

Abstract
Dynamic analysis of the uptake and metabolism of polycyclic aromatic hydrocarbons (PAHs) and their metabolites within live cells in real time has the potential to provide novel insights into genotoxic and non-genotoxic mechanisms of cellular injury caused by PAHs. The present work, combining the use of metabolite spectra generated from metabolite standards using multiphoton spectral analysis and an “advanced unmixing process”, identifies and quantifies the uptake, partitioning, and metabolite formation of one of the most important PAHs (benzo[a]pyrene, BaP) in viable cultured rat liver cells over a period of 24 hr. The application of the advanced unmixing process resulted in the simultaneous identification of 8 metabolites in live cells at any single time. The accuracy of this unmixing process was verified using specific microsomal epoxide hydrolase inhibitors, glucuronidation and sulfation inhibitors as well as several mixtures of metabolite standards. Our findings prove that the two-photon microscopy imaging surpasses the conventional fluorescence imaging techniques and the unmixing process is a mathematical technique that seems applicable to analysis of BaP metabolites in living cells especially for analysis of changes of the ultimate carcinogen benzo[a]pyrene-r-7,t-8-dihydrodiol-t-9,10-epoxide. Therefore, the combination of the two-photon acquisition with the unmixing process should provide important insights into the cellular and molecular mechanisms by which BaP and other PAHs alter cellular homeostasis.
Keywords: Benzo[a]pyrene, microsomal epoxide hydrolase inhibitor, aryl hydrocarbon receptor, EROD activity, BaP metabolites, multiphoton spectral analysis
Introduction
PAHs are persistent organic environmental contaminants formed as by-products of incomplete combustion of fossil fuels. These compounds have been identified in ground and rain water, tap water, waste water, sewage sludge and foodstuffs ((Ramesh et al. 2004), (Samanta et al. 2002)). Due to their ubiquitous presence and toxicity, PAHs are among the most important environmental pollutants; and several PAHs such as benzo[a]pyrene (BaP) and PAH mixtures are carcinogenic. BaP is an extensively studied prototype carcinogen; exposure to BaP by inhalation results in rapid uptake and distribution to several tissues with the highest levels found in liver, esophagus, small intestine, and blood within 30 min to 1 hr after exposure ((Ramesh et al. 2002), (Weyand and Bevan 1986)). BaP exerts its toxic and mutagenic effects through bioactivation and generation of reactive metabolites (Phillips 2005) and hepatobiliary excretion is a major route for elimination of hydroxylated metabolites and other conjugates.
The metabolism of BaP is complex and numerous metabolites are generated in the biotransformation process including hydroxylated intermediates, epoxides, quinones, dihydrodiols, dihydrodiol epoxides (see supplemental figure S1) and various metabolite-conjugates in cells ((Bolton et al. 2000), (Shimada et al. 2002)). Some of these metabolites such as benzo[a]pyrene-r-7,t-8-dihydrodiol-t-9,10-epoxide (BPDE) can alkylate DNA to form BaP-DNA adducts which have been associated with BaP-induced carcinogenesis (Gammon et al. 2004). In addition, many of the major metabolites can be conjugated to glucuronic acid, sulfate and glutathione to become more water soluble facilitating excretion (Zhu et al. 2008).
An interesting and useful property of several PAHs and their metabolites is that they fluoresce efficiently in solution (Dabestani 1999). The multiple fused aromatic ring systems are highly fluorescent due to their delocalized pi bonds which permit relatively low-energy photons to excite their electrons into excited states that return to the ground state with the emission of fluorescence. This property has been exploited to detect and evaluate environmental PAH contamination (e.g., (Wild et al. 2007), (Weston et al. 1993), (Goryacheva et al. 2005)), tissue accumulation of PAHs and their metabolites, and DNA and protein adducts by high performance liquid chromatography and fluorescence detection ((Gmur and Varanasi 1982), (Xu and Jin 1984), (Boysen and Hecht 2003)). The fluorescence properties of BaP have also been used to investigate the processes of tissue penetration and metabolism in vivo in a mouse skin model (Lopp et al. 1986). At the cellular level, the characteristic fluorescence of BaP has previously been exploited to monitor mixed function oxygenase activity in cell populations by flow cytometry (Miller and Whitlock 1982) and in individual anchored cells in culture ((Plant et al. 1985), (Moore et al. 1994)).
