

Abstract
Dysfunctional mTORC1 signaling is associated with a number of human pathologies owing to its central role in controlling cell growth, proliferation, and metabolism. Regulation of mTORC1 is achieved by the integration of multiple inputs, including those of mitogens, nutrients, and energy. It is thought that agents that increase the cellular AMP/ATP ratio, such as the anti-diabetic biguanides metformin and phenformin, inhibit mTORC1 through AMPK activation of TSC1/2-dependent or -independent mechanisms. Unexpectedly, we found that biguanides inhibit mTORC1 signaling, not only in the absence of TSC1/2, but also in the absence of AMPK. Consistent with these observations, in two distinct pre-clinical models of cancer and diabetes, metformin acts to suppress mTORC1 signaling in an AMPK-independent manner. We found that the ability of biguanides to inhibit mTORC1 activation and signaling is, instead, dependent on the Rag GTPases.
Introduction
Increased mammalian Target of Rapamycin (mTOR) signaling has been implicated in a number of specific human pathologies, including obesity, type 2 diabetes, and cancer (Dann et al., 2007; Marshall, 2006; Um et al., 2006; Wullschleger et al., 2006). mTOR exists in two complexes that consist of distinct sets of protein binding partners (Dann et al., 2007; Sabatini, 2006). The first, mTORC1, is rapamycin sensitive and is thought to mediate many of its downstream effects through S6 Kinase 1 (S6K1) and initiation factor 4E binding protein 1 (4E-BP1) (Um et al., 2006; Wullschleger et al., 2006). In contrast, mTORC2 is largely rapamycin resistant and mediates phosphorylation of protein kinase B (PKB/Akt) S473, one of two phosphorylated residues required for full PKB/Akt activation (Sarbassov et al., 2005). Although both complexes respond to hormones and growth factors, only mTORC1 responds positively to nutrients and cellular energy status (Um et al., 2006), metabolic parameters intimately linked to obesity, diabetes, and cancer (Dann et al., 2007; Um et al., 2006).
Many mitogens initiate mTORC1 signaling by the sequential activation of class 1 phosphatidylinositol kinase (PI3K) and PKB/Akt, which reverses the inhibitory effects of Tuberous Sclerosis Complex proteins 1 and 2 (TSC1/2) (Manning and Cantley, 2007) and proline-rich PKB/Akt substrate 40 kDa (PRAS40) on mTORC1 (Haar et al., 2007; Sancak et al., 2007). This allows activation of Ras homolog enriched in brain (Rheb), required for mTORC1 activation (Garami et al., 2003; Long et al., 2005), whereas PRAS40 suppression relieves its inhibitory effects on mTORC1 (Haas and Hagedorn, 1992; Sancak et al., 2007). Nutrients, unlike insulin, have been shown to mediate mTORC1 signaling through class 3 PI3K, hVps34 (Byfield et al., 2005; Nobukuni et al., 2005) and, more recently, Rag GTPases (Kim et al., 2008; Sancak et al., 2008). Apart from insulin and nutrients, cellular energy status is a key regulator of mTORC1 signaling (Dennis et al., 2001). Initially, we demonstrated that mTORC1 signaling is blunted in response to the inhibition of glycolytic flux and ATP production (Dennis et al., 2001). Moreover, employing S6K1 or 4E-BP1 as in vitro substrates revealed that mTOR possessed a high Km for ATP in the presence of Mg2+ (Dennis et al., 2001). We hypothesized that direct sensing of ATP by mTOR could be one mechanism whereby intracellular ATP levels were maintained in a homeostatic narrow range. However, inhibition of glycolytic flux also leads to increased intracellular AMP levels and activation of AMP-activated kinase (AMPK). (Hardie, 2008; Steinberg and Kemp, 2009). In turn AMPK phosphorylates TSC2, attenuating downstream signaling, by an as yet unknown mechanism (Inoki et al., 2003).
Elucidating the role of mTORC1 in response to energy depletion is critical because of the broad clinical use of the anti-diabetic drug metformin (Libby et al., 2009; Shackelford and Shaw, 2009). Metformin, and its more hydrophobic analogue, phenformin, are biguanides. They inhibit mitochondrial respiratory chain complex I, which reduces ATP production, thus resulting in the activation of AMPK (Hardie, 2008). The major action of metformin in the treatment of Type 2 diabetes is to decrease hepatic glucose output and to increase intestinal glycolytic lactate production as well as peripheral tissue insulin-dependent glucose uptake (Bailey and Turner, 1996). Moreover, AMPK activation by metformin leads to the inhibition of mTORC1 (Tzatsos and Kandror, 2006) in a TSC1/2-dependent manner (Dowling et al., 2007), seemingly consistent with the earlier findings described above for other energy-depleting agents (Inoki et al., 2003). The observation that metformin inhibits mTORC1 signaling has drawn attention to its potential use as an anti-cancer agent (Dann et al., 2007; Shackelford and Shaw, 2009). Clinically, this concept is supported by retrospective record-linkage case-control studies demonstrating that metformin treatment of diabetic patients reduced the risk of certain cancers (Bowker et al., 2006; Libby et al., 2009), whereas insulin mimetics could increase such risks.
Although a role for TSC1/2 in mediating the inhibitory input to mTORC1 in response to energy depletion in mammalian cells is well documented (Inoki et al., 2003; Dowling et al., 2007), the AMPK phosphorylation sites are not conserved in Drosophila TSC2, dTsc2 (Um et al., 2006). Moreover, in direct contradiction to the findings of Dowling et al. (Dowling et al., 2007), others have shown that the biguanides inhibit this response via a TSC2-independent pathway, through AMPK phosphorylation of raptor, an mTORC1 constituent (Gwinn et al., 2008). Based on these observations, we set-out to examine the role of TSC1/2 and AMPK in mediating the inhibition of mTORC1 signaling by AMPK activators, particularly phenformin and metformin. We found that mTORC1 inhibition by biguanides is independent of TSC1/2 and, unexpectedly, independent of AMPK. Instead, we found that biguanides abolish mTORC1 activation in a Rag GTPase-dependent manner.
Results
dTSC1/dTSC2 are not essential for dTORC1 inhibition by energy-depletion
mTORC1 signaling is highly conserved from Drosophila to man (Montagne and Thomas, 2004; Pan and Hardie, 2002). However, Drosophila dTsc2 lacks the AMPK phosphorylation sites equivalent to mammalian TSC2 T1227 and S1345, as well as the encompassing sequence (Fig. 1A). Therefore, we investigated the ability of oligomycin, an inhibitor of mitochondrial oxidative phosphorylation, to inhibit dTORC1 signaling in Drosophila Kc167 cells, as compared to rapamycin. Basal and insulin-induced activation of dTORC1 signaling, as measured by dS6K T398 phosphorylation or dS6K activity, were inhibited to the same extent by oligomycin as they were by rapamycin (Fig. 1B), although in the former case, dTORC1 activity began to recover with time (see below). The glycolytic inhibitor 2-deoxyglucose (2DG), like oligomycin, also inhibited dS6K T398 phosphorylation (data not shown). To determine whether the effects of oligomycin were mediated through dTsc1/dTsc2, we treated Kc167 cells with double-stranded RNA (dsRNA) to deplete their protein levels. As previously reported (Gao et al., 2002; Radimerski et al., 2002), depletion of either protein enhanced dS6K activity, but did not alter inhibition by oligomycin (Fig. 1C). Depletion of dTsc1 was detected with antibodies, and though there are no appropriate antibodies to detect endogenous dTsc2 (Fig. 1C), consistent with others (Gao et al., 2002), dTsc1 protein levels decreased in cells treated with dsRNA against dTsc2 (Fig. 1C). These findings indicated that energy depletion can inhibit dTOR in a dTsc1/dTsc2-independent manner.
Figure 1. Oligomycin-induced dS6K dephosphorylation does not require dTSC1/2.
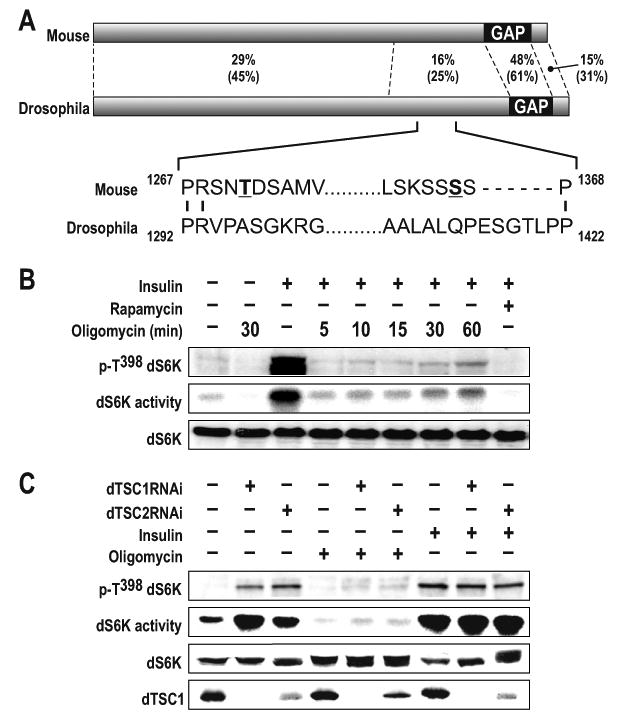
(A) Alignment of Drosophila TSC2 (accession number NP_524177) with the mouse TSC2, isoform 1 (accession number NP_035777). Percentage of sequence identity and similarity (parentheses) of different regions of TSC2 are indicated. The major AMPK phosphorylation sites in TSC2 reported to be affected by energy depletion are in bold and underlined. The mouse 1271 and 1366 phosphorylation sites correspond to the respective rat 1227 and 1345 sites described by Inoki et al. GAP denotes the GTPase-activating domain (B) Analysis of dS6K activity following treatment of Kc167 cells with 10μM oligomycin for 30min or 100nM insulin for 30min. In samples containing 100nM insulin plus 10μM oligomycin, the inhibitor was added for the indicated time prior to the 30min insulin treatment. 20nM rapamycin was added 15min prior to 30min insulin treatment. Western blot analysis of dS6K T398 phosphorylation and dS6K are as indicated (C) Analysis of dS6K activity following dTsc1, dTsc2 dsRNA treatment in the absence or presence of either 100nM insulin or 10μM oligomycin for 30min. Western blot analysis of dS6K T398 phosphorylation, dS6K activity, dS6K and dTSC1 are as indicated. The data shown are representative of two independent experiments.
