

Abstract
The ability to monitor and characterize DNA mismatch repair activity in various mammalian cells is important for understanding mechanisms involved in mutagenesis and tumorigenesis. Since mismatch repair proteins recognize mismatches containing both normal and chemically altered or damaged bases, in vitro assays must accommodate a variety of mismatches in different sequence contexts. Here we describe the construction of DNA mismatch substrates containing G:T or O6meG:T mismatches, the purification of recombinant native human MutSα (MSH2-MSH6) and MutLα (MLH1-PMS2) proteins, and in vitro mismatch repair and excision assays that can be adapted to study mismatch repair in nuclear extracts from mismatch repair proficient and deficient cells.
Keywords: DNA repair, Mismatch repair, MutS, MutL
Introduction
DNA mismatch repair (MMR) is a highly conserved DNA repair pathway that plays a key role in the correction of DNA mismatches generated during replication and recombination. Mutations in MMR genes are associated with hereditary nonpolyposis colorectal cancer (HNPCC), and epigenetic silencing of MMR genes has been implicated in sporadic cancers [1; 2; 3]. In eukaryotes, the first step of MMR is the recognition of mismatches by MutSα, a heterodimer of MSH2 and MSH6. MutSα together with MutLα, a heterodimer of MLH1 and PMS2, directs excision of the newly synthesized, error-containing strand and subsequent gap repair by DNA polymerase and DNA ligase.
Several methods have been developed for monitoring in vitro MMR activity in cell extracts. These utilize phagemid DNA substrates that are assayed in E. coli [4] or plasmid-based assays that are monitored by restriction endonuclease cleavage [5]. A GFP reporter system for monitoring MMR in living cells has also been reported [6]. Here we describe improved methods to monitor mismatch repair in vitro including the creation of mismatched DNA substrates that can accommodate both natural and modified bases, a robust and streamlined purification protocol for untagged recombinant human MutSα and MutLα proteins using baculovirus expression systems and assays to monitor mismatch repair and mismatch-directed excision in nuclear extracts. These assays and reagents are useful for mechanistic studies of MMR and for assessing MMR competency in a variety of mammalian cell lines.
Methods and Materials
1. Preparation of mismatched DNA substrates
All plasmids were propagated in TOP10 E. coli cells (Invitrogen) and isolated using a Qiagen Plasmid Maxi kit (Qiagen) followed by CsCl/ethidium bromide equilibrium centrifugation.
pSCW01 and pSCW02 were derived from pUC19CPDrev and follow the original numbering system with the four Nt.BstNBI endonuclease sites residing on the sense strand as defined by the pUC19 bla gene in pSCW01 and the antisense strand in the case of pSCW02 (Fig. 1A) [5]. A BbvC1 restriction endonuclease site was introduced at position 55 (Stratagene QuikchangeXL) in pUC19CPDrev and an existing BbvC1 site removed between positions 331-348. The resulting plasmid was digested with AatII and SapI, gel purified to remove the 76 bp intervening sequence, and ligated to synthetic 76 bp duplexes to create two 2030 bp plasmids, pSCW01 and pSCW02, that served as homoduplex DNA controls in MMR assays. The 76 bp sequence was created from two complementary oligonucleotides (Integrated DNA Technologies) that were heated at 85°C for 10 mins followed by slow cooling and then purification by native PAGE. Top strand: 5′- (p)CAT CGA GTC GGA TCC GAG TCA TTC CTG CAG CGA GTC CAT GGG AGT CAA ATA GAC AAC GAT TTG AAT TCG CTC TTC C -3′ (76mer) and bottom strand: 5′- (p)AGC GGA AGA GCG AAT TCA AAT CGT TGT CTA TTT GAC TCC CAT GGA CTC GCT GCA GGA ATG ACT CGG ATC CGA CTC GAT GAC GT -3′ (83mer) in the case of pSCW01 or top strand: 5′- (p)CAT CGA CTC GGA TCC GAC TCA TTC CTG CAG CGA CTC CAT GGG ACT CAA ATA GAC AAC GAT TTG AAT TCG CTC TTC C -3′ (76mer) and bottom strand: 5′- (p)AGC GGA AGA GCG AAT TCA AAT CGT TGT CTA TTT GAG TCC CAT GGA GTC GCT GCA GGA ATG AGT CGG ATC CGA GTC GAT GAC GT -3′ (83mer) in the case of pSCW02.
Figure 1.


DNA mismatch repair substrates. (A) Map of essential features of pSCW01 and pSCW02 plasmids. Numbering is with respect to start of coding sequence of bla gene with sense strand on top. Red denotes nucleotide that is replaced to create a mismatch. Blue asterisk indicates positions of 4 × Nt.BstNBI nicking sites. (B) Scheme for the creation of 5′ or 3′ nick-directed mismatched DNA substrates harboring G:T or O6meG:T (meG:T) mismatches.