One of the challenges in correlating exposure levels and adduct formation for the purpose of quantitative exposure assessments (Gammon et al. 2004) is the lack of analytical methods that can directly identify the presence and levels of BaP and its reactive metabolites in real time within living cells and tissues. Using laser cytometry, we previously evaluated the rapid uptake and partitioning of BaP into the plasma membrane and membranes of intracellular organelles within minutes after addition of the fluorescent genotoxicant (Barhoumi et al. 2000) and analyzed a number of non-genotoxic effects of BaP on cell signaling in cultured cells ((Barhoumi et al. 2002), (Barhoumi et al. 2006)). Because BaP is photosensitive and breaks down when exposed to UV light, single cell assessment of BaP uptake and metabolism using conventional fluorescence microscopes or continuous wavelength laser confocal microscopes has been limited to BaP identification in cells without quantification. However, integration of pulsed femtosecond infrared laser systems in multiphoton microscopes provide high detection sensitivity and minimal fluorophore excitation volumes to reduce photobleaching, thereby providing new opportunities for investigating BaP uptake and metabolism in situ ((Hornung et al. 2007), (Barhoumi et al. 2009)) as well as the consequences of BaP exposure within individual cells.
The objective of the current study was to extend the previous single cell multiphoton microscopic analysis of cellular BaP uptake and partitioning in order to better evaluate BaP metabolism in situ using: 1) multiphoton microscopy spectral acquisition to minimize the effect of UV exposure on BaP autofluorescence and metabolite generation, and 2) a spectral unmixing process that uses the unique spectral properties of metabolites generated from standards in order to detect their presence and levels in cells treated with BaP. The normal rat liver cell line (Clone 9) used in this study exhibits an increase in ethoxyresorufin-O-deethylase (EROD) activity in response to PAH treatment and has been utilized as a model system for investigating the nongenotoxic effects of BaP (Barhoumi et al. 2002). These liver cells were used to identify BaP parent compound and reactive metabolites generated within cells in real time, to quantify these metabolites using database reference spectra generated with reference standards, and to use specific metabolic inhibitors to modify metabolite profiles. The development of this real time approach for investigating BaP metabolism in living cells will provide insights on the rates of metabolite formation and persistence and also facilitate development of agents that will block critical mutagenic/carcinogenic metabolic pathways (Hecht 2002).
Materials and Methods
Materials
Ham’s F-12 culture media, Dulbecco’s phosphate buffered saline (PBS), Janus green, β-glucuronidase (keyhole limpet, #G8132), triclosan (Irgasan), pyrene (Pyr), resorufin ethyl ether and 3,3′-methylene-bis(4-hydroxycoumarin) (dicumarol) were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO). Fetal bovine serum was obtained from Equitech-Bio (Kerrville, TX). Benzo[a]pyrene, benzo[a]pyrene-r-7,t-8-dihydrodiol-t-9,10-epoxide(±),(anti) (BPDE), benzo[a]pyrene-3,6-dione (3,6BPQ), 3-hydroxybenzo[a]pyrene (3OH), 9-hydroxybenzo[a]pyrene (9OH), benzo(a)pyrene-trans-7, 8- dihydrodiol(+/−) (t7,8), benzo[a]pyrene-r-7,t-8,t-9,c-10-tetrahydrotetrol(+/−) (c-tetrol), Benzo[a]pyrene-r-7,t-8,t-9,t-10-tetrahydrotetrol (±) (t-tetrol), benzo(a)pyrene-3-sulfate, potassium salt (3-S), benzo(a)pyrene-9-sulfate (9-S), 9-benzo(a)pyrene-β-D-glucopyranosiduronic acid (9-G) were purchased from Midwest Research Institute (Kansas City, MO) which operates the Chemical Carcinogen Reference Standard Repository. Analytical data provided with each standard was reported as >99% pure by HPLC and UV/visible spectra. These UV/visible spectra were also confirmed prior to use in cells. Soluble epoxide hydrolase (sEH) inhibitors used included 1-(1-acetyl-piperidin-4-yl)-3-(4-trifluoromethoxy-phenyl)-urea (TUPS, 1709, (Rose et al. 2010)) and trans-4-[4-(3- trifluoromethoxyphenyl −1-yl-ureido)-cyclohexyloxy]-benzoic acid (t-TUCB, 1728, (Hwang et al. 2007)). Microsomal epoxide hydrolase (mEH) inhibitors used included 2-nonylsulfanyl-propionamide (mEH #16) and 10-hydroxy-octadecanamide (mEH #29) (Morisseau et al. 2008). Tissue culture flasks, 2-well Lab-Tek chambered coverglass slides and 96 well Greiner glass bottom multi-well plates were purchased from Thermo Fisher Scientific (Waltham, MA). BaP, Pyr, 3,6BPQ, 3OH, 9OH, 3-S, 9-S, 9-G, t7,8, and BPDE were each prepared as 10 mM stocks in DMSO. Resorufin ethyl ether was prepared as a 7 mM stock in methanol and diluted to 7 μM for EROD activity measurement. Janus green was prepared in PBS at 1 mg/ml.