TSC2 is not required for mTORC1 inhibition by energy-depleting agents
As previously reported (Gao et al., 2002; Jaeschke et al., 2002), TSC2-/- mouse embryonic fibroblasts (MEFs) exhibited elevated levels of S6K1 T389 phosphorylation as compared to TSC2+/+ MEFs (Fig. 2A). However, the addition of oligomycin was as efficient in inducing S6K1 T389 dephosphorylation, and AMPK T172 phosphorylation, in TSC2-/- MEFs as in TSC2+/+ MEFs (Fig. 2A). Moreover, as in Drosophila cells (Fig. 1B), the effect of oligomycin appeared to be transient (Fig. 2A). Although mitochondrial oxidative phosphorylation produces ATP more efficiently than glycolysis, most cultured cells depend on both processes (Schieke et al., 2006). Consistent with this, 100mM 2DG inhibited mTORC1 to a similar extent as oligomycin, with its activity recovering over time (Fig. 2B). Others have shown that TSC2-/- MEFs are resistant to lower concentrations of 2DG, and have attributed the sensitivity of higher concentrations to osmotic shock (Inoki et al., 2003). Although we also found that mTORC1 is more resistant to lower concentrations of 2DG in TSC2-/- versus TSC2+/+ MEFs (Fig. 2C), this was not the case for low concentrations of oligomycin (Fig. 2A). Because oligomycin and 2DG use distinct mechanisms to inhibit ATP production this raised the possibility that in TSC2-/- MEFs there was an inherent genetic alteration that confers 2G resistance. It has been reported that mTORC1 activation in TSC2-/- MEFs leads to the increased expression of HIF-1α and its glycolytic targets, including hexokinase-II (HKII) (Brugarolas and Kaelin, 2004). Importantly, HKII binds glucose, which is competitively inhibited by 2DG (Manuel y Keenoy et al., 1992). Thus, increases in HKII levels could be responsible for conferring resistance to inhibition of glycolysis by 2DG (Maher et al., 2007). We find that as TSC2-/-, versus TSC2+/+, MEFs became confluent HKII levels increased ∼ two-fold, as compared to either S6K1 or tubulin, which may explain their resistance to 2DG (Fig. 2D and Suppl. Fig. S1). In parallel, we asked whether mitochondrial oxidative phosphorylation and glycolysis act together to maintain mTORC1 signaling. We found that treatment with oligomycin or 2DG, followed by addition of the other inhibitor sixty minutes later, blunted the recovery in mTORC1 signaling, whereas a second addition of the same inhibitor had little effect (Figs. 2E and 2F, respectively). Together, the results show that both glycolysis and mitochondrial oxidative phosphorylation are utilized to maintain mTORC1 activation, and that the acute inhibition of mTORC1 signaling by blocking either ATP-producing process is largely independent of TSC1/TSC2.
Figure 2. TSC2 is not required for downregulation of S6K1 T389 by energy-depleting agents.
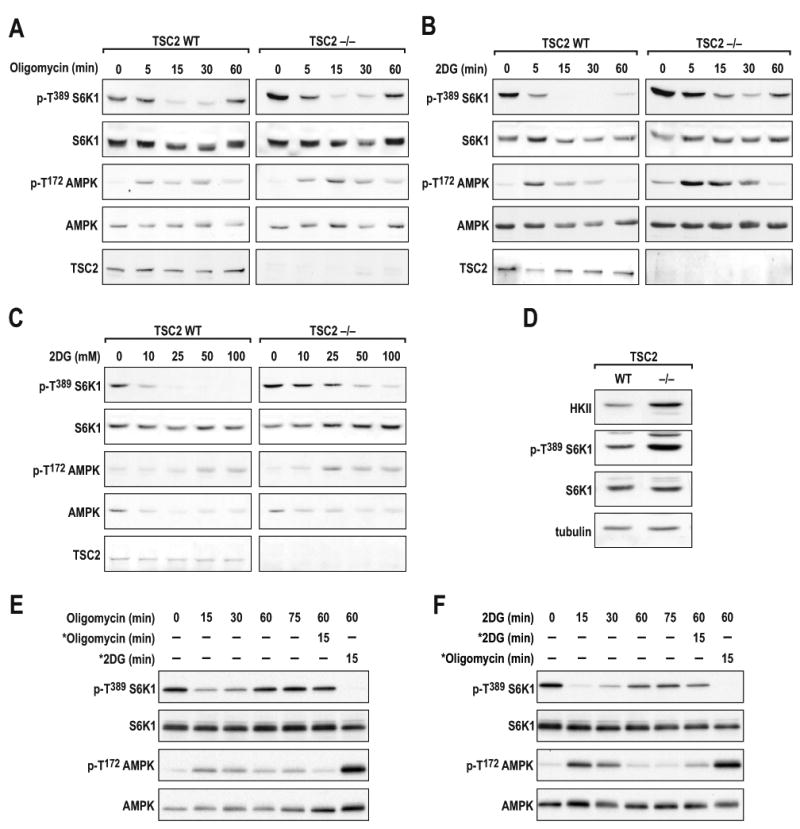
(A) TSC2+/+ or TSC2-/- MEFs were treated with either 10μM oligomycin or (B) 100mM 2DG for the times indicated. (C) TSC2+/+ or TSC2-/- MEFs were treated with the indicated concentrations of 2DG for 30min. (D) Western blot analysis of the indicated proteins in TSC2+/+ and TSC2-/- MEFs. (E and F) TSC2+/+ MEFs were treated with either 10μM oligomycin or 25mM 2DG at the indicated times. * denotes the addition of a fresh aliquot of either ATP-depleting agent following a 60min incubation with the initial inhibitor. The data shown are representative of at least three independent experiments.
Effect of metformin and phenformin on mTORC1 signaling in TSC2-/- MEFs
As oligomycin inhibits mTORC1 signaling in TSC2-/- MEFs, such cells would be expected to be sensitive to metformin, as reported for its hydrophobic derivative phenformin (Gwinn et al., 2008). However, Dowling and colleagues showed that TSC2-/- MEFs were resistant to metformin inhibition of mTORC1 signaling (Dowling et al., 2007). In contrast, we found that treating TSC2+/+ or TSC2-/- MEFs with metformin for 24hr, or phenformin for 1hr, led to inhibition of mTORC1 (Figs. 3A and 3B). In parallel, AMPK T172 phosphorylation and phosphorylation of its substrate acetyl coenzyme A carboxylase (ACC) at S79 increased (Figs. 3A and 3B). To understand the basis for the discrepancy between our findings and those of Dowling et al. (Dowling et al., 2007), we explored a number of parameters including metformin dosage, culture conditions, and cell density. We found that as TSC2-/- MEFs become confluent, mTORC1 signaling becomes sensitive to metformin, whereas metformin's ability to induce ACC S79 phosphorylation was enhanced (Fig. 3C). Neither mTORC1 inhibition nor AMPK activation was influenced by media constituents, as replenishment of fresh media at the time of metformin addition did not alter either response in confluent cells (Fig. 3D). It is known that oxygen consumption increases with cell density, leading to a hypoxic milieu and induction of HIF-1α (Dayan et al., 2009). Moreover, the induction of HIF-1α by hypoxia is amplified in TSC2-/- MEFs (Brugarolas et al., 2003), consistent with the increase in HKII, a HIF-1α target, that we saw as these cells become more confluent (Suppl. Fig. S1). Such a rise in HKII would be expected to increase a tumor cell's dependence on mitochondrial oxidative phosphorylation for ATP (Mathupala et al., 2006). These findings raised the possibility that metformin would inhibit the ability of TSC2-/- cells to form colonies in soft agar, a measure of malignantly transformed cells. Metformin was as effective in blocking colony formation of TSC2+/-ang1 cells (Fig. 3E), derived from a Tsc2+/− mouse spontaneous sarcoma (Govindarajan et al., 2003), and mTORC1 signaling (Figs. 3F), as rapamycin. Thus, metformin inhibits mTORC1 in a TSC-independent manner.
Figure 3. Metformin and phenformin inhibit mTORC1 signaling independent of TSC2.
