CL, Rosi NL. Angew Chem, Int Ed. 2010;49:1924–1942. doi: 10.1002/anie.200903572. [DOI] [PubMed] [Google Scholar]
- 2.(a) Akhtar S, Hughes MD, Khan A, Bibby M, Hussain M, Nawaz Q, Double J, Sayyed P. Adv Drug Delivery Rev. 2000;44:3–21. doi: 10.1016/s0169-409x(00)00080-6. [DOI] [PubMed] [Google Scholar]; (b) Crooke ST. Annu Rev Med. 2004;55:61–95. doi: 10.1146/annurev.med.55.091902.104408. [DOI] [PubMed] [Google Scholar]
- 3.(a) Petersen M, Wengel J. Trends Biotechnol. 2003;21:74–81. doi: 10.1016/S0167-7799(02)00038-0. [DOI] [PubMed] [Google Scholar]; (b) Shangguan D, Tang ZW, Mallikaratchy P, Xiao ZY, Tan WH. ChemBioChem. 2007;8:603–606. doi: 10.1002/cbic.200600532. [DOI] [PubMed] [Google Scholar]
- 4.(a) Mirkin CA, Letsinger RL, Mucic RC, Storhoff JJ. Nature. 1996;382:607–609. doi: 10.1038/382607a0. [DOI] [PubMed] [Google Scholar]; (b) Alivisatos AP, Johnsson KP, Peng XG, Wilson TE, Loweth CJ, Bruchez MP, Schultz PG. Nature. 1996;382:609–611. doi: 10.1038/382609a0. [DOI] [PubMed] [Google Scholar]; (c) Park SY, Lytton-Jean AKR, Lee B, Weigand S, Schatz GC, Mirkin CA. Nature. 2008;451:553–556. doi: 10.1038/nature06508. [DOI] [PubMed] [Google Scholar]; (d) Nykypanchuk D, Maye MM, van der Lelie D, Gang O. Nature. 2008;451:549–552. doi: 10.1038/nature06560. [DOI] [PubMed] [Google Scholar]
- 5.(a) He L, Musick MD, Nicewarner SR, Salinas FG, Benkovic SJ, Natan MJ, Keating CD. J Am Chem Soc. 2000;122:9071–9077. [Google Scholar]; (b) Liu JW, Lu Y. J Am Chem Soc. 2003;125:6642–6643. doi: 10.1021/ja034775u. [DOI] [PubMed] [Google Scholar]; (c) Nam JM, Thaxton CS, Mirkin CA. Science. 2003;301:1884–1886. doi: 10.1126/science.1088755. [DOI] [PubMed] [Google Scholar]; (d) Seferos DS, Giljohann DA, Hill HD, Prigodich AE, Mirkin CA. J Am Chem Soc. 2007;129:15477–15479. doi: 10.1021/ja0776529. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Prigodich AE, Lee OS, Daniel WL, Seferos DS, Schatz GC, Mirkin CA. J Am Chem Soc. 2010;132:10638–10641. doi: 10.1021/ja104859j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Agbasi-Porter C, Ryman-Rasmussen J, Franzen S, Feldheim D. Bioconjugate Chem. 2006;17:1178–1183. doi: 10.1021/bc060100f. [DOI] [PubMed] [Google Scholar]; (b) Rosi NL, Giljohann DA, Thaxton CS, Lytton-Jean AKR, Han MS, Mirkin CA. Science. 2006;312:1027–1030. doi: 10.1126/science.1125559. [DOI] [PubMed] [Google Scholar]; (c) Liu YL, Franzen S. Bioconjugate Chem. 2008;19:1009–1016. doi: 10.1021/bc700421u. [DOI] [PubMed] [Google Scholar]; (d) Prigodich AE, Seferos DS, Massich MD, Giljohann DA, Lane BC, Mirkin CA. ACS Nano. 2009;3:2147–2152. doi: 10.1021/nn9003814. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Giljohann DA, Seferos DS, Daniel WL, Massich MD, Patel PC, Mirkin CA. Angew Chem, Int Ed. 2010;49:3280–3294. doi: 10.1002/anie.200904359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Storhoff JJ, Lazarides AA, Mucic RC, Mirkin CA, Letsinger RL, Schatz GC. J Am Chem Soc. 2000;122:4640–4650. [Google Scholar]; (b) Zhu M, Aikens CM, Hollander FJ, Schatz GC, Jin R. J Am Chem Soc. 2008;130:5883–5885. doi: 10.1021/ja801173r. [DOI] [PubMed] [Google Scholar]
- 8.(a) Huang CC, Huang YF, Cao ZH, Tan WH, Chang HT. Anal Chem. 2005;77:5735–5741. doi: 10.1021/ac050957q. [DOI] [PubMed] [Google Scholar]; (b) Lytton-Jean AKR, Mirkin CA. J Am Chem Soc. 2005;127:12754–12755. doi: 10.1021/ja052255o. [DOI] [PubMed] [Google Scholar]
- 9.Giljohann DA, Seferos DS, Patel PC, Millstone JE, Rosi NL, Mirkin CA. Nano Lett. 2007;7:3818–3821. doi: 10.1021/nl072471q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Massich MD, Giljohann DA, Seferos DS, Ludlow LE, Horvath CM, Mirkin CA. Mol Pharm. 2009;6:1934–1940. doi: 10.1021/mp900172m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Giljohann DA, Seferos DS, Prigodich AE, Patel PC, Mirkin CA. J Am Chem Soc. 2009;131:2072–2073. doi: 10.1021/ja808719p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Seferos DS, Prigodich AE, Giljohann DA, Patel PC, Mirkin CA. Nano Lett. 2009;9:308–311. doi: 10.1021/nl802958f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Macfarlane RJ, Jones MR, Senesi AJ, Young KL, Lee B, Wu JS, Mirkin CA. Angew Chem, Int Ed. 2010;49:4589–4592. doi: 10.1002/anie.201000633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Gopalakrishnan V, Peliska JA, Benkovic SJ. Proc Natl Acad Sci U S A. 1992;89:10763–10767. doi: 10.1073/pnas.89.22.10763. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lima WF, Wu HJ, Nichols JG, Prakash TP, Ravikumar V, Crooke ST. J Biol Chem. 2003;278:49860–49867. doi: 10.1074/jbc.M306543200. [DOI] [PubMed] [Google Scholar]
- 14.(a) Halford SE, Marko JF. Nucleic Acids Res. 2004;32:3040–3052. doi: 10.1093/nar/gkh624. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) von Hippel PH. Annu Rev Biophys Biomol Struct. 2007;36:79–105. doi: 10.1146/annurev.biophys.34.040204.144521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Katayanagi K, Miyagawa M, Matsushima M, Ishikawa M, Kanaya S, Ikehara M, Matsuzaki T, Morikawa K. Nature. 1990;347:306–309. doi: 10.1038/347306a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.