

Abstract
Pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-amine derivatives such as SCH 442416 display high affinity and selectivity as antagonists for the human A2A adenosine receptor (AR). We extended ether-linked chain substituents at the p-position of the phenyl group using optimized O-alkylation. The conjugates included an ester, carboxylic acid and amines (for amide condensation), an alkyne (for click chemistry), a fluoropropyl group (for 18F incorporation), and fluorophore reporter groups (e.g. BODIPY conjugate 14, Ki 15 nM). The potent and A2AAR-selective N-aminoethylacetamide 7 and N-[2-(2-aminoethyl)-aminoethyl]acetamide 8 congeners were coupled to polyamidoamine (PAMAM) G3.5 dendrimers, and the multivalent conjugates displayed high A2AAR affinity. Theoretical docking of an AlexaFluor conjugate to the receptor X-ray structure highlighted the key interactions between the heterocyclic core and the binding pocket of the A2AAR as well as the distal anchoring of the fluorophore. In conclusion, we have synthesized a family of high affinity functionalized congeners as pharmacological probes for studying the A2AAR.
Keywords: SCH 442416, G protein-coupled receptor, fluorescence, dendrimer, radioligand binding
Adenosine acts as a neuromodulator and has been termed as an endogenous cerebroprotective agent with respect to epilepsy and ischemia. Selective ligands for the adenosine receptors (ARs) are considered to have potential in the treatment of conditions of the central nervous system (CNS) [1,2]. Adenosine activates four receptor subtypes - A1, A2A, A2B, and A3 – the first two of which have been assigned specific roles in the central nervous system [3]. The A2AAR is coupled to stimulation of adenylate cyclase and in the brain is highly expressed in the striatum and olfactory tubercle. A2AAR antagonism is suggested to mediate many of the CNS stimulatory effects of caffeine in man [4]. Its blockade by potent and selective antagonists is under development as an approach to the symptomatic treatment of Parkinson's disease (PD), and such antagonists might be useful against progressive neurodegeneration [2,3,5]. The selective A2AAR antagonists 1a, a 1,3,7-trialkylxanthine derivative, and 1b, a triazolopyrimidine derivative (Chart 1), have been in clinical trials [6,7]. Triazolo[1,5-c]pyrimidin-5-amine antagonists 2 (SCH 442416) and Preladenant (SCH 420814), have also shown promise for the treatment of PD [8,9]. Another envisioned disease application of A2AAR antagonists is in cancer treatment [10].
Chart 1.

Representative selective A2AAR antagonists that have been in clinical trials for PD: Xanthine derivative 1 and nonxanthine heterocycle 2.
The recently reported X-ray structure of the A2AAR [11] has been utilized for the in silico screening of chemical libraries for the identification of new leads for selective antagonists of this receptor, based on novel chemotypes [12,13]. The screening of in silico hits is routinely carried out using radioligand binding assays. It would be desirable to have new pharmacological probes for this receptor that would not require the use of radioactivity and instead would be based on fluorescence and other spectroscopic methods. We have explored chain functionalization of the A2AAR antagonist 2 for this purpose to arrive at a series of functionalized congeners that provide a means of covalent attachment of sterically bulky groups without losing the ability to bind to the A2AAR with high affinity. Also, a biotin handle was included in this approach for complexation to streptavidin. In continuation of our efforts to develop receptor imaging agents for positron emission tomography (PET) that selectively target the most relevant subtype of ARs that is highly expressed in the striatum, i.e. the A2AAR, we developed a O-(3-fluoropropyl) analogue, intended for incorporation of 18F. It was essential to identify an insensitive position on 2 that would accommodate a chain. The methoxy group of the phenyl ring of 2 was selected for this purpose because it has been shown to display flexibility of substitution and conformation with respect to binding to the A2AAR, as deduced from structure activity relationship (SAR) studies in conjunction with molecular modeling and ligand docking [14].
Table 1 lists the structures of the pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-amine derivatives that were assayed for binding affinity at human ARs. The various 4-ether derivatives were chemically functionalized with amino, carboxylic acid, fluoropropyl or extended alkynyl chains, making them functionalized congeners for conjugation to other biologically active moieties or to carriers [15].
Table 1.
Affinity of a series of pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-amine derivatives at three subtypes of human ARs.