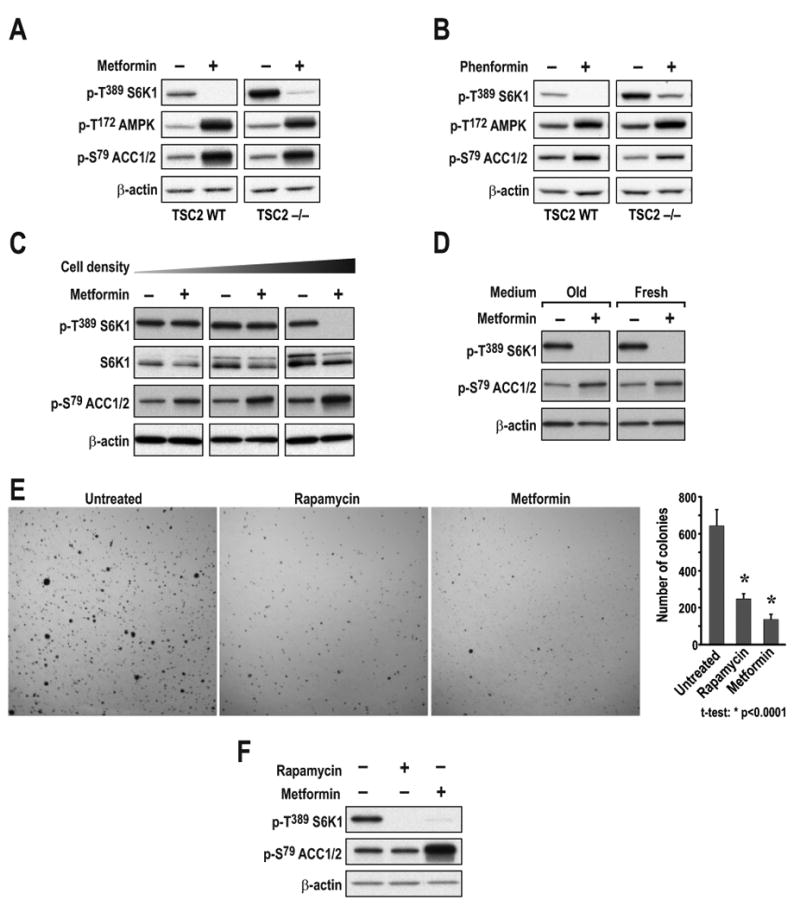
TSC2+/+ or TSC2-/- MEFs were treated with either (A) 10mM metformin for 24h or (B) 6mM phenformin for 1h. (C) TSC2-/- MEFs seeded at increasing density (1.25 ×104, 5 ×104, or 20 ×104 cells/well) were treated as in A. (D) TSC2-/- MEFs seeded at 20 ×104 cells/well and treated with 10mM metformin for 24h in either the initial or fresh medium. (E) TSC2+/-ang1 cells were allowed to grow in soft agar for 3 weeks, with or without 20nM rapamycin or 10mM metformin. Colonies were stained with MTT and counted as described in Experimental Procedures. (F) TSC2+/-ang1 cells were treated with either 20nM rapamycin for 15min or 10mM metformin for 24h. Western blots were performed as described in Experimental Procedures and probed with the indicated antibodies. The data shown are representative of at least two independent experiments.
AMPK is sufficient, but not necessary for mTORC1 inhibition by energy-depleting agents
Recent studies have shown that 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside (AICAR), through activation of AMPK, induces raptor phosphorylation, causing it to be sequestered by 14-3-3 proteins and inactivating mTORC1 (Gwinn et al., 2008). AICAR is metabolized to ZMP, an AMP analog that mediates the activation of AMPK (Hardie, 2008). Consistent with these findings, ectopic expression of either of two constitutively active AMPK mutants, γ1 R70Q or γ1 R152Q, strongly inhibited insulin-induced S6K1 T389 phosphorylation, and this inhibition was paralleled by AMPK T172 phosphorylation (Fig. 4A). However, such studies, including those of pharmacological intervention, are usually performed over extended times, raising the possibility of indirect AMPK effects on mTORC1 signaling. To test this possibility we used C2C12 cells, a non-transformed myoblast cell line, which responds acutely to both AICAR and energy-depleting agents (Jakobsen et al., 2001). We observed that AICAR induced rapid phosphorylation of AMPK T172 and ACC S79, but that there was a significant delay in S6K1 T389 dephosphorylation (Fig. 4B), which was not evident following 2DG treatment (Fig. 4C). To test AMPK dependence, we took advantage of AMPKα1-/-:α2-/- MEFs (AMPK dKO), which lack both catalytic subunits of AMPK (Laderoute et al., 2006). DKO and WT MEFs showed an equivalent dose-dependent dephosphorylation of S6K1 T389 upon 2DG treatment (Fig. 4D), whereas AMPK T172 and ACC S79 phosphorylation were only observed in WT MEFS. This effect was also observed in Drosophila Kc167 cells, where dsRNA depletion of dAMPK did not protect dTOR signaling from energy depletion (Suppl. Fig. S2). Unlike TSC2-/- MEFs, there was no difference in the dephosphorylation of S6K1 T389 at lower 2DG concentrations, supporting the argument that the resistance seen in the TSC2-/- MEFs is a TSC2-specific phenomenon (Fig. 2C). Similar inhibition of mTORC1 signaling was found in AMPK dKO MEFs following glucose withdrawal (Fig. 4E). These findings strongly support a model in which AMPK is dispensable for the inhibition of mTORC1 by glycolytic blockage.
Figure 4. AMPK is sufficient to inhibit mTORC1 signaling but it is not required for mTORC1 inhibition by energy depletion.
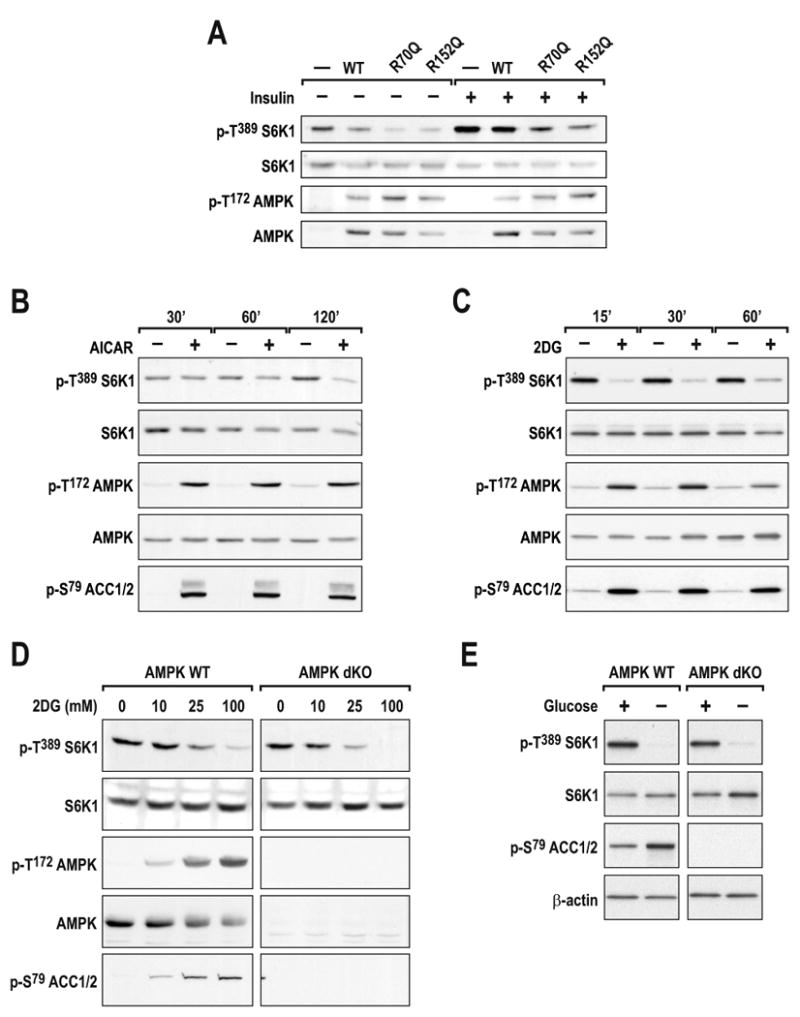
(A) WT indicates HEK293 cells in which α, β, and γ subunits of AMPK were co-expressed for 40h. R70Q and R172Q indicate that the WT γ subunit was substituted with the respective γ mutants. After 24h of ectopic expression, cells were deprived of serum overnight and stimulated with 200nM insulin for 30min. C2C12 myoblasts were treated with either (B) 2mM AICAR or (C) 25mM 2DG for the indicated times. (D) Primary AMPK α1 and α2 double knock-out (AMPK dKO) and AMPK WT MEFS were treated with 2DG for 30min at the indicated concentrations. (E) Immortalized AMPK dKO and AMPK WT cells were starved of glucose for 1h. Western blots were performed as described in Experimental Procedures and using the indicated antibodies. The data shown are representative of at least two independent experiments
Metformin and phenformin inhibit mTORC1 signaling independent of AMPK
Others have reported that phenformin inhibits mTORC1 signaling through AMPK phosphorylation of raptor (Gwinn et al., 2008). These findings suggest that the biguanides blunt mTORC1 signaling in a manner that is distinct from the effects of 2DG and glucose deprivation. However, we found that treatment of AMPK dKO MEFs with either biguanide, abolished S6K1 T389 phosphorylation to the same extent as in AMPK WT MEFs (Figs. 5A and 5B). In AMPK dKO MEFs, as compared to AMPK WT MEFs, neither agent had an effect on ACC S79 phosphorylation (Figs. 5A and 5B), supporting the notion that they operate through an AMPK-independent mechanism to inhibit mTORC1 signaling. It should be noted that, despite the absence of AMPK, such treatment did not have a more pronounced effect on the energy status of AMPK dKO versus AMPK WT MEFs, (Suppl. Fig. S3A). Unlike the biguanides, AICAR had no effect on S6K1 T389 dephosphorylation in AMPK dKO MEFs, verifying the specificity of AICAR for AMPK, as well as the complete loss of AMPK function in these cells (Fig. 5C). Moreover, this effect was not confined to S6K1 T389 phosphorylation as similar results were observed for 4E-BP1, as measured by its electrophoretic mobility (Fig. 5C). Consistent with the findings of Gwinn et al. (Gwinn et al., 2008), we found that phenformin induced raptor S792 phosphorylation, which paralleled inhibition of S6K1 T389 phosphorylation in AMPK WT MEFs (Fig. 5D). However, phenformin did not induce raptor S792 phosphorylation in AMPK dKO MEFs, despite abolishing S6K1 T389 and 4E-BP1 phosphorylation (Fig. 5D). Finally, mTORC1 derived from insulin-stimulated AMPK dKO MEFs, as compared to AMPK WT MEFs, showed elevated in vitro S6K1 T389 phosphorylation activity, which was unaffected by AICAR pretreatment but was abolished with phenformin (Fig. 5E). This led to the finding that metformin was just as efficient as rapamycin in blocking colony formation of AMPK dKO MEFs in soft agar (Suppl. Fig. S3B). Taken together, the results show that the inhibitory effect of biguanides on mTORC1 activity is AMPK independent.