For the construction of mismatched substrates, 400 μg of pSCW01 or pSCW02 was incubated with 300 U of Nt.BstNBI endonuclease (New England BioLabs) at 55°C for 2 hr in 1.0 ml final volume in 1 × Buffer 3 (New England BioLabs) followed by the addition of another 300 U of Nt.BstNBI endonuclease and further incubation at 55°C for 2 hr. Nicking was monitored by gel electrophoresis. Displacement of the short, nicked fragments was facilitated by the addition of a 50-fold molar excess of displacer oligonucleotides (for pSCW01: 5′- ATT TGA CTC C-3′, 5′- CAT GGA CTC GCT GCA G-3′ and 5′-GAA TGA CTC GG-3′; for pSCW02: 5′-CAT CGA CTC GG-3′, 5′-ATC CGA CTC ATT CCT G-3′, and 5′-CAG CGA CTC C-3′) followed by 5 min at 85°C and then slow cooling to room temperature over 1.5 hr (Fig. 1B). The resulting gapped DNA was precipitated in the presence of 13% (w/v) PEG 8000 and 10 mM MgCl2 by the addition of an equal volume of 26% (w/v) PEG 8000 and 20 mM MgCl2. Immediately after mixing, the sample was centrifuged at 12,000 × g at RT for 10 mins. The supernatants were discarded and the pellets were washed twice with 70% ethanol and resuspended in 50 μl of 10 mM Tris-HCl, pH 8.0. Gapped DNA was stored at -20°C.
A 10-fold molar excess of mismatched 37 nt oligonucleotides (For a G:T mismatch in pSCW01: 5′-(p)CCG AGT CAT TCC TGT AGC GAG TCC ATG GGA GTC AAA T-3′; for G:T in pSCW02: 5′-(p)GGA GTC GCT GCG GGA ATG AGT CGG ATC CGA GTC GAT G-3′; for meG:T in pSCW02: 5′-(p)GGA GTC GCT GCmeG GGA ATG AGT CGG ATC CGA GTC GAT G-3′) were annealed to the gapped pSCW01 or pSCW02 in 1 ml ligation buffer by incubation at 85°C for “G” oligonucleotide and 65°C for “O6meG” oligonucleotide for 10 min followed by slow cooling to room temperature. Ligation was monitored by gel electrophoresis after addition of 1 mM each DTT and ATP and incubation overnight at 16°C with 100 U of T4 DNA ligase (New England BioLabs). The mismatched DNA was extracted twice with phenol/chloroform and chloroform followed by ethanol precipitation. The DNA, resuspended in TE, was purified by CsCl/ethidium bromide equilibrium centrifugation followed by extraction in water-saturated n-butanol to remove ethidium bromide and dialysis against three 1-liter changes of TE buffer, pH 7.4. Covalently closed mismatched DNA was stored at -20°C. Nicked substrates were prepared by incubation with 20 U of Nt.BbvCI (5′ nick) or Nt.BspQI (3′ nick) (New England BioLabs) followed by phenol-chloroform extraction and ethanol precipitation (Fig. 1B). The nicked mismatched substrates, pSCW01_GT, pSCW02_GT or pSCW02_mGT, were stored in TE, pH7.4, at -20°C.
2. Recombinant hMutSα and hMutLα
Recombinant hMutSα and hMutLα were produced in baculovirus-infected insect cells. Coding sequences for hMSH2 and hMSH6 were amplified by PCR from Image Clones (#3629489 and #4110354, respectively, Open Biosystem). The coding sequences for hMLH1 and hPMS2 were amplified by PCR from pFBdual_MutLα plasmid, a gift from Guo-Min Li at University of Kentucky Medical Center. Coding sequences for hMSH2, hMSH6, and hMLH1 were inserted into the pFastBac1 vector (Invitrogen) at BamHI and XhoI sites, and for hPMS2 at BamHI and SalI sites to yield pFB1_hMSH6, pFB1_hMSH2, pFB1_hMLH1, and pFB1_hPMS2. Constructs were verified by DNA sequencing and match Genbank accession BC004246 (hMSH6), BC021566 (hMSH2), U07343 (hMLH1) and U14658 (hPMS2). Baculovirus stocks were created (Kinnakeet Biotechnology) and propagated and titered in Sf9 cells (Invitrogen).
High Five insect cells (Invitrogen) were cultured in serum-free Express Five SFM medium (Invitrogen) supplemented with 20 mM L-glutamine at 27°C. Cells were coinfected at a density of 1×106/ml with baculovirus stocks encoding hMSH2 and hMSH6 at a multiplicity of infection (MOI) of 8 to produce MutSα, or with baculovirus constructs encoding hMLH1 and hPMS2 at a MOI of 2 to generate MutLα. One liter of infected cells was collected 60 hr later by centrifugation at 6,000 rpm for 10 min. For MutSα, cell pellets were dissolved in 80 ml of buffer A (25 mM HEPES, pH7.5; 0.1 mM EDTA; 10% glycerol; 1 mM DTT; 1 × Complete proteinase inhibitor cocktail (Roche); 0.1% PMSF). After swelling on ice for 10 min, cells were lysed with 20 strokes using a tight-fitting Dounce homogenizer on ice. After adjusting the salt concentration to 150 mM KCl, the suspension was clarified by centrifugation at 16,000 rpm for 30 min. The supernatant was then filtered through a 0.45μM filter unit (Corning) and loaded at 4°C onto a 6-ml Resource™ Q anion exchange column (GE Healthcare) equilibrated in Buffer A containing 150 mM KCl with a flow rate of 1.5 ml/min. The column was first washed with 60-ml buffer A plus 150 mM KCl, and then developed with a 90-ml gradient of KCl (150-650 mM) in buffer A. hMutSα eluted at approximately 250 mM KCl. The hMutSα-containing fractions were diluted to 150 mM KCl with buffer A and loaded onto a 5-ml HiTrap™ Heparin column (GE Healthcare) at 4°C which was developed with a 90-ml gradient of KCl, 150-650 mM, in buffer A; MutSα eluted at approximately 350 mM KCl. After concentration to 2 ml with an Amicon Ultracel-10K (Millipore), hMutSα-containing fractions were loaded on a HiLoad 16/60 Superdex 200 column (GE Healthcare) equilibrated with buffer A containing 100 mM KCl. The pooled hMutSα fractions were aliquoted, frozen in liquid nitrogen and stored at -80 °C.