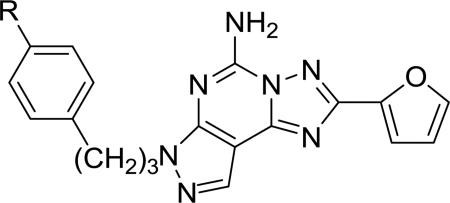
| Affinity, Ki, nM (or %inhib) | ||||
|---|---|---|---|---|
| Compd | Structure | A1a | A2Aa | A3a |
| 2 | R = -OCH3 | (35±5%) c | 4.1b | (67±1%) c |
| 3 | R = -OH | 4440±810 | 48±28b | (34±3%) c |
| 4 | R = -O(CH2)3F | 1000±40 | 53.6±25.5 | 1320±200 |
| 5 | R = -OCH2CO2CH3 | 4840±430 | 96±48 | (50±6%) c |
| 6 | R = -OCH2CO2H | 1700±250 | 10.2±2.2 | (29±2%) c |
| 7 | R = -OCH2CONH(CH2)2NH2 | 1270±140 | 6.8±1.1 | 3970±120 |
| 8 | R = -OCH2CONH(CH2)2NH(CH2)2NH2 | (20±4%) c | 10.7±4.6 | 3290±1500 |
| 9b,e | AlexaFluor conjugate of 7 | (20±3%) c | 111±16 | (4±2%) c |
| 10 | TAMRA conjugate of 7 | (7±5%) c | 357±130 | (24±1%) c |
| 11 | R = -OCH2CONH(CH2)2NHCO(CH2)4C≡CH | 1100±130 | 75.4±23.3 | 1280±80 |
| 12 | biotin-PEG4 conjugate of 11 | 1300±330 | 330±15 | >1000 |
| 13 | N-(ethylene)acetamide conjugate of 11 | 1700±290 | 220±14 | 302±50 |
| 14 e | BODIPY conjugate of 8 | 4180±920 | 15.1±1.8 | (28±3%) c |
| 16 e | PAMAM conjugate of 7 (13% substitution) | (0%) | 31.7±1.0 | 1250±160 |
| 17 e | PAMAM conjugate of 8 (16% substitution) | (7±5%) d | 38.1±4.3 | (26±4%) d |
Competition radioligand binding assays were conducted with membranes prepared from mammalian cells expressing recombinant human A1, A2A, or A3AR. The radioligands used were: A1, [3H]8-cyclopentyl-1,3-dipropylxanthine; A2AAR, [3H]2-[p-(2-carboxyethyl)phenyl-ethylamino]-5′-N-ethylcarboxamidoadenosine; A3AR, [125I]N6-(4-amino-3-iodobenzyl)adenosine-5′-N-methyl-uronamide. Values for the multivalent conjugates 16 and 17 are calculated on the basis of dendrimer concentration. Values are expressed as the mean ± SEM. ND, not determined.
Percent inhibition at 10 μM.
Percent inhibition at 1 μM.
9, MRS5346; 14, MRS5418; 16, MRS5475; 17, MRS5383.
Synthetic routes to various pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-amine derivatives (3–17) in which extended chain substituents have replaced the methoxy group of SCH 442416 are described in Schemes 1–3. Shinkre et al. reported the removal of the methyl ether of 2 as a means of further derivatizing this series of A2AAR antagonists [14]. Commercially available SCH 442416 was demethylated using BBr3 in CH2Cl2 to yield phenol 3, which was used in the next reaction without further purification. Various conditions to chemoselectively alkylate phenol 3 in the presence of the exocyclic amine functionality were compared. Reaction with methyl 2-bromoacetate and Cs2CO3 as a base in MeOH at room temperature and addition of 5 equiv. of electrophile at 1 h intervals formed 5 as the major product and considerably decreased the formation of di- and trialkylated products from ~25% to ~10% and the formation of polar byproducts. However, our attempts to first protect the amine of 3 as a tert-butyl carbamate (Boc) using Boc2O and triethylamine in DMF resulted in the exclusive formation of the Boc-protected phenol (data not shown).
Scheme 1.