Figure 5. Metformin and phenformin, but not AICAR, inhibit mTORC1 independent of AMPK.
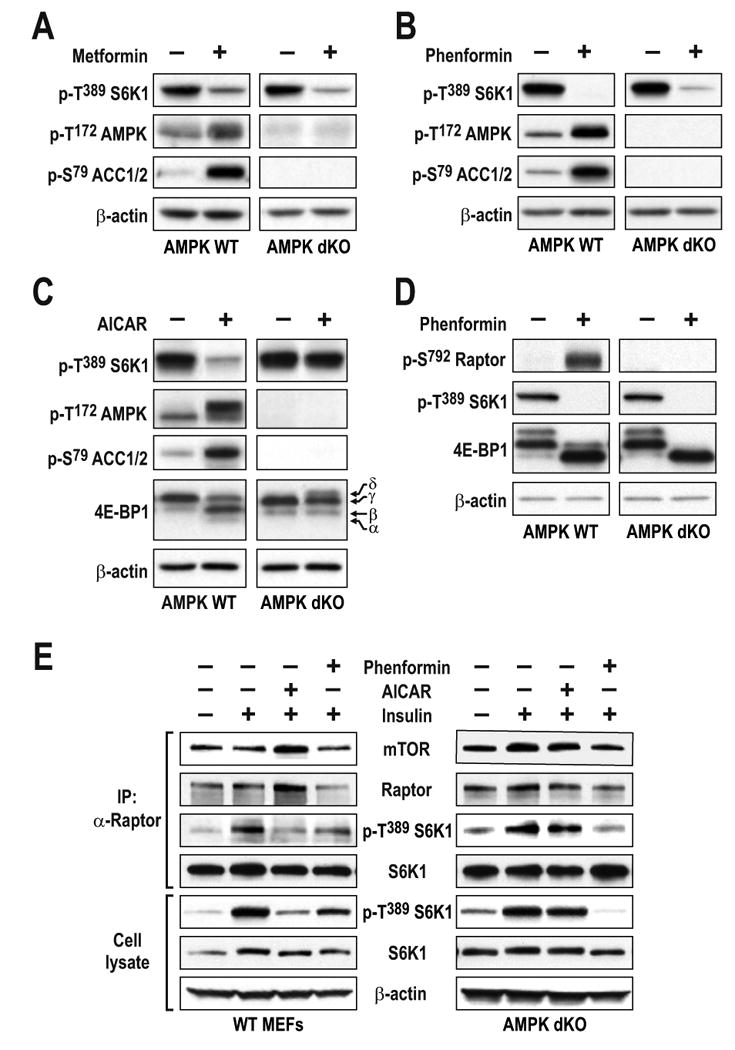
Western blot analysis of extracts from AMPK dKO or AMPK WT MEFs treated either with (A) 10mM metformin for 24h, (B and D) 6mM phenformin for 1h, or (C) 2mM AICAR for 2h. (E) Serum-starved AMPK dKO or AMPK WT MEFs treated for 1h with phenformin or AICAR were stimulated with insulin for 15 min. Raptor was immunoprecipitated, and in vitro mTORC1 activity was assayed as described previously (Gulati et al., 2008; Sancak et al., 2007). In parallel, western blot analyses were performed on cell lysates. The data shown are representative of multiple independent experiments.
Absence of AMPK does not affect metformin-mediated glucose uptake
The findings above have particular relevance in the treatment of Type 2 diabetes, where mTORC1/S6K1 hyperactivation has been shown to induce insulin resistance through IRS1 phosphorylation (Harrington et al., 2004; Tremblay et al., 2007; Tzatsos and Kandror, 2006; Um et al., 2006; Um et al., 2004). As metformin treatment is known to induce glucose uptake in skeletal muscle (Sakamoto et al., 2005; Zhou et al., 2001), it was hypothesized that, at least in part, these effects are mediated by metformin-induced activation of the AMPK-TSC1/2 axis and inhibition of mTORC1/S6K1 phosphorylation of IRS1 (Tzatsos and Kandror, 2006). To examine the role of AMPK in glucose uptake in L6 myotubes, the AMPKα1/α2 catalytic subunits were depleted by siRNA treatment, with such treatment abolishing AICAR-induced ACC S79 phosphorylation and S6K1 T389 dephosphorylation (Fig. 6A). However, metformin treatment still inhibited S6K1 T389 phosphorylation, but had no effect on ACC S79 phosphorylation in L6 myotubes depleted of AMPKα1/α2 (Fig. 6A). Importantly, depletion of AMPKα1/α2 had no measurable effect on glucose uptake induced by metformin, whereas such treatment largely abolished the effect of AICAR on this response (Fig. 6B). The data presented here suggest that AMPK is not essential for the effects of metformin on glucose uptake.
Figure 6. Absence of AMPK does not affect metformin-mediated glucose uptake.
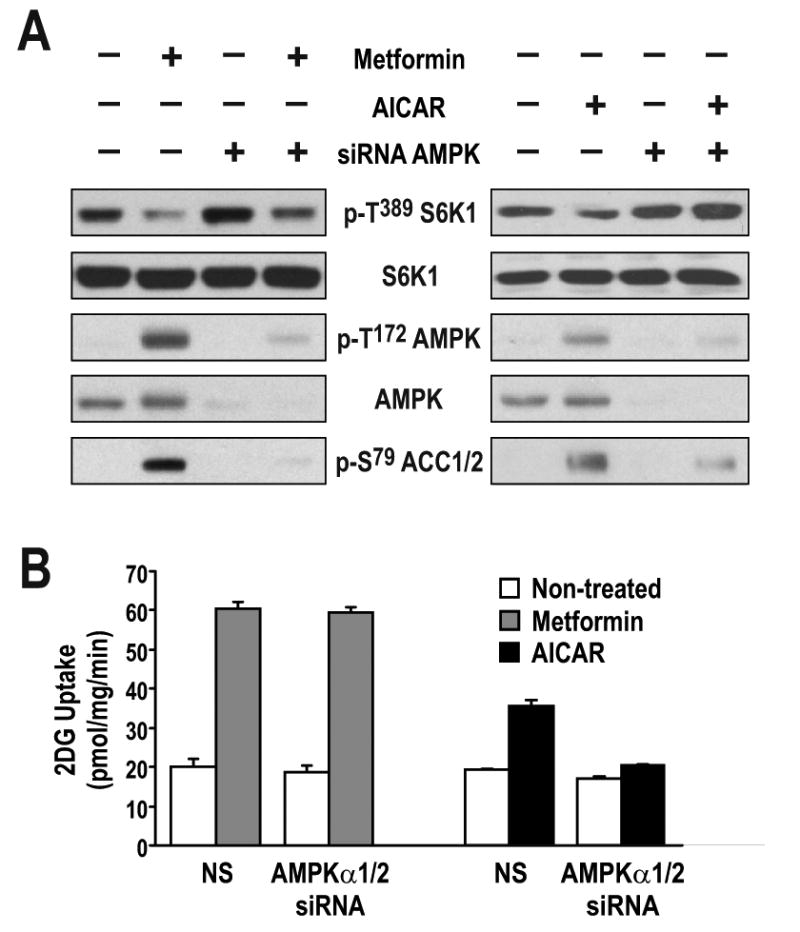
(A) L6 myotubes were transfected with either non-silencing dsRNA (NS) or with two different dsRNAs directed against either the α1 or the α2 catalytic subunit of AMPK and incubated for 72 hrs. Cells were then treated with either 5mM metformin for the last 24 hrs or 2mM AICAR for the last 2 hrs. Parallel plates were either extracted for western blotting with the indicated antibodies or (B) further processed for glucose-uptake measurements, as described in Experimental Procedures. Results are representative of three independent experiments conducted in triplicate.