hMutLα was purified over Resource Q, Heparin and Superdex 200 columns as described for hMutSα with the following modifications. The lysis buffer contained 80 mM KCl; the 6-ml Resource™ Q anion exchange column and heparin column were equilibrated in buffer A containing 80 mM and 100 mM KCl, respectively. Columns were developed in a gradient of 100-450 mM KCl. hMutLα eluted at approximately 200 mM KCl on both Resource Q and heparin columns. The HiLoad 16/60 Superdex 200 column was equilibrated in buffer A containing 200 mM KCl. Concentrations of MutSα and MutLα were determined with a modified Bradford protein assay (Bio-Rad) using BSA as standard.
3. Mammalian cell culture and nuclear extracts
HeLaS3, MSH2-deficient LoVo, and MLH1-deficient HCT116 cells (American Type Culture Collection) were maintained at 37°C in DMEM (Invitrogen) with 10% fetal bovine serum (Invitrogen), 50 U penicillin, and 50 μg/ml streptomycin (Invitrogen) in a 5% CO2, humidified atmosphere. Nuclear extracts were isolated following the previous studies but with some modifications [7; 8]. 4-5 × 108 cells were collected by centrifugation at 3000 × g for 5 min and washed once with 40 ml of cold hypotonic buffer (20 mM Hepes, pH 7.5; 5 mM KCl; 0.5 mM MgCl2; 0.1% PMSF; 2 mM DTT; 1 μg/ml of each aprotinin, leupeptin, pefabloc, and E-64 (Roche)) containing 0.2 M sucrose. The cell pellet was resuspended in 10 ml of cold hypotonic buffer without sucrose, incubated on ice for 10 min, and then homogenized by 10 strokes of a loose-fitting Dounce homogenizer on ice. The resulting solution was centrifuged at 2000 × g for 5 min. The nuclear pellet was resuspended in 2.5 ml of extract buffer (50 mM Hepes, pH 7.5; 10% sucrose; 0.1% PMSF; 2 mM DTT; 1 μg/ml of each aprotinin, leupeptin, pefabloc, and E-64). After addition of 0.03 vol of 5 M NaCl, the nuclear suspension was mixed on a rotator for 1 hr at 4 °C and centrifuged at 15,000 × g for 30 min. The supernatant was concentrated to 5-8 mg/ml with an Amicon Ultracel-10K (Millipore), frozen in liquid nitrogen and stored at -80 °C.
4. Mismatch repair reactions (MMR)
In vitro MMR assays were performed with minor modification as previously described [5] in a 40 μl volume containing 75 fmol (100 ng) of nicked pSCW01_GT DNA substrate, 100 μg of nuclear extract, 0.1 mM each of four dNTPs, in the standard MMR buffer condition of 20 mM Tris-HCl, pH 7.6; 1.5 mM ATP; 1 mM glutathione; 5 mM MgCl2; 50 mg/ml BSA; the salt concentration was adjusted to 110 mM NaCl. After incubation at 37 °C for 15 min, the reaction was terminated by the addition of 80 μl of stop solution (25 mM EDTA, 0.67% sodium dodecyl sulfate, and 90 μg/ml proteinase K), then incubated at 37°C for another 15 min. DNA was extracted twice with an equal volume of phenol/chloroform and twice with chloroform. After precipitation with 2.5 volume of ethanol, DNA was dissolved in H2O, digested with 4 units each of PstI and AseI endonuclease (New England Biolabs) and 1 μg of RNAase (Sigma) at 37°C for 1 hr, and then separated by electrophoresis on a 1.5% agarose gel. Alphaview 2.0 software (AlphaInnotek) was used to analyze the repair yield, which equals the ratio of the summed intensities of the 0.8- and 1.2-kb fragments to the total intensities of the 0.8-, the 1.2-kb, and the 2.0-kb bands. MMR assays for nicked pSCW02_GT substrate were performed as for pSCW01_GT. The recovered DNA was first cleaved with FauI (New England Biolabs) plus RNAase at 55°C for 1 hr, then digested with AseI at 37°C for another 1 hr. Repair products yield 0.8- and 1.2-kb fragments.
5. Analysis of excision gaps
To measure the extent of excision generated during MMR, we omitted exogenous dNTPs from a standard MMR reaction mixture containing nicked mismatched DNA substrate or various control substrates, nuclear extract and 20-40 nM hMutSα and 20-40 nM hMutLα as indicated. Incubation was at 37 °C for 7 min. The reaction was terminated by stop solution. DNA was extracted with phenol/chloroform and precipitated with ethanol as described above. Excision was measured in one of three ways employing adaptations of previously described excision assays [5; 9; 10].