Cell Culture
The normal rat liver cell line, Clone 9 (CRL 1439) was obtained at passage 17 from American Type Culture Collection (Manassas, VA) and used between passages 25 to 35. Cultures were approximately 80% confluent at the time of analysis.
Ethoxyresorufin-O-deethylase (EROD) Activity
EROD activity is a biomarker of exposure to planar halogenated and polycyclic aromatic hydrocarbons (PHHs and PAHs, respectively) and provides evidence of aryl hydrocarbon receptor-mediated induction of cytochrome P450-dependent monooxygenases (Donato et al. 1993). To identify the contribution of BaP and each of its metabolites to the induction of EROD activity, cells were plated on 96 well-plates at 25,000/well for 48 hr prior to treatment with different concentrations of BaP, 3OH, 9OH, t7,8, BPDE, 3-S, 9-S, 9-G, or 3,6BPQ. Following treatments for 3 hr, cells were then washed twice with PBS and loaded with 7 μM resorufin ethyl ether and 10 μM dicumarol for 30 min. EROD activity was measured using a BioTek Synergy 4 plate reader (Biotek Instruments Inc., Winooski, VT, USA) with an excitation wavelength of 540 nm and an emission wavelength of 590 nm. For comparison of EROD activity between different concentrations within the same treatment, cell number per well was determined using the Janus green assay (as described below) and EROD fluorescence intensities measured were corrected accordingly. Eight samples per concentration were collected and at least 3 experiments were preformed on different days.
Cell Count / Janus Green Assay
Cells were incubated in 96 well plates, and after treatments were washed with PBS (2x) and fixed with 100% methanol for 30 min at room temperature. Methanol was then completely removed and 1 mg/ml Janus green was added to the cultures for 3 min. Following removal of Janus green, cultures were washed twice with PBS and 100 μl of 50% methanol was added to each well. Cell counts were then determined with a BioTek Synergy 4 plate reader set to an absorbance of 630 nm (Raspotnig et al. 1999).
Single Cell Multiphoton Spectral Analysis of BaP and BaP Metabolite Standards
Clone 9 cells were cultured for 24 hr in Ham’s Nutrient Mixture F-12 with 10% fetal bovine serum on 2-well Lab-Tek slides following plating at a density of 105 cells per well. Cells were then washed and incubated for 24 hr with 2 μM of one of the following treatments: BaP, 3OH, 9OH, 3-S, 9-S, 9-G, 3,6BPQ, t7,8, BPDE, and DMSO or ethanol solvent carrier. Treatments were then removed by washing cells with serum- and phenol red-free culture medium and slides were transferred to the stage of a Zeiss 510 META NLO laser scanning microscope (Carl Zeiss Microimaging, Thornwood, NY). Directly after washing, spectral analysis of an area of 225 × 225 μm (typically containing 25 to 40 cells) was performed while irradiating cells with a Chameleon tunable Ti:Sapphire laser (Coherent Inc., Santa Clara, CA) at an excitation wavelength of 740 nm (which is roughly equivalent to 370 nm in single photon excitation with a continuous wavelength laser system). Using the lambda stack algorithm software available with the Zeiss 510 META NLO instrument, a fluorescence emission spectrum ranging from 399–600 nm with a 10.7 nm bandwidth was recorded for each scanned area. Eight to ten areas were scanned per treatment.
For generation of BaP spectra at different time points, Clone 9 cells were cultured for 48 hr, placed on the stage of the microscope and scanned directly after addition of 2 μM BaP and at different time intervals (0, 0.25, 1, 1.5, and 3 hr). Spectra were then collected as described above.
Generation of a Spectra Database from BaP and Metabolite Standards
Multiphoton fluorescence spectra were generated for each reference standard in culture medium in the absence of cells (BaP, 3OH, 9OH, t7,8, BPDE, 3-S, 9-S, 9-G, 3,6 BPQ or Pyr) under the same experimental conditions used to generate BaP spectra within cells. These spectra were saved in the spectra database and were labeled as reference spectra for later use to identify metabolites of BaP and/or metabolite standards present in Clone 9 cells at specific time points following incubation with BaP or specific metabolite standards. A background autofluorescence spectrum from untreated Clone 9 cells was generated using the same experimental conditions as used for BaP and standards, in order to separate BaP and metabolite spectra from the background spectra.
Linear Unmixing
Once the database reference spectra (BaP parent compound and metabolite standards) were generated, advanced spectral linear unmixing, available with the Zeiss 510 META instrument, was used. This unmixing process uses the fact that the total signal (S) detected in every channel wavelength (λj) is a linear