Metformin and phenformin inhibit RAG GTPase-mediated mTORC1 signaling
In the search for potential mitochondria-dependant mechanisms by which biguanides reduce mTORC1 signaling, we analyzed the role of redox status (Sarbassov and Sabatini, 2005) and FKBP38 (Bai et al., 2007), a member of the FK506-binding protein (FKBP) family. Although oxidizing agents, such as phenylarsine oxide (PAO), raise basal levels of mTORC1 signaling, they did not protect this response from the effects of either glycolytic or mitochondrial oxidative phosphorylation inhibitors (Suppl. Fig. S4A). Similarly, siRNA depletion of FKBP38 did not prevent such inhibitors from abolishing mTORC1 signaling (Suppl. Fig. S4B). Recently, it was shown that amino acids (AAs) activate a novel family of GTPases, termed Rags, to stimulate mTORC1 signaling (Kim et al., 2008; Sancak et al., 2008). The ability of Rag GTPases to mediate this response is based on their capacity to induce translocation of mTORC1 to a perinuclear intracellular compartment occupied by Rheb (Sancak et al., 2008). We found in growing cells that mTOR localized to large perinuclear aggregates, whereas, upon removal of AAs, mTOR diffused throughout the cytoplasm, an effect reversed by re-addition of AAs (Fig. 7A). The specificity of the mTOR polyclonal antibody signal was confirmed by deleting mTOR in Cre+-mTORfl/fl MEFs, which largely abolished mTOR staining by immunofluorescence or western blot analysis (Suppl. Fig. S4C). Similar to the effect of AA withdrawal, treatment of cells growing in complete media with phenformin caused mTOR to disperse throughout the cytoplasm (Fig. 7A and Suppl. Fig. S4D). Although phenformin phenocopied AA withdrawal, it had no effect on AA steady state levels (Suppl. Fig. S4E & F). To test whether the inhibitory effects of phenformin on mTORC1 signaling are mediated by the Rag GTPases, we compared the ability of a transiently expressed WT HA-GST-Rag B versus a constitutively activated HA-GST-Rag B construct to protect phosphorylation of a GST-tagged S6K1 T389 reporter from either AA withdrawal or phenformin treatment. We found that AA withdrawal or phenformin treatment almost totally abolished reporter S6K1 T389 phosphorylation in cells transfected with the empty vector (Fig. 7B). Co-transfection of either constitutively active or WT HA-GST-Rag B slightly raised basal levels of reporter S6K1 T389 phosphorylation; however, only constitutively activated HA-GST-Rag B protected against AA withdrawal and phenformin treatment (Fig. 7B). Similar results were obtained by treatment with oligomycin (Suppl. Fig. S4G). These findings support a role for biguanides in suppressing mTORC1 signaling through inhibition of Rag function, as depicted in Fig. 7C.
Figure 7. Rag GTPases mediate the inhibition of mTORC1 by Phenformin.
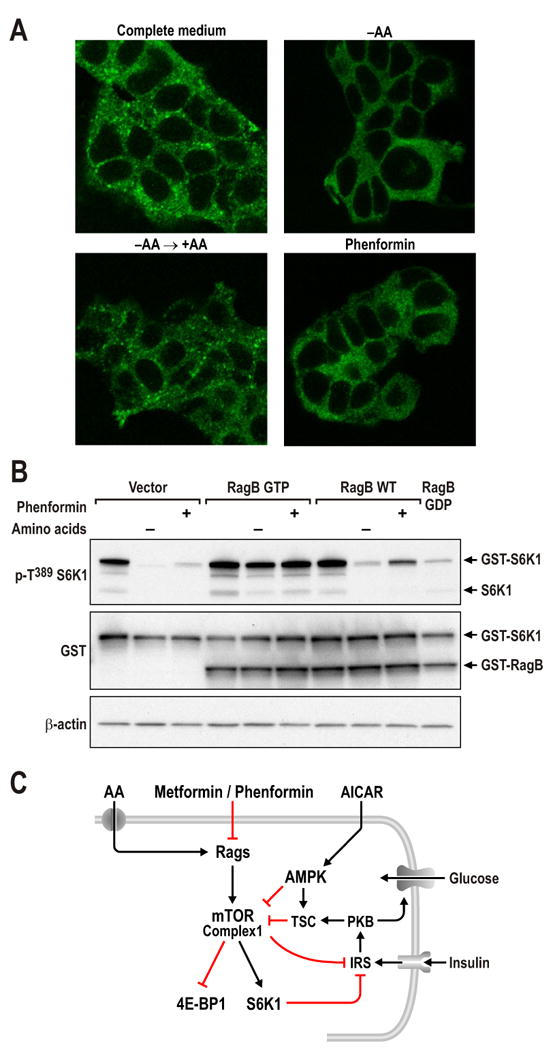
(A) HEK293T cells were either starved for serum and AAs for 50 min (-AA) and re-stimulated with AAs for 10min (-AA→+AA), or cells in full media treated with 5mM phenformin for 1hr. Cells were processed as described in Experimental Procedures and immunofluorescence assays were performed to detect mTOR (green) (B) HEK293T cells with PRK5 empty vector, used as a transfection control, or expressing epitope-tagged HA-GST-RagB GTP or WT HA- GST-RagB were deprived of AAs for 1hr, or treated with 5mM phenformin for 1hr. Western blots were performed as described in Experimental Procedures and proteins were detected with the indicated antibodies. These studies are representative of at least two independent experiments (C) Model for metformin/phenformin action on mTORC1.
Discussion
The acute and chronic response to energy depletion
The acute response to energy depletion is transient in Drosophila Kc167 cells and in mammalian cells (Figs. 1 and 2). Although mitochondrial oxidative phosphorylation, as compared to glycolysis, is the most efficient mechanism to produce ATP, most cultured cells depend equally on both processes to generate energy (Schieke et al., 2006). We found that re-addition of the same energy-depleting agent during the recovery period had little effect on mTORC1 signaling or on AMPK activation, whereas addition of the alternative inhibitor had immediate effects on both responses (Figs 2C and 2D). These data imply that mTORC1 signaling is sensitive to both sources of energy production and that inhibition by either is restored by the upregulation of the alternate energy-generating system. Although the rise in the AMP/ATP ratio did not directly reflect the extent of mTORC1 signaling inhibition, the addition of both inhibitors together led to a dramatic rise in this response (A. K. and G. T., unpublished). In addition to the acute response, a second, chronic energy-depletion response, which is mediated by the tumor suppressor, Regulated in Development and DNA Damage responses (REDD1) (Sofer et al., 2005) in a TSC1/TSC2-dependent manner, has been described. REDD1 suppresses mTORC1 activity by releasing TSC2 from 14-3-3 protein inhibition (DeYoung et al., 2008). Unlike TSC1/TSC2, the role of AMPK in the chronic energy response has been ambiguous, because a dominant-interfering AMPK blocks long-term 2DG inhibition of mTORC1 signaling, but an inducible REDD1 construct is able to bypass this block (Sofer et al., 2005). It does not appear that AMPK acts upstream to regulate REDD1, as Sofer et al. could detect no changes in REDD1 levels or other modifications of the protein following 2DG treatment in the presence of a dominant-interfering AMPK (Sofer et al., 2005). The use of AMPK dKO MEFs will be valuable in addressing the chronic energy response.
HKII upregulation in TSC2-deficient MEFs
We first proposed a model by which mTORC1 was controlled by ATP levels (Dennis et al., 2001). However, later studies showed that AMPK phosphorylates TSC2, potentially increasing TSC2 GAP activity towards Rheb (Inoki et al., 2003). In these latter studies, 25mM 2DG had little inhibitory effect on mTORC1 signaling in TSC2-/- MEFs (Inoki et al., 2003). Here we also noted that treatment with lower concentrations of 2DG attenuated inhibition of mTORC1 signaling in TSC2-/- MEFs. However, such cells are known to express elevated levels of HIF-1α (Brugarolas et al., 2003), which can drive the expression of HKII, whose ability to bind glucose is inhibited by 2DG (Wick et al., 1957). This inhibition is the basis of Phase 1 clinical trials employing 2DG in the treatment of tumors that exhibit increased glycolysis (Maher et al., 2007). Thus, the increase in HKII levels may explain the requirement for a higher concentration of 2DG to achieve inhibition of mTORC1 signaling (see below), as has been shown in resistant tumor cell lines under conditions where HIF-1α is induced by hypoxia (Maher et al., 2007). Such substrate-induced resistance is observed for other chemotherapeutic agents, including methotrexate, where increased expression of dihydrofolate reductase leads to drug resistance (Bertino et al., 1983). This model may also explain why decreasing glucose in the culture media increases the sensitivity of TSC2-/- MEFs to lower concentrations of 2DG (Smith et al., 2005).
The effect of metformin on mTORC1 signaling in TSC2-deficient cells
We find that biguanides inhibit mTORC1 signaling in a TSC2-independent manner. Efforts to reconcile this observation with that of Dowling et al. (Dowling et al., 2007) led to our finding that, as the density of cells in culture increases, mTORC1 signaling in TSC2-deficient cells acquires sensitivity to metformin inhibition (Figs. 3C and 3D, respectively). The increased demand for oxygen as a function of cell density leads to a hypoxic environment and the induction of HIF-1α (Dayan et al., 2009; Paltoglou and Roberts, 2005), an effect readily reversed by increasing the available oxygen or inhibiting oxygen consumption by oligomycin treatment (Dayan et al., 2009). In TSC2-/- MEFs, basal levels of HIF-1α are elevated, as are a number of HIF-1α target genes, an effect which is strongly potentiated by hypoxia (Brugarolas et al., 2003). We found that as TSC2-/- MEFs became confluent, the levels of HKII, a HIF-1α target, increased approximately 2-fold (Suppl. Fig. S1). In most tumor cells, under aerobic conditions, glycolysis is upregulated to generate glycolytic intermediates, required as biosynthetic building blocks (Mathupala et al., 2006, 2009). An early step in tumorigenesis is the switch from the use of HK-IV (glucokinase) to HKII (Mathupala et al., 2006, 2009), the latter having a much higher affinity for glucose, increasing the rate of glucose-6 phosphate production. The increased ATP required for the production of glucose-6 phosphate is channeled from the mitochondrial ATP synthase to the porin-like protein voltage-dependent anion channel (VDAC), which localizes HKII to the outer mitochondrial membrane (Mathupala et al., 2006). Under these conditions the tumor cell would be predicted to become more sensitive to inhibitors of mitochondrial oxidative phosphorylation. The increase in HKII may explain the acquired sensitivity to metformin of mTORC1 signaling as TSC2-/- MEFs become confluent (Fig 3C). In preliminary studies we found that partial siRNA depletion of HKII in TSC2-/- MEFs, afforded mTORC1 some protection from inhibition by metformin (A. K. and G. T., unpublished); however, a more in-depth analysis will be required to understand the role of HKII on density-dependent sensitivity. The difference in metformin sensitivity prompted us to test the efficacy of the drug in a TSC-deficient setting, which we found inhibits colony-formation to the same extent as rapamycin. It would be of interest to determine, through clinical registries, whether TSC patients being treated for diabetes with metformin showed any alleviation of TSC syndromes. This may be of clinical importance as extended rapamycin treatment is known to suppress pancreatic β-cell adaptation to hyperglycemia and to exacerbate Type 2 diabetes (Fraenkel et al., 2008).