5.1 Restriction endonuclease cleavage
The gapped DNA was digested with BamHI and AseI (New England Biolabs) and RNAase at 37 °C for 1 hr and then separated by electrophoresis on a 1.5% agarose gel. Since the BamHI endonuclease cleavage site is located between the mismatch site and the 5′ nick site, excision initiating at the nick and extending beyond the mismatch will result in loss of the BamHI site resulting in a 2 kb linearized DNA.
5.2 Annealing to oligonucleotide probes
The recovered DNA was digested with AseI plus RNAase, annealed with 0.2 pmol of a particular 32P-labeled oligomer probe (5′-GTC ATT CCT GCA GCG AGT CCA TGG GAG TCA-3′), and then separated from free oligomers by electrophoresis. DNA bands were measured quantitatively by ethidium bromide staining to normalize DNA recovery and loading. The gels were dried and the radioactivity was measured with a Fuji phosphoimager BSA-2500.
5.3 Southern blot analysis
The excision products were digested with AflII (New England BioLabs) plus RNAase and mixed with 2 × formamide loading buffer (95% formamide, 10 mM NaOH, 0.25% Bromophenol blue, 0.25% Xylene cyanol). The mixture was heated at 85°C for 5 min and cooled on ice followed by electrophoresis on 6% denaturing polyacrylamide gels and transfer to Hybond-NX membranes (Amersham). Membranes were cross-linked with a UV crosslinker (Stratagene), and then pre-hybridized at 37 °C for 1 hr with hybridization solution (0.2% SDS, 0.5% PVP, 0.2% Heparin, 0.05 M Tris pH 7.5, 1 M NaCl, 1 mM EDTA). After incubation with a 32P-5′ end-labeled oligonucleotide probe (5′-TTG GAG CGA ACG ACC TAC ACC GA-3′) overnight at 37°C, the membranes were washed twice with washing solution I (2 × SSC, 0.1% SDS) at 37°C, and twice with washing solution II (1 × SSC, 0.1% SDS). Membranes containing reaction products were analyzed using a Fuji phosphoimager BAS-2500 and ImageGauge V4.22.
Results and Discussion
DNA substrates for in vitro mismatch repair and excision assays
DNA substrates for in vitro MMR and excision assays are circular plasmids containing a mismatch in a defined position and a single nick positioned either 5′ or 3′ to the mismatch. MMR is monitored by the gain or loss of a diagnostic restriction endonuclease site at the mismatch. To create a mismatched substrate, the starting plasmid, denoted pSCW01 or pSCW02, is nicked at 4 closely positioned sites with a site-specific nicking enzyme and heated to create a single-strand gap. The mismatch, at position 185 or 183 for pSCW01_GT or pSCW02_GT, respectively, is introduced by annealing and ligating an oligonucleotide that spans the gapped region and contains a sequence heterology or modified base, e.g. O6-meG (Fig. 1A). pUC19CPDrev, a prototype for such substrates [5], was modified to confer the following improvements. A BbvC1 restriction endonuclease site was introduced at position 55 (Stratagene) resulting in a unique nicking site on either the sense strand 5′ to the mismatch at position 55 (Nb.BbvCI, New England BioLabs) or the antisense strand 3′ to the mismatch at position 58′ (Nt.BbvCI, New England BioLabs). An Nt.BspQI nicking site (New England BioLabs) at position 233 on the sense strand affords 3′ excision on the sense strand. Sequence context immediately surrounding the mismatch can be altered by means of a removable 76 bp cassette utilizing unique restriction endonuclease sites flanking the mismatch, AatII (position 157) and SapI (position 233). To facilitate the formation of a gapped intermediate in the creation of the final mismatched substrate, we introduced four Nt.BstNBI nicking sites flanking the mismatch position on the sense strand for pSCW01 or the antisense strand for pSCW02 (Fig. 1A). This is essential for efficient displacement of the endogenous DNA strand and its replacement by the heterologous oligonucleotide especially in the case of G-C rich flanking sequence. We have previously shown that a G-C-rich sequence context is desirable for binding of MutSα to O6meG:T mismatches [11]. Recognition of O6meG:T mismatches does not occur in the sequence context of the pUC19CPDrev plasmid (data not shown). Mapping excision tracts leading to the formation of gapped DNA can be monitored qualitatively by cleavage with unique BamHI, NcoI and EcoRI sites that flank the mismatch.
Purity and yield of recombinant hMutSα and hMutLα
Recombinant human MutSα and MutLα proteins produced in High Five insect cells following co-infection with baculovirus stocks encoding hMSH2, hMSH6, hMLH1 or hPMS2 followed by purification are displayed by SDS-PAGE (Fig. 2A-B). Native proteins were in excess of 95% pure (Fig. 2B). From a starting culture of 1×109 High Five cells approximately 6 mg of hMuSα and 5 mg hMutLα were obtained. We previously observed that N-terminal His6-tagged murine MutSα and MutLα were labile upon long-term storage at -80°C [11]. Production of native hMuSα and hMutLα resulted in robust activity that was retained after 6 months at -80°C. Successful purification of untagged recombinant proteins benefits from high level expression as apparent in Fig. 2A. Unlike a previously published purification scheme for hMutSα, this procedure can be used to purify mutant mismatch repair proteins that are defective for DNA binding or ATP binding and hydrolysis [12].
Figure 2.
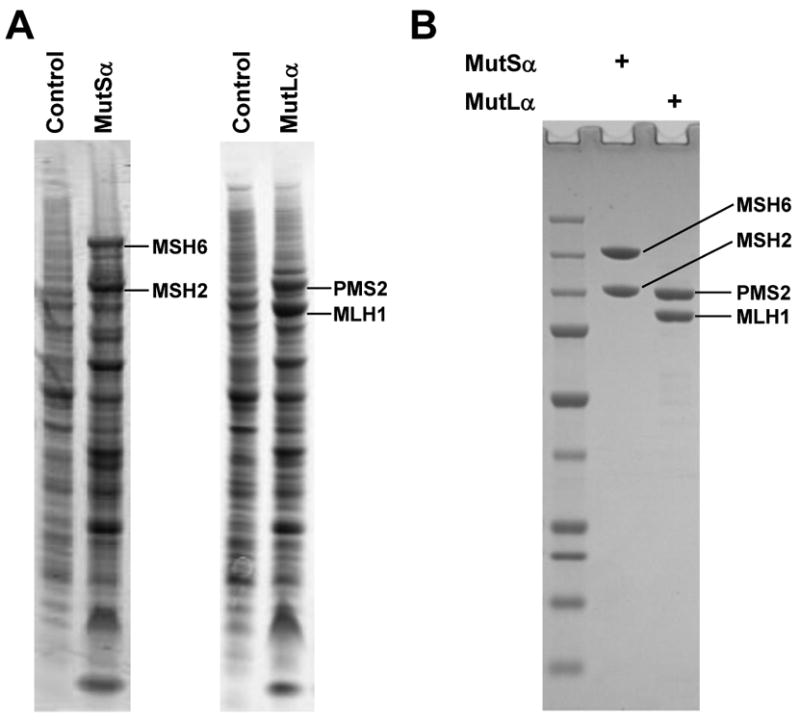
Recombinant hMutSα and hMutLα. (A) Expression of hMutSα and hMutLα in whole cell lysates of control (uninfected) and infected (MutSα and MutLα) High Five insect cells. (B) Purified hMutSα and hMutLα, 1.5 μg each, were electrophoresed on SDS-PAGE and stained with Coomassie blue.
Specificity of MMR excision
Correction of pSCW01_GT mismatches to G:C leads to the restoration of a cryptic PstI cleavage site. Thus, when MMR is successful, the corrected DNA substrate yields 0.8- and 1.2-kb fragments when cleaved with PstI and AseI. The uncorrected G:T substrate is linearized by AseI yielding a 2.0-kb band. Correction of pSCW02_GT mismatches to G:C introduces a FauI cleavage site. After MMR, the corrected pSCW02_GT DNA substrate can generate 0.8- and 1.2-kb fragments when digested with FauI and AseI (Table 1). As indicated in Fig. 3A, there was little if any MMR activity with the pSCW01_GT in MSH2-deficient LoVo cells and MLH1-deficient HCT116 cells, while HeLa cell extracts yielded close to 60% MMR efficiency. The addition of recombinant MutSα or MutLα proteins restored MMR efficiency in LoVo cells and HCT116 cells, respectively, yielding as much as 40% repaired substrate. As shown in Fig. 3B, repair efficiency of HeLa extracts for pSCW02_GT was roughly comparable to that for pSCW01_GT. As noted previously, repair does not approach 100% due in large part to ligation of the nicked substrate by ligases in the nuclear extracts rendering the covalently closed plasmid resistant to MMR [13].
Table 1. DNA Mismatch Substrates.
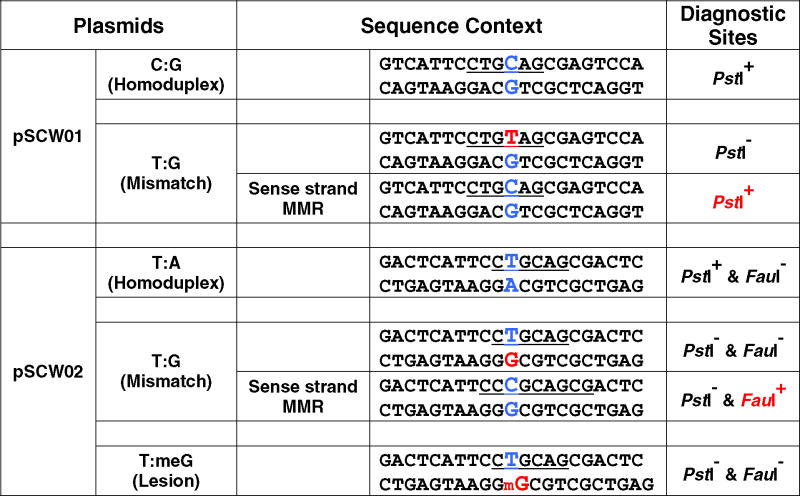 |
Figure 3.
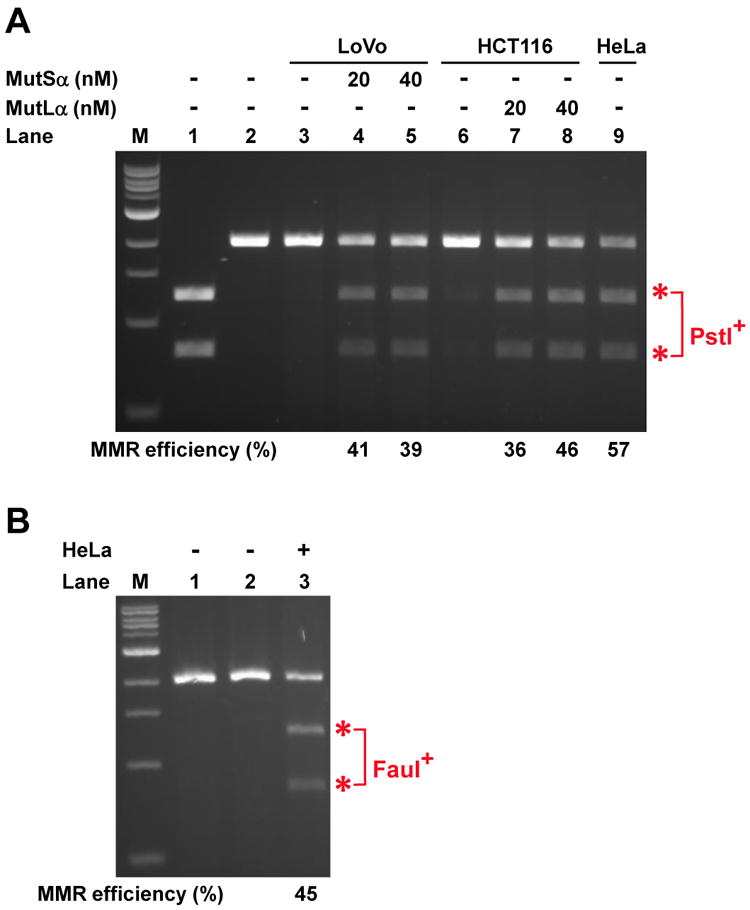
Nick-directed mismatch repair assay. (A) Repair assays were performed in 40 μl reactions containing 75 fmol (100 ng) of pSCW01_GT DNA substrate nicked by Nb.BbvCI, 100 mg of nuclear extract, 20-40 nM of MutSα and 20-40 nM of MutLα proteins as indicated. Recovered DNA was digested with PstI and AseI endonucleases and separated by electrophoresis. The repair yield equals the ratio of the summed intensities of the 0.8- and 1.2-kb fragments to the total DNA. Lane 1 - control with covalently closed pSCW01 homoduplex in the absence of nuclear extract. Lane 2 - control with 5′-nicked pSCW01_GT in the absence of nuclear extract. Red asterisk denotes PstI and AseI cleavage products indicating repair. (B) MMR assay was performed as in (A), but with pSCW02_GT substrate. Recovered DNA was digested with FauI and AseI. Lane 1 - control with covalently closed pSCW02 homoduplex in the absence of nuclear extract. Lane 2 - control with 5′-nicked pSCW02_GT in the absence of nuclear extract. Lane 3 - 5′-nicked pSCW02_GT with HeLa nuclear extract. Red asterisk denotes FauI and AseI cleavage products indicating repair.
A simple and rapid measure of excision utilizes restriction endonuclease digestion at one of several unique sites near the mismatch. A BamHI cleavage site is located 25 bp away from the mismatch on the 5′ nick site. If the MMR excision occurs, the BamHI digestion site will be lost. BamHI and AseI double digestion can only generate a 2 kb linear DNA in the case of excision that extends from the nick to a point past the BamHI site whereas nonexcised DNA will yield two bands of 0.8 kb and 1.2 kb. As shown in Fig. 4A, there was only very weak excision detected in the MSH2-deficient LoVo cells. Addition of recombinant MutSα protein largely restored mismatch-directed excision.
Figure 4.
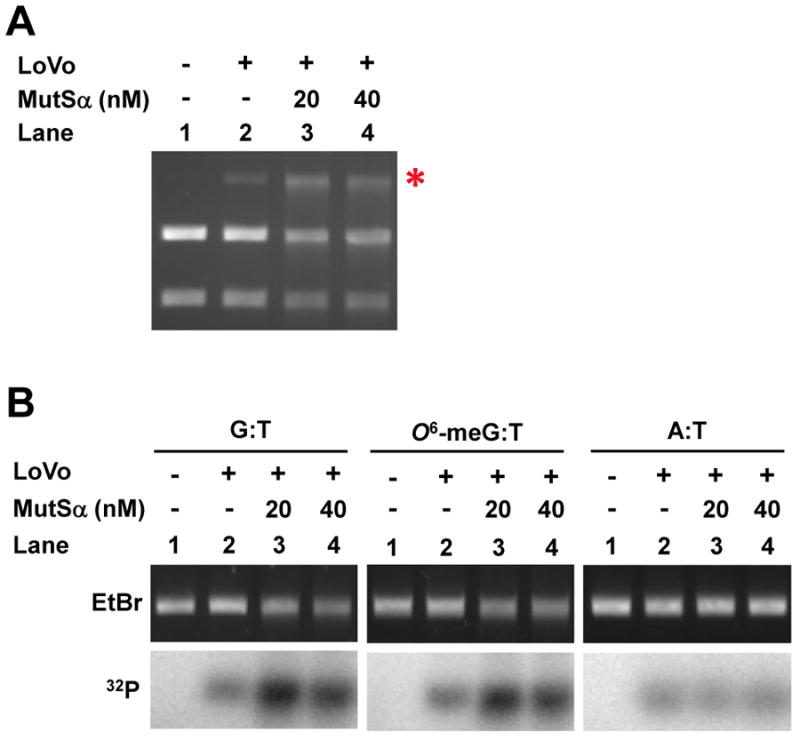
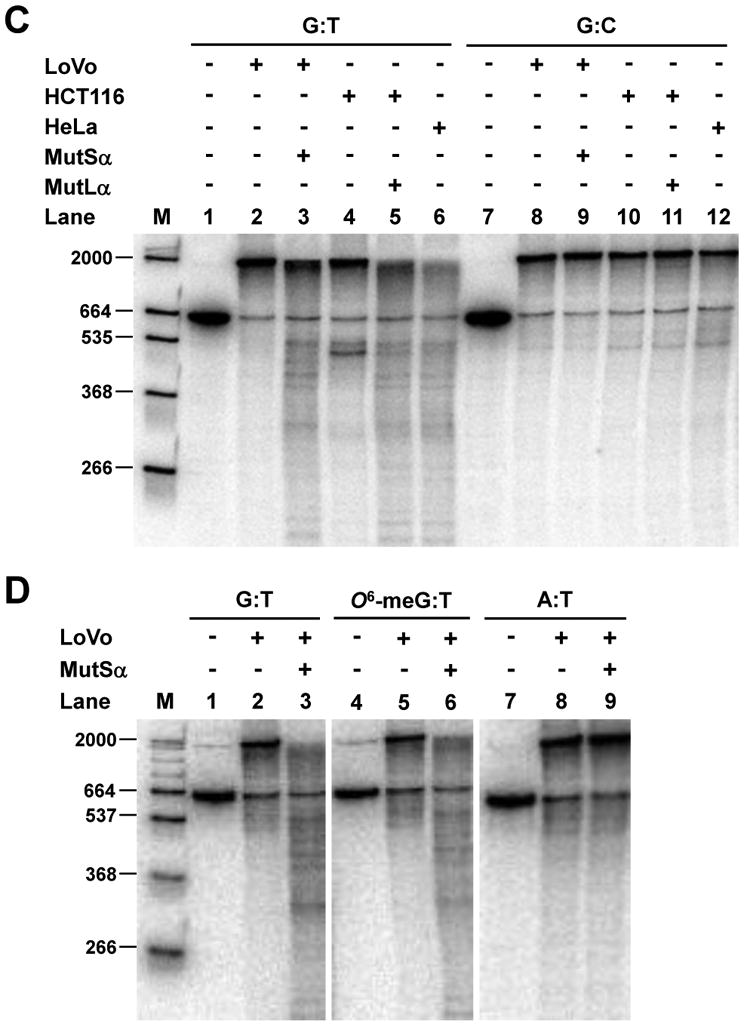
Nick-directed mismatch-provoked excision. (A) Excision assay measured by restriction endonuclease sensitivity was performed with MSH2-deficient LoVo nuclear extract and pSCW01_GT substrate in the absence of exogenous dNTPs. Recovered DNA was digested with BamHI & AseI endonucleases and separated by electrophoresis. Red asterisk denotes BamHI-resistant gapped DNA after excision. (B) Excision assay measured by annealing of an oligonucleotide probe using pSCW02_GT, pSCW02_O6meGT, and pSCW02 homoduplex substrates. Gapped DNA was digested with AseI, and annealed to a 32P-oligonucleotide probe that spans the mismatch site. (C) Excision assay utilizing Southern blotting was with pSCW01_GT DNA substrate. Nicked homoduplex pSCW01 was used to measure ExoI random excision. Lane 1 and 7 are controls with nicked pSCW01_GT or pSCW01 in the absence of nuclear extract. (D) Experiments were performed as in (C) but with the pSCW02_GT, pSCW02_O6-meGT and pSCW02 homoduplex substrates in a Southern blotting assay.
The extent of excision can also be measured using a set of 32P-labeled oligonucleotide probes that correspond to sequences at varying distances from the initiating nick. Following incubation with a nuclear extract that can carry out mismatch-directed excision, the mismatch substrate is cleaved by AseI to generate a 2 kb linear gapped DNA which can be annealed to a 32P-labeled probe. As shown in Fig. 4B, A LoVo nuclear extract gives rise to low levels of background anneal of the 32P-labeled probe due to random nuclease activity. However, the addition of recombinant MutSα protein can greatly increase the efficiency of excision. In this in vitro assay, O6-meG:T mismatches are as effective as G:T mismatches in triggering excision. A:T homoduplex substrate showed low levels of random excision that was not dependent on MutSα.
The MMR substrate can also be used to monitor the length and extent of the excision tracts that give rise to a gapped DNA intermediate. The distance from the AflII cleavage site to the Nb.BbvCI nick site on the top strand is 664 nt. If no 5′ nick-directed excision takes place, the intact strand will be 664 bp. Excision will generate a family of shorter fragments; e.g. excision to the mismatched base would generate a 129 bp gap and a radiolabeled product that is 535 nt in length. As shown in Fig. 4C, when nicked homoduplex DNA (G:C) was used as substrate, all samples yield a low background level of random excision. In the presence of G:T mismatched DNA, MSH2-deficient LoVo extracts supported only very weak excision. MLH1-deficient HCT116 nuclear extracts supported some shortened excision tracts that extended past the mismatch site. This is consistent with previous observations demonstrating that 5′ -directed MMR excision mediated by EXO1 can proceed without a requirement for MutLα in vitro [9]. When we reconstituted LoVo or HCT116 nuclear extracts with recombinant hMutSα or hMutLα, respectively, excision was restored to levels observed in the HeLa nuclear extract. When we used pSCW02_GT and pSCW02_O6-meGT as DNA substrates, similar extents of mismatch-provoked excision were detected in this in vitro assay, as indicated in Fig. 4D. Homoduplex DNA elicited much lower levels of excision.
In summary, we describe methods for monitoring MMR and mismatch-directed excision using defined plasmid mismatch substrates, MMR-proficient and MMR-deficient nuclear extracts supplemented with recombinant native hMutSα or hMutLα proteins. These modified protocols facilitate efficient measures of mismatch-directed excision and repair using a variety of different mismatched substrates containing normal and modified DNA bases in different sequence contexts. The protein purification protocol uses commercially available chromatography columns and allows the reproducible purification of milligram quantities of native, recombinant MMR proteins with good stability. These methods will be useful for assessing MMR competency in a variety of cell lines and for studying detailed mechanism using both native and mutant MMR proteins. In addition, these substrates can be utilized for microscopy studies of protein-mismatch DNA complexes. Finally, the pSCW01 and 02-based substrates accommodate a variety of non-native DNA bases that can be used for in vitro studies of other repair pathways that target chemically altered bases, abasic sites and UV photoproducts.
Acknowledgments
We thank John Hays for the gift of pUC19CPDrev and Steve Matson for helpful discussions regarding nicking enzymes. We are grateful to Nelson Chan and Guo-Min Li for helpful discussions regarding excision assays. Funding was provided by the Intramural Research Program of NIDDK.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hsieh P, Yamane K. DNA mismatch repair: molecular mechanism, cancer, and ageing. Mech Ageing Dev. 2008;129:391–407. doi: 10.1016/j.mad.2008.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kunkel TA, Erie DA. DNA mismatch repair. Annu Rev Biochem. 2005;74:681–710. doi: 10.1146/annurev.biochem.74.082803.133243. [DOI] [PubMed] [Google Scholar]
- 3.Iyer RR, Pluciennik A, Burdett V, Modrich PL. DNA mismatch repair: functions and mechanisms. Chem Rev. 2006;106:302–23. doi: 10.1021/cr0404794. [DOI] [PubMed] [Google Scholar]
- 4.Thomas DC, Roberts JD, Kunkel TA. Heteroduplex repair in extracts of human HeLa cells. J Biol Chem. 1991;266:3744–51. [PubMed] [Google Scholar]
- 5.Wang H, Hays JB. Mismatch repair in human nuclear extracts. Quantitative analyses of excision of nicked circular mismatched DNA substrates, constructed by a new technique employing synthetic oligonucleotides. J Biol Chem. 2002;277:26136–42. doi: 10.1074/jbc.M200357200. [DOI] [PubMed] [Google Scholar]
- 6.Zhou B, Huang C, Yang J, Lu J, Dong Q, Sun LZ. Preparation of heteroduplex enhanced green fluorescent protein plasmid for in vivo mismatch repair activity assay. Anal Biochem. 2009;388:167–9. doi: 10.1016/j.ab.2009.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Challberg MD, Kelly TJ., Jr Adenovirus DNA replication in vitro. Proc Natl Acad Sci U S A. 1979;76:655–9. doi: 10.1073/pnas.76.2.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Holmes J, Jr, Clark S, Modrich P. Strand-specific mismatch correction in nuclear extracts of human and Drosophila melanogaster cell lines. Proc Natl Acad Sci U S A. 1990;87:5837–41. doi: 10.1073/pnas.87.15.5837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Genschel J, Modrich P. Mechanism of 5′-directed excision in human mismatch repair. Mol Cell. 2003;12:1077–86. doi: 10.1016/s1097-2765(03)00428-3. [DOI] [PubMed] [Google Scholar]
- 10.Zhang Y, Yuan F, Presnell SR, Tian K, Gao Y, Tomkinson AE, Gu L, Li GM. Reconstitution of 5′-directed human mismatch repair in a purified system. Cell. 2005;122:693–705. doi: 10.1016/j.cell.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 11.Yoshioka K, Yoshioka Y, Hsieh P. ATR kinase activation mediated by MutSalpha and MutLalpha in response to cytotoxic O6-methylguanine adducts. Mol Cell. 2006;22:501–10. doi: 10.1016/j.molcel.2006.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Drummond JT, Li GM, Longley MJ, Modrich P. Isolation of an hMSH2-p160 heterodimer that restores DNA mismatch repair to tumor cells. Science. 1995;268:1909–12. doi: 10.1126/science.7604264. [DOI] [PubMed] [Google Scholar]
- 13.Fang WH, Li GM, Longley M, Holmes J, Thilly W, Modrich P. Mismatch repair and genetic stability in human cells. Cold Spring Harb Symp Quant Biol. 1993;58:597–603. doi: 10.1101/sqb.1993.058.01.066. [DOI] [PubMed] [Google Scholar]