Synthesis of fluoropropyl, amine and carboxylic acid functionalized congeners derived from SCH 442416. Reagents and conditions: a) BBr3, CH2Cl2, rt, 2 h; b) 1-bromo-3-fluoropropane, Cs2CO3, an. MeOH, rt, 18 h; c) methyl 2-bromoacetate, Cs2CO3, an. MeOH, rt, 16 h; d) Aq. 1N NaOH, MeOH:THF (1 mL, 1:1, v/v), rt, 2 h (for 6); ethylenediamine:MeOH (9:1, v/v), rt, 36 h (for 7); diethylenetriamine, an. DMF, rt, 32 h (for 8).
Scheme 3.

Synthesis of conjugates of pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-amines with a PAMAM dendrimer. Reagents and conditions: a) EDC·HCl, DMSO, freshly prepared 0.1M MES aq. buffer (pH = 5, adjusted with 1N NaOH or 10% HCl).
Fluoropropyl derivative 4 was synthesized in satisfactory yields with the optimized chemo-selective alkylation conditions using 1-bromo-3-fluoropropane and Cs2CO3 in MeOH (Scheme 1). Hydrolysis of the methyl ester of the O-alkylated product 5 with aqueous 1N NaOH in MeOH and THF provided the corresponding carboxylic acid derivative 6 (Scheme 1). Alternatively, methyl phenoxyacetate 5 could be reacted with either neat ethylenediamine or diethylenetriamine in DMF at room temperature (Scheme 1) to form N-aminoethylacetamide 7 (55%) and N-[2-(2-aminoethyl)-aminoethyl]acetamide 8 (56%), respectively.
N-Aminoethylacetamide 7 was used as a common key intermediate to anchor two fluorophore moieties viz. AlexaFluor® 488 in 9 or 6-carboxytetramethylrhodamine in 10 by active ester coupling in a mixture of DMF and aqueous Na2B4O7 at pH 8.5 with purification by semi-preparative HPLC (Scheme 2). Similarly, diamine 8 was labeled with BODIPY® 650/665 [23] to yield compound 14.
Scheme 2.

Synthesis of conjugates of pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-amines with fluorescent moieties (9, 10, and 14) and other acyl derivatives using standard amide coupling protocols and click chemistry. Reagents and conditions: a) activated fluorophore (AlexaFluor 488 carboxylic acid, 2,3,5,6-tetrafluorophenyl ester for 9; 6-carboxytetramethylrhodamine succinimidyl ester for 10), DMF, 0.1M aq. solution of Na2B4O7 (pH = 8.5), rt, 20 h; 6-heptynoic acid, EDC·HCl, an. DMF : an. CH2Cl2 (1:1, v/v), rt, 16 h for 11; b) corresponding azide (PEG4 carboxamide-6-azidohexanyl biotin for 12, N-(2-azidoethyl)acetamide for 13, CuSO4·5H2O, sodium ascorbate, DMSO:H2O (1:1, v/v), rt 16 h; c) 6-(((4,4-difluoro-5-(2-pyrrolyl)-4-bora-3a,4a-diaza-s-indacene-3-yl)styryloxy)acetyl)aminohexanoic acid, succinimidyl ester (BODIPY 650/665-X), DMF, 0.1 M aq. Na2B4O7 (pH = 8.5), rt, 18 h.
As an alternative coupling approach, the primary amine of N-aminoethylacetamide 7 was regio-selectively coupled to 6-heptynoic acid using EDC***HCl in DMF to afford compound 11 with a terminal alkyne, which is suitable for the derivatization using a click cycloaddition reaction [20]. The alkyne 11 reacted with PEG4 carboxamide-6-azidohexanyl biotin and N-(2-azidoethyl)acetamide [16] using the Cu(I)-catalyzed 2+3 cyclization to form corresponding triazole derivatives 12 and 13, respectively.
Finally, the amine congeners 7 and 8 were conjugated to the peripheral carboxylic acids of polyamidoamine (PAMAM) G3.5 dendrimer 15 (containing a butane core) using the EDC-mediated coupling protocol in MES buffer to obtain 16 (35%) and 17 (69%), respectively (Scheme 3). Mass analysis of the conjugates 16 and 17 (MW of 16 – 17K) indicated an average loading of 8.1 and 10 monomer moieties, respectively, out of a possible 64 sites. Consistently, UV spectroscopic analysis indicated the loading of approximately 6.8 and 7.8 ligands on 16 and 17, respectively (Supporting Information).
Binding assays were carried out using standard radioligands in Chinese hamster ovary (CHO) cells expressing the human A1 or A3ARs and in HEK293 cells expressing the human A2AAR [15, 17, 18]. High selectivity for the A2AAR was generally achieved (e.g. amine congeners 7 and 8 at the A3AR), with binding at the A1 or A3ARs in the micromolar range at best. This selectivity was evidently a function of the robust pharmacophore present in SCH 442416 and the insensitivity in receptor binding at the phenolic position. However, some modulation of the A2AAR affinity, depending on the terminal groups, was observed. For example, the fluoropropyl derivative bound to the A2AAR with a Ki value of 53.6 nM. An ester derivative 5 was less potent at the A2AAR than either carboxylic 6 or amino derivatives 7 and 8. There was no preference at the A2AAR for cationic vs. anionic terminal groups in 6 – 8, and the addition of a second amine in the chain in 8 did not increase the affinity. The introduction of sterically bulky groups in conjugates 9, 10, 12, and 14 had variable effects on the affinity at the A2AAR. 14 retained nearly full affinity with respect to the precursor amine 8, supporting the expectation that this chain extends into an external region of the receptor. Interestingly, 14 contained a secondary amine in the chain, suggesting this positively charged group as a source of the relatively high affinity of the conjugate. Amide conjugates 11 – 13, including 12 and 13 with a triazole linker from click coupling, showed reduced A2AAR affinity relative to precursor amine 7.
The three fluorescent conjugates 9, 10, and 14 were considered for use as fluorescent probes in A2AAR assays. Compound 9 proved to be a useful probe in fluorescence polarization (FP) assays [19]. The fluorescent dye, AlexaFluor 488 is a suitable fluorophore for FP because of its prolonged lifetime of 4.1 ns, which proved to be appropriate in our experiments and in other studies [19,21,22]. If the fluorescent lifetime is either too short or too long, a significant FP signal is unattainable. Compounds 10 and 14 contained as fluorophores tetramethylrhodamine (TAMRA) and BODIPY 650/665-X, respectively, which are not often applied in FP experiments because of their short lifetime. In preliminary experiments, it was not possible to get a significant FP signal with compound 14 in our assay system. Thus, compounds 10 and 14 were mainly designed for fluorescence microscopy experiments to detect the A2AAR in transfected or control cells with the advantage of a long wavelength of emission. In fluorescent microscopy, one of the main problems is the autofluorescence of the cells in the green wavelength range obscuring the signal from the noise, which can be eliminated with a fluorophore emitting at a longer wavelength. These two fluorophores are in this range, and compound 14 with a Ki value of 15 nM is likely suitable for cell labeling experiments.
The multivalent dendrimer conjugates 16 and 17 were highly potent and selective in binding to the A2AAR. The two spacer chains were indistinguishable by A2AAR affinity. Previously, various AR agonists were tethered from PAMAM to form GPCR ligand-dendrimer (GLiDe) conjugates that retained high affinity, selectivity, and efficacy as agonists [24]. This is the first example of PAMAM dendrimer conjugates of a strategically designed, functionalized AR antagonist.
A molecular modeling study was carried out to explore the recognition of the AlexaFluor-labeled antagonist 9 in binding to the X-ray crystal structure of the A2AAR. The impeded free rotation of the molecule, which leads to a fluorescence polarization signal [19], could be explained by the anchoring of the AlexaFluor moiety to the receptor through interactions with specific residues in the extracellular region of the A2AAR. A flexible molecular docking was performed to find a likely binding pose of the antagonist in the A2AAR binding cavity and to predict its putative interactions. The recently released 2.4 Å resolution structure of the A2AAR in complex with the triazolotriazine antagonist ZM241385 (PDB code: 1EML) was used as the starting structure [11].
The AlexaFluor-labeled antagonist 9 was built and energy minimized in MacroModel and then automatically docked in the binding pocket of the unoccupied receptor by means of Glide. The docking box was centered on the bicyclic core of ZM241385, and the box dimensions were sufficiently large to accommodate 9, which is of greater volume than ZM241385. The most favorable poses from the Glide docking results were chosen as representative binding modes for the antagonist, and the ligand-receptor complex was further subjected to conformational search via Monte Carlo Multiple Minima (MCMM) in MacroModel. Both the ligand and the amino acid side chains in the binding pocket were considered fully flexible during the MCMM conformational search calculations in order to extensively explore the conformational space. The lowest energy conformation of the ligand-receptor complex was chosen as the preferred binding mode of the antagonist.
As expected, the pyrazolotriazolopyrimidine core of our antagonists was oriented within the A2AAR binding cavity, with the triazolopyrimidine moiety binding in a manner similar to the triazolotriazine ring of ZM241385. The ligand-receptor binding mode corresponding to 9 is shown in Figure 1. In detail, the N3 of the pyrazolotriazolopyrimidine accepted an H-bond from the CONH2 of N253 in transmembrane helical domain (TM) 6, designated residue 6.55 [25]. Moreover, the exocyclic amino group of the antagonist was anchored by strong H-bonding to the side chain carbonyl group of N253 and to the carboxyl group of E169 in the second extracellular loop (EL2). The pyrazolotriazolopyrimidine moiety was further stabilized by a π-stacking interaction with the aromatic side chain of F168 located in EL2. Hydrophobic residues such as M174(5.35), M177(5.38), I252(6.54), L267(7.32), and M270(7.35) created a favorable environment for the heterocyclic moiety of the ligand. The furan substituent common to both antagonist classes interacted with the NH2 group of N253 and was also coordinated by favorable aromatic or hydrophobic residues such as H250(6.52), L249(6.51), W246(6.48), L85(3.31), and V84(3.32). The docking pose of the antagonist showed the phenoxy extension protruding toward the extracellular region of the receptor, allowing the linker chain and the AlexaFluor moiety to be situated at the top of the receptor with the xanthene ring system lying almost parallel to the plane of the cell membrane. The phenoxy ring was close to hydrophobic side chains, including L167 in EL2 and L267 at the extracellular portion of TM7. The two carboxamido groups in the linker chain between the phenoxy group and the xanthene were solvent exposed and located between EL2 and EL3. The negatively charged carboxyl group of the phenyl ring pointed up, presumably interacting with the solvent molecules. Figure 1B shows, from a top view, 9 docked in the binding site of A2AAR, depicting the strong interactions that anchor the AlexaFluor xanthene core to the extracellular region of the receptor. The charged sulfonate groups of the AlexaFluor engaged in interactions with N145 in EL2 on one side and with N175(5.36), at the top of TM5, on the other side of the xanthene ring. In addition, the backbone NH of M174(5.35) acted as a H-bond donor to one of the sulfonate groups, while the hydrophobic side chain of the same residue was oriented in a favorable position underneath the xanthene ring. The hydrophilic backbone carbonyl group of F257(6.59) was in proximity to the uncharged amino group of the AlexaFluor moiety, while the positively charged ketimine group formed an ionic interaction with the carboxyl group of D170 in EL2.
Figure 1.

Molecular modeling of ligand binding to the human A2AAR. Details of the binding site of the X-ray structure of the receptor complexed with the AlexaFluor-labeled antagonist 9 as obtained after a fully flexible MCMM conformational search. A) View from the plane of the phospholipid bilayer. B) View from the extracellular side (putative H-bonds between the ligand and receptor are shown). The ligand is represented as a ball and stick structure, colored by element (carbon atoms colored in cyan). For clarity, the residues located in proximity to the ligand are labeled and represented as sticks, colored by element (carbon atoms colored in green).
In conclusion, we have identified a path to design novel pharmacological probes for the A2AAR. The introduction of a chemically funtionalized chain at an insensitive site on a pyrazolotriazolopyrimidine antagonist allows the incorporation of sterically large reporter groups, such as fluorophores that avoid the use of radioactivity in drug screening. Some of these analogues have retained the selectivity and high affinity of the parent compound, e.g. fluorescent BODIPY conjugate 14. An analogue of moderate affinity contained a fluoroalkyl chain for PET imaging. The potent and A2AAR-selective N-aminoethylacetamide 7 and N-[2-(2-aminoethyl)-aminoethyl]acetamide 8 congeners were coupled to polyamidoamine (PAMAM) G3.5 dendrimers, and the multivalent conjugates displayed high A2AAR affinity. The modeling results highlighted the key interactions between the heterocyclic core and the binding pocket of the receptor. The results also predicted the binding mode of a fluorescent AlexaFluor moiety attached to the pharmacophore in conjugate 9 to find putative stabilizing interactions with residues of the ELs, consistent with the experimental data indicating reduced rotation of the fluorophore when bound to the receptor. Moreover, with high molecular weight dendrimer conjugates that bound potently and selectively at the A2AAR, we have confirmed experimentally that the functionalized chain is accessing the extracellular regions of the receptor.
Supplementary Material
Acknowledgements
This research was supported in part by the Intramural Research Program of the NIH, National Institute of Diabetes and Digestive and Kidney Diseases. We thank Dr. Dale Kiesewetter (NIBIB, NIH) for helpful discussions.
ABBREVIATIONS
- AR
adenosine receptor
- BODIPY
4,4-difluoro-4-bora-3a,4adiaza-s-indacene
- CHO
Chinese hamster ovary
- CNS
central nervous system
- DMF
N,N-dimethylformamide
- EDC
N-(3-dimethylaminopropyl)-N'-ethylcarbodiimide
- FP
fluorescence polarization
- HEK
human embryonic kidney
- KW6002
(E)-1,3-diethyl-8-(3,4-dimethoxystyryl)-7-methyl-3,7-dhydro-1H-purine-2,6-dione
- MCMM
Monte Carlo Multiple Minima
- MES
2-(N-morpholino)-ethanesulfonic acid
- NECA
5'-N-ethylcarboxamidoadenosine
- PAMAM
polyamidoamine dendrimer
- PD
Parkinson's disease
- PET
positron emission tomography
- SCH 442416
5-amino-7-(3-(4-methoxy)phenylpropyl)-2-(2-furyl)pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidine
- TAMRA
tetramethylrhodamine
- THF
tetrahydrofuran
- TLC
thin layer chromatography
- TM
transmembrane helical domain
- ZM241385
4-[2-[7-amino-2-(2-furyl)-1,2,4-triazolo[1,5-a][1,3,5]triazin-5-yl-amino]ethylphenol
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supporting information Supplementary data (chemical synthesis, characterization data, HPLC purification and purity analysis procedures, Cell culture and membrane preparation, Radioligand membrane binding studies, UV determination of the number of amine monomers 7 on G3.5 dendrimer 17 and additional modeling information) associated with this article can be found, in the online version, at doi:xxxxxxx/bmcl/10.
References and Notes
- [1].Haselkorn ML, Shellington DK, Jackson EK, Vagni VA, Janesko-Feldman K, Dubey RK, Gillespie DG, Cheng D, Bell MJ, Jenkins LW, Homanics GE, Schnermann J, Kochanek PM. J. Neurotrauma. 2010;27:901. doi: 10.1089/neu.2009.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Ferguson AL, Stone TW. Eur. J. Neurosci. 2010;31:1208. doi: 10.1111/j.1460-9568.2010.07157.x. [DOI] [PubMed] [Google Scholar]
- [3].Jacobson KA, Gao ZG. Nature Rev. Drug Disc. 2006;5:247. doi: 10.1038/nrd1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Yu L, Coelho JE, Zhang X, Fu Y, Tillman A, Karaoz U, Fredholm BB, Weng Z, Chen JF. Physiol. Genomics. 2009;37:199. doi: 10.1152/physiolgenomics.90353.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Pinna A. Expert Opin. Investig. Drugs. 2009;18:1619. doi: 10.1517/13543780903241615. [DOI] [PubMed] [Google Scholar]
- [6].LeWitt PA, Guttman M, Tetrud JW, Tuite PJ, Mori A, Chaikin P, Sussman NM. Ann. Neurol. 2008;63:295. doi: 10.1002/ana.21315. [DOI] [PubMed] [Google Scholar]
- [7].Brooks DJ, Papapetropoulos S, Vandenhende F, Tomic D, He P, Coppell A, O'Neill G. Clin. Neuropharmacol. 2010;33:55. doi: 10.1097/WNF.0b013e3181d137d2. [DOI] [PubMed] [Google Scholar]
- [8].Todde S, Moresco RM, Simonelli P, Baraldi PG, Cacciari B, Spalluto G, Varani K, Monopoli A, Matarrese M, Carpinelli A, Magni F, Kienle MG, Fazio F. J. Med. Chem. 2000;43:4359. doi: 10.1021/jm0009843. [DOI] [PubMed] [Google Scholar]
- [9].Hodgson RA, Bertorelli R, Varty GB, Lachowicz JE, Forlani A, Fredduzzi S, Cohen-Williams ME, Higgins GA, Impagnatiello F, Nicolussi E, Parra LE, Foster C, Zhai Y, Neustadt BR, Stamford AW, Parker EM, Reggiani A, Hunter JC. J. Pharm. Exp Therap. 2009;330:294. doi: 10.1124/jpet.108.149617. [DOI] [PubMed] [Google Scholar]
- [10].Ohta A, Kjaergaard J, Sharma S, Mohsin M, Goel N, Madasu M, Fradkov E, Sitkovsky M. Br. J. Pharmacol. 2009;156:297. doi: 10.1111/j.1476-5381.2008.00019.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EYT, Lane JR, IJzerman AP, Stevens RC. Science. 2008;322:1211. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Katritch V, Jaakola VP, Lane JR, Lin J, IJzerman AP, Yeager M, Kufareva I, Stevens RC, Abagyan R. Structure- based discovery of novel chemotypes for adenosine A2A receptor antagonists. J. Med. Chem. 2010;53:1799. doi: 10.1021/jm901647p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Carlsson J, Yoo L, Gao ZG, Irwin J, Shoichet B, Jacobson KA. Structure-based discovery of adenosine A2A receptor ligands. J. Med. Chem. 2010;53:3748. doi: 10.1021/jm100240h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Shinkre BA, Kumar TS, Gao ZG, Deflorian F, Jacobson KA, Trenkle WC. Bioorg. Med. Chem. Lett. 2010;20:5690. doi: 10.1016/j.bmcl.2010.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Olah ME, Gallo-Rodriguez C, Jacobson KA, Stiles GL. Mol. Pharmacol. 1994;45:978. [PMC free article] [PubMed] [Google Scholar]
- [16].Saito S, Tamai H, Usui Y, Inaba M, Moriwake T. Chem. Letters. 1984;7:1243. [Google Scholar]
- [17].(a) Schwabe U, Trost T. Naunyn-Schmiedeberg's Arch. Pharmacol. 1980;313:179. doi: 10.1007/BF00505731. [DOI] [PubMed] [Google Scholar]; (b) Perreira M, Jiang JK, Klutz AM, Gao ZG, Shainberg A, Lu C, Thomas CJ, Jacobson KA. J. Med. Chem. 2005;48:4910. doi: 10.1021/jm050221l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Jarvis MF, Schutz R, Hutchison AJ, Do E, Sills MA, Williams M. J. Pharmacol. Exp. Ther. 1989;251:888. [PubMed] [Google Scholar]
- [19].Kecskés M, Kumar TS, Yoo L, Gao ZG, Jacobson KA. Biochem. Pharmacol. 2010;80:506. doi: 10.1016/j.bcp.2010.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kolb HC, Sharpless KB. Drug Discov. Today. 2003;8:1128. doi: 10.1016/s1359-6446(03)02933-7. [DOI] [PubMed] [Google Scholar]
- [21].Martin H, Murray C, Christeller J, McGhie T. Anal. Biochem. 2008;381:107. doi: 10.1016/j.ab.2008.06.025. [DOI] [PubMed] [Google Scholar]
- [22].Albizu L, Teppaz G, Seyer R, Bazin H, Ansanay H, Manning M, Mouillac B, Durroux T. J. Med. Chem. 2007;50:4976. doi: 10.1021/jm061404q. [DOI] [PubMed] [Google Scholar]
- [23].Loudet A, Burgess K. Chem. Rev. 2007;107:4891. doi: 10.1021/cr078381n. [DOI] [PubMed] [Google Scholar]
- [24].Jacobson KA. Trends Pharmacol. Sci. 2010;31:575. doi: 10.1016/j.tips.2010.09.002. doi: 10.1016/j.tips.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ballesteros JA, Weinstein H. Methods Neurosci. 1995;25:366. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.