The effect of biguanides on mTORC1 signaling in AMPK-deficient cells
Our findings in C2C12 cells showed that AICAR-induced S6K1 T389 dephosphorylation is modest and delayed, as compared to the rapid phosphorylation of AMPK T172 and ACC S79 (Fig. 4C), suggesting that the effects of AMPK on mTORC1 are indirect. Moreover, that the kinetics of 2DG-induced inhibition of mTORC1 signaling were so much more rapid than those induced by AICAR (Fig. 4D), despite similar effects on ACC S79 phosphorylation, led to the notion of an alternative AMPK-independent mechanism for acute energy depletion. This was substantiated in AMPK dKO MEFs, where energy-depleting agents, but not AICAR, blocked S6K1 T389 phosphorylation (Figs. 5 and 6). Moreover, although metformin induced raptor S792 phosphorylation in AMPK WT MEFs, this response was not detected in AMPK dKO MEFS. On the basis of these observations, we conclude that biguanides do not require TSC1/TSC2 to inhibit mTORC1 signaling and, although AMPK activation is sufficient to attenuate mTORC1 signaling, it is not essential for these responses.
Inhibition of mTORC1 by biguanides is dependent on the Rag GTPases
A search for alternative mechanisms for biguanide action led to a study of redox status, which was shown to affect the interaction between raptor and mTOR, in a manner similar to nutrients, including AAs and glucose (Kim et al., 2002; Sarbassov and Sabatini, 2005). Although the cell-permeable oxidant phenylarsine oxide (PAO), increased basal levels of S6K1 T389 phosphorylation, treatment of cells with either oligomycin or 2DG induced dephosphorylation to the same extent as in non-PAO-treated cells (Suppl. Fig. S4A). This argued against the involvement of redox status in mediating inhibition of mTORC1 by energy depletion. We also examined the potential role of FKBP38, which contains a mitochondrial targeting sequence and has recently been described as a negative regulator of mTORC1 by sequestering Rheb (Bai et al., 2007). However, siRNA depletion of FKBP38 did not protect mTORC1 signaling from either the glycolytic or mitochondrial oxidative phosphorylation inhibitors, suggesting that it does not convey the energy-depletion signal to mTORC1 (Suppl. Fig. S4B). Given that glucose deprivation affects the interaction between raptor and mTOR and the kinase activity of the raptor-mTOR complex in a manner similar to AA withdrawal (Kim et al., 2002; Sarbassov and Sabatini, 2005), we reasoned that the Rag GTPases might be involved in the metformin inhibitory response. Rag GTPases have been shown to direct mTORC1 to a cellular compartment that contains Rheb, leading to kinase activation (Sancak et al., 2008). Consistent with this possibility, we found that, similar to AA withdrawal, phenformin treatment caused mTOR to disperse from perinuclear aggregates and diffuse throughout the cytoplasm (Fig. 7A). Moreover, although Sancak et al. concluded that energy-deprivation agents, such as 2DG, appeared not to mediate their inhibitory affects through the Rag GTPases (Sancak et al., 2008), we found that ectopic expression of a constitutively active, but not WT, Rag GTPase protected mTORC1 signaling from inhibition by phenformin and oligomycin (Fig. 7B and Suppl. Fig. S4G).
Metformin, mTORC1 signaling, and glucose uptake
Metformin is argued to lower hyperglycemia by inhibiting hepatic gluconeogenesis and increasing glucose uptake in skeletal muscle (Zhou et al., 2001). These effects are attributed to activation of AMPK and, in the case of hepatic gluconeogenesis, recent studies in LKB1-/--deficient mice support such a model (Shaw et al., 2005). However, it should be noted that the effects of metformin on glucose phosphorylation in AMPK dKO hepatocytes are AMPK independent (Guigas et al., 2006). In the case of increased glucose uptake in skeletal muscle, both mTORC1 and S6K1 have been implicated in suppressing insulin signaling by the phosphorylation of IRS1 (Um et al., 2006). Metformin, as well as 2DG, have been shown to reverse the effects of mTORC1 phosphorylation on two IRS1 sites known to antagonize PI3K binding to IRS1 (Tzatsos and Kandror, 2006). Given that both agents induced increased AMPK T172 phosphorylation, it was concluded that the effects of metformin and 2DG were mediated by activation of LKB1 and AMPK (Tzatsos and Kandror, 2006). However, we found that both metformin and phenformin inhibited mTORC1 signaling in the absence of AMPK (Figs. 6A & B). Additionally, we found that depletion of AMPKα1/α2 catalytic subunits did not block the metformin-enhanced increase in glucose uptake (Fig. 7). Taken together, these studies reveal the existence of an alternative TSC1/2-AMPK-independent pathway, potentially mediated by RAG GTPase, which responds rapidly to perturbations in energy status to maintain homeostatic mTORC1 signaling.
In summary, the studies presented here reveal a novel mechanism by which metformin inhibits mTORC1 signaling. Given that greater than 100 million individuals are treated daily for Type 2 diabetes with metformin, and that its use may be extended to the treatment of specific cancers (Shackelford and Shaw, 2009), it is critical to determine its mode of action. Therefore, elucidating the mechanism by which inhibition of Rag GTPases by metformin suppress mTORC1 signaling could be beneficial in the treatment of particular human pathologies.
Experimental Procedures
Materials
Reagents were from the following sources: bovine insulin, 2DG, metformin, and phenformin from Sigma Aldrich; rapamycin from Calbiochem; oligomycin from Calbiochem and Acros; AICAR from Toronto Research Chemicals; antibodies to phospho-T389 S6K1, phospho-T172 AMPK, AMPK, phospho-S79 ACC1/2, TSC2, and phospho-T398 dS6K from Cell Signaling Technology and antibodies to S6K1, hexokinase II, and 4E-BP1 (R113) from Santa Cruz Biotechnology. The monoclonal tubulin E7 antibody was from the Developmental Studies Hybridoma Bank, NICHD, maintained by The University of Iowa. The Drosophila TSC1 antibody was from D. Pan (Johns Hopkins). The expression vectors for the WTα, β, and γ subunits of AMPK and the γ subunit mutants R70Q and R172Q, have been described (Adams et al., 2004). The expression vectors pRK5-HA GST RagB 99L and pRK5-HA GST RagB 54L were from D. Sabatini and the wt pRK5-HA GST RagB vector was from Addgene. 2-deoxy-D-[3H]glucose was from NEN Life Science. Calcium phosphate-based transfection reagent CellPhect was from GE Healthcare. AMPK α1 and α2 siRNA sequences were GCAUAUGCUGCAGGUAGAUdtdt (nucleotides 738-756, accession no NM_019142) and CGUCAUUGAUGAUGAGGCUdtdt (nucleotides 865-883, accession no NM_023991), respectively.
Cell culture, treatments, and transfections
TSC2+/+/P53-/- and TSC2-/-/P53-/- were from D. Kwiatkowski (Harvard Medical School). AMPK WT MEFs and AMPK dKO, have been previously described (Laderoute et al., 2006). All mammalian cell lines were grown in DMEM high glucose supplemented with 10% fetal bovine serum (FBS) and 50μg/ml of a Pen/Strep mix, except for L6 cells, which were grown in α-MEM with the same additives. Typically 6-well plates were seeded at 105 cells per well and inhibitors were added directly in the medium after 48h growth. Glucose starvation was performed using glucose and pyruvate free medium. All exogenously expressed cDNAs were transfected using Fugene 6 (Roche) according to the manufacturer's instructions. AMPK α1, α2 siRNA treatments were carried out on L6 myoblasts seeded in 12-well plates by using CellPhect according to the manufacturer's instructions. 6h post-transfection, the medium was replaced with α-MEM supplemented with 2% FBS to induce differentiation, and treatments were performed 72h post-transfection. Glucose uptake measurements were performed as described previously (Bedard et al., 1997).
Cell extraction and western blotting
All mammalian cell extracts were prepared with the ELB extraction buffer: 50mM Tris pH 7.5, 0.1% NP40, 120mM NaCl, 1mM EDTA, 6mM EGTA, 20mM NaF, 1mM Na pyrophosphate, 30mM 4-nitrophenyl phosphate, 1mM benzamidine, and one tablet of EDTA-free protease inhibitor cocktail (Roche) per 25ml of buffer. Cells were rinsed twice with ice-cold PBS and extracted on ice with ice-cold ELB buffer. Typically, cell extracts were kept at -80°C overnight and the soluble fractions cleared of debris the following day with a 14000 rpm spin for 10min at 4°C. A stock of the extract was made in 1× Laemmli buffer and either stored at -20°C 10-30μg of protein, was used for western blotting.
Drosophila
Drosophila Kc167 cells were maintained as described (Radimerski et al., 2002). Protein extracts of cells were prepared and kinase activity of dS6K was measured as previously described (Radimerski et al., 2002). H2B was used as a substrate for the assay. Treatment with dsRNA was performed essentially as described (Radimerski et al., 2002), with an incubation time of 7 days. All primers were designed starting with the T7 RNA polymerase binding site as follows: 5′-TTAATACGACTCACTATAGGGAGA-3′ dTsc1, accession no. AF173560, sense-primer 436-453, anti-sense-primer 1081-1098; dTsc2, accession no. AF172995, sense primer 591-608, antisense primer 1371-1388.
Confocal microscopy
HEK-293T cells were seeded on 6cm dish with fibronectin-coated glass coverslips. Cells were treated as indicated 36hrs post seeding and processed as described (Sancak et al., 2008). The coverslips were mounted on glass slides using Vectashield, (Vector Laboratories) and imaged using a Zeiss LSM 510 Meta confocal with the appropriate filter settings.
Anchorage-independent growth assay
Colony formation assays were performed in 6-well plate format in 1ml of DMEM/10% FBS containing 0.5% Difco™ Agar Noble (Becton Dickinson). Trypsinized cells were then re-suspended in 1ml of DMEM/10% FBS containing 0.3% agar at either 40000 (TSC2+/-ang1) or 60000 cells/ml (WT and AMPK dKO), supplemented with the indicated inhibitor. Plates were incubated overnight at 37°C, 5%CO2 and 0.5ml of DMEM/10% containing the desired inhibitor and then overlaid on the agar and replaced every 3 days. Colonies were stained by washing plates in 37°C PBS, then incubation with PBS containing 0.5mg/ml MTT for 4h at 37°C. Colony number was determined using the AxioVision software (Carl Zeiss Vision).
Sequence analysis
Alignment of the TSC2 sequences and the AMPK phosphorylation motif search were carried out using Vector-NTI software (InforMax).
Supplementary Material
Acknowledgments
We are indebted to all the members of the Kozma/Thomas laboratory for discussions and critical reading of the manuscript. We also thank M. Daston and G. Doerman for editing of the manuscript and computer graphics, respectively. We thank Birgit Ehmer for assistance in microscopy, D. Pan for the Drosophila TSC1 antibody, D. Sabatini laboratory for RagB constructs and useful discussions. A. K. was supported by the Krebsliga, Basel. A.K and A.S are supported by appointments to the Research Participation Program at the Air Force Research Laboratory, Human Effectiveness Directorate, Bioscience and Protection, Wright Patterson AFB administered by the Oak Ridge Institute for Science and Education. B.V. is supported by funding from the Association Nationale de la Recherche (Muscle bioenergetics R06428KS), Association de Langue Française pour l'Etude du Diabète et des Maladies Métaboliques and by the EXGENESIS Integrated Project (LSHM-CT-2004-005272) funded by the European Commission. B. E. K. is supported by the NHMRC and ARC. A.M is supported by a grant from the Canadian Institutes of Health Research (CIHR). G.T. and S. K. are supported by the NIH Mouse Models for Human Cancer Consortium, U01 CA84292-06, and NIH NIDDK grant DK73802. G. T. is supported by the Strauss Chair in Cancer Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams J, Chen ZP, Van Denderen BJ, Morton CJ, Parker MW, Witters LA, Stapleton D, Kemp BE. Intrasteric control of AMPK via the gamma1 subunit AMP allosteric regulatory site. Protein Sci. 2004;13:155–165. doi: 10.1110/ps.03340004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai X, Ma D, Liu A, Shen X, Wang QJ, Liu Y, Jiang Y. Rheb activates mTOR by antagonizing its endogenous inhibitor, FKBP38. Science. 2007;318:977–980. doi: 10.1126/science.1147379. [DOI] [PubMed] [Google Scholar]
- Bailey CJ, Turner RC. Metformin. N Engl J Med. 1996;334:574–579. doi: 10.1056/NEJM199602293340906. [DOI] [PubMed] [Google Scholar]
- Bedard S, Marcotte B, Marette A. Cytokines modulate glucose transport in skeletal muscle by inducing the expression of inducible nitric oxide synthase. Biochem J. 1997;325(Pt 2):487–493. doi: 10.1042/bj3250487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertino JR, Carman MD, Weiner HL, Cashmore A, Moroson BA, Srimatkandada S, Schornagel JH, Medina WD, Dube SK. Gene amplification and altered enzymes as mechanisms for the development of drug resistance. Cancer treatment reports. 1983;67:901–904. [PubMed] [Google Scholar]
- Bowker SL, Majumdar SR, Veugelers P, Johnson JA. Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin: Response to Farooki and Schneider. Diabetes care. 2006;29:1990–1991. doi: 10.2337/dc06-0997. [DOI] [PubMed] [Google Scholar]
- Brugarolas J, Kaelin WG., Jr Dysregulation of HIF and VEGF is a unifying feature of the familial hamartoma syndromes. Cancer Cell. 2004;6:7–10. doi: 10.1016/j.ccr.2004.06.020. [DOI] [PubMed] [Google Scholar]
- Brugarolas JB, Vazquez F, Reddy A, Sellers WR, Kaelin WG., Jr TSC2 regulates VEGF through mTOR-dependent and -independent pathways. Cancer Cell. 2003;4:147–158. doi: 10.1016/s1535-6108(03)00187-9. [DOI] [PubMed] [Google Scholar]
- Byfield MP, Murray JT, Backer JM. hVps34 Is a Nutrient-regulated Lipid Kinase Required for Activation of p70 S6 Kinase. J Biol Chem. 2005;280:33076–33082. doi: 10.1074/jbc.M507201200. [DOI] [PubMed] [Google Scholar]
- Dann SG, Selvaraj A, Thomas G. mTOR Complex1-S6K1 signaling: at the crossroads of obesity, diabetes and cancer. Trends Mol Med. 2007 doi: 10.1016/j.molmed.2007.04.002. [DOI] [PubMed] [Google Scholar]
- Dayan F, Bilton RL, Laferriere J, Trottier E, Roux D, Pouyssegur J, Mazure NM. Activation of HIF-1alpha in exponentially growing cells via hypoxic stimulation is independent of the Akt/mTOR pathway. Journal of cellular physiology. 2009;218:167–174. doi: 10.1002/jcp.21584. [DOI] [PubMed] [Google Scholar]
- Dennis PB, Jaeschke A, Saitoh M, Fowler B, Kozma SC, Thomas G. Mammalian TOR: a homeostatic ATP sensor. Science. 2001;294:1102–1105. doi: 10.1126/science.1063518. [DOI] [PubMed] [Google Scholar]
- DeYoung MP, Horak P, Sofer A, Sgroi D, Ellisen LW. Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling. Genes Dev. 2008;22:239–251. doi: 10.1101/gad.1617608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowling RJ, Zakikhani M, Fantus IG, Pollak M, Sonenberg N. Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res. 2007;67:10804–10812. doi: 10.1158/0008-5472.CAN-07-2310. [DOI] [PubMed] [Google Scholar]
- Fraenkel M, Ketzinel-Gilad M, Ariav Y, Pappo O, Karaca M, Castel J, Berthault MF, Magnan C, Cerasi E, Kaiser N, Leibowitz G. mTOR inhibition by rapamycin prevents beta-cell adaptation to hyperglycemia and exacerbates the metabolic state in type 2 diabetes. Diabetes. 2008;57:945–957. doi: 10.2337/db07-0922. [DOI] [PubMed] [Google Scholar]
- Gao X, Zhang Y, Arrazola P, Hino O, Kobayashi T, Yeung RS, Ru B, Pan D. Tsc tumour suppressor proteins antagonize amino-acid-TOR signalling. Nat Cell Biol. 2002;4:699–704. doi: 10.1038/ncb847. [DOI] [PubMed] [Google Scholar]
- Garami A, Zwartkruis FJ, Nobukuni T, Joaquin M, Roccio M, Stocker H, Kozma SC, Hafen E, Bos JL, Thomas G. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol Cell. 2003;11:1457–1466. doi: 10.1016/s1097-2765(03)00220-x. [DOI] [PubMed] [Google Scholar]
- Govindarajan B, Mizesko MC, Miller MS, Onda H, Nunnelley M, Casper K, Brat D, Cohen C, Arbiser JL. Tuberous sclerosis-associated neoplasms express activated p42/44 mitogen-activated protein (MAP) kinase, and inhibition of MAP kinase signaling results in decreased in vivo tumor growth. Clin Cancer Res. 2003;9:3469–3475. [PubMed] [Google Scholar]
- Guigas B, Bertrand L, Taleux N, Foretz M, Wiernsperger N, Vertommen D, Andreelli F, Viollet B, Hue L. 5-Aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside and metformin inhibit hepatic glucose phosphorylation by an AMP-activated protein kinase-independent effect on glucokinase translocation. Diabetes. 2006;55:865–874. doi: 10.2337/diabetes.55.04.06.db05-1178. [DOI] [PubMed] [Google Scholar]
- Gulati P, Gaspers LD, Dann SG, Joaquin M, Nobukuni T, Natt F, Kozma SC, Thomas AP, Thomas G. Amino acids activate mTOR complex 1 via Ca2+/CaM signaling to hVps34. Cell Metab. 2008;7:456–465. doi: 10.1016/j.cmet.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haar EV, Lee SI, Bandhakavi S, Griffin TJ, Kim DH. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol. 2007 doi: 10.1038/ncb1547. [DOI] [PubMed] [Google Scholar]
- Haas DW, Hagedorn CH. Protein kinase C phosphorylates both serine and threonine residues of the mRNA cap binding protein eIF-4E. Second Messengers and Phosphoproteins. 1992;14:55–63. [PubMed] [Google Scholar]
- Hardie DG. Role of AMP-activated protein kinase in the metabolic syndrome and in heart disease. FEBS Lett. 2008;582:81–89. doi: 10.1016/j.febslet.2007.11.018. [DOI] [PubMed] [Google Scholar]
- Harrington LS, Findlay GM, Gray A, Tolkacheva T, Wigfield S, Rebholz H, Barnett J, Leslie NR, Cheng S, Shepherd PR, Gout I, Downes CP, Lamb RF. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J Cell Biol. 2004;166:213–223. doi: 10.1083/jcb.200403069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–590. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- Jaeschke A, Hartkamp J, Saitoh M, Roworth W, Nobukuni T, Hodges A, Sampson J, Thomas G, Lamb R. Tuberous sclerosis complex tumor suppressor-mediated S6 kinase inhibition by phosphatidylinositide-3-OH kinase is mTOR independent. J Cell Biol. 2002;159:217–224. doi: 10.1083/jcb.jcb.200206108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobsen SN, Hardie DG, Morrice N, Tornqvist HE. 5′-AMP-activated protein kinase phosphorylates IRS-1 on Ser-789 in mouse C2C12 myotubes in response to 5-aminoimidazole-4-carboxamide riboside. J Biol Chem. 2001;276:46912–46916. doi: 10.1074/jbc.C100483200. [DOI] [PubMed] [Google Scholar]
- Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163–175. doi: 10.1016/s0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- Kim E, Goraksha-Hicks P, Li L, Neufeld TP, Guan KL. Regulation of TORC1 by Rag GTPases in nutrient response. Nat Cell Biol. 2008;10:935–945. doi: 10.1038/ncb1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laderoute KR, Amin K, Calaoagan JM, Knapp M, Le T, Orduna J, Foretz M, Viollet B. 5′-AMP-activated protein kinase (AMPK) is induced by low-oxygen and glucose deprivation conditions found in solid-tumor microenvironments. Mol Cell Biol. 2006;26:5336–5347. doi: 10.1128/MCB.00166-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby G, Donnelly LA, Donnan PT, Alessi DR, Morris AD, Evans JM. New users of metformin are at low risk of incident cancer: A cohort study among people with type 2 diabetes. Diabetes care. 2009 doi: 10.2337/dc08-2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long X, Lin Y, Ortiz-Vega S, Yonezawa K, Avruch J. Rheb Binds and Regulates the mTOR Kinase. Curr Biol. 2005;15:702–713. doi: 10.1016/j.cub.2005.02.053. [DOI] [PubMed] [Google Scholar]
- Maher JC, Wangpaichitr M, Savaraj N, Kurtoglu M, Lampidis TJ. Hypoxia-inducible factor-1 confers resistance to the glycolytic inhibitor 2-deoxy-D-glucose. Mol Cancer Ther. 2007;6:732–741. doi: 10.1158/1535-7163.MCT-06-0407. [DOI] [PubMed] [Google Scholar]
- Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manuel y Keenoy B, Zahner D, Malaisse WJ. Dissociated effects of 2-deoxy-D-glucose on D-[2-3H]glucose and D-[5-3H]glucose conversion into 3HOH in rat erythrocytes. Biochem J. 1992;288(Pt 2):433–438. doi: 10.1042/bj2880433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall S. Role of insulin, adipocyte hormones, and nutrient-sensing pathways in regulating fuel metabolism and energy homeostasis: a nutritional perspective of diabetes, obesity, and cancer. Sci STKE 2006. 2006:re7. doi: 10.1126/stke.3462006re7. [DOI] [PubMed] [Google Scholar]
- Mathupala SP, Ko YH, Pedersen PL. Hexokinase II: cancer's double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene. 2006;25:4777–4786. doi: 10.1038/sj.onc.1209603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathupala SP, Ko YH, Pedersen PL. Hexokinase-2 bound to mitochondria: cancer's stygian link to the “Warburg Effect” and a pivotal target for effective therapy. Semin Cancer Biol. 2009;19:17–24. doi: 10.1016/j.semcancer.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagne J, Thomas G. S6K Integrates Nutrient anf Mitogenic Signals to Control Cell Growth. (Cold Spring Laboratory Press); 2004. [Google Scholar]
- Nobukuni T, Joaquin M, Roccio M, Dann SG, Kim SY, Gulati P, Byfield MP, Backer JM, Natt F, Bos JL, Zwartkruis FJ, Thomas G. Amino acids mediate mTOR/raptor signaling through activation of class 3 phosphatidylinositol 3OH-kinase. Proc Natl Acad Sci U S A. 2005;102:14238–14243. doi: 10.1073/pnas.0506925102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paltoglou SM, Roberts BJ. Role of the von Hippel-Lindau tumour suppressor protein in the regulation of HIF-1alpha and its oxygen-regulated transactivation domains at high cell density. Oncogene. 2005;24:3830–3835. doi: 10.1038/sj.onc.1208531. [DOI] [PubMed] [Google Scholar]
- Pan DA, Hardie DG. A homologue of AMP-activated protein kinase in Drosophila melanogaster is sensitive to AMP and is activated by ATP depletion. Biochem J. 2002;367:179–186. doi: 10.1042/BJ20020703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radimerski T, Montagne J, Hemmings-Mieszczak M, Thomas G. Lethality of Drosophila lacking TSC tumor suppressor function rescued by reducing dS6K signaling. Genes Dev. 2002;16:2627–2632. doi: 10.1101/gad.239102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer. 2006;6:729–734. doi: 10.1038/nrc1974. [DOI] [PubMed] [Google Scholar]
- Sakamoto K, McCarthy A, Smith D, Green KA, Grahame Hardie D, Ashworth A, Alessi DR. Deficiency of LKB1 in skeletal muscle prevents AMPK activation and glucose uptake during contraction. Embo J. 2005;24:1810–1820. doi: 10.1038/sj.emboj.7600667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, Sabatini DM. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008;320:1496–1501. doi: 10.1126/science.1157535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E, Carr SA, Sabatini DM. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell. 2007;25:903–915. doi: 10.1016/j.molcel.2007.03.003. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Sabatini DM. Redox regulation of the nutrient-sensitive raptor-mTOR pathway and complex. J Biol Chem. 2005;280:39505–39509. doi: 10.1074/jbc.M506096200. [DOI] [PubMed] [Google Scholar]
- Schieke SM, Phillips D, McCoy JP, Jr, Aponte AM, Shen RF, Balaban RS, Finkel T. The mammalian target of rapamycin (mTOR) pathway regulates mitochondrial oxygen consumption and oxidative capacity. J Biol Chem. 2006;281:27643–27652. doi: 10.1074/jbc.M603536200. [DOI] [PubMed] [Google Scholar]
- Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009;9:563–575. doi: 10.1038/nrc2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, Montminy M, Cantley LC. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;310:1642–1646. doi: 10.1126/science.1120781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith EM, Finn SG, Tee AR, Browne GJ, Proud CG. The tuberous sclerosis protein TSC2 is not required for the regulation of the mammalian target of rapamycin by amino acids and certain cellular stresses. J Biol Chem. 2005;280:18717–18727. doi: 10.1074/jbc.M414499200. [DOI] [PubMed] [Google Scholar]
- Sofer A, Lei K, Johannessen CM, Ellisen LW. Regulation of mTOR and cell growth in response to energy stress by REDD1. Mol Cell Biol. 2005;25:5834–5845. doi: 10.1128/MCB.25.14.5834-5845.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg GR, Kemp BE. AMPK in Health and Disease. Physiol Rev. 2009;89:1025–1078. doi: 10.1152/physrev.00011.2008. [DOI] [PubMed] [Google Scholar]
- Tremblay F, Brule S, Hee Um S, Li Y, Masuda K, Roden M, Sun XJ, Krebs M, Polakiewicz RD, Thomas G, Marette A. Identification of IRS-1 Ser-1101 as a target of S6K1 in nutrient- and obesity-induced insulin resistance. Proc Natl Acad Sci U S A. 2007;104:14056–14061. doi: 10.1073/pnas.0706517104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzatsos A, Kandror KV. Nutrients suppress phosphatidylinositol 3-kinase/Akt signaling via raptor-dependent mTOR-mediated insulin receptor substrate 1 phosphorylation. Mol Cell Biol. 2006;26:63–76. doi: 10.1128/MCB.26.1.63-76.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Um SH, D'Alessio D, Thomas G. Nutrient overload, insulin resistance, and ribosomal protein S6 kinase 1, S6K1. Cell Metab. 2006;3:393–402. doi: 10.1016/j.cmet.2006.05.003. [DOI] [PubMed] [Google Scholar]
- Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, Fumagalli S, Allegrini PR, Kozma SC, Auwerx J, Thomas G. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature. 2004;431:200–205. doi: 10.1038/nature02866. [DOI] [PubMed] [Google Scholar]
- Wick AN, Drury DR, Nakada HI, Wolfe JB. Localization of the primary metabolic block produced by 2-deoxyglucose. J Biol Chem. 1957;224:963–969. [PubMed] [Google Scholar]
- Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N, Hirshman MF, Goodyear LJ, Moller DE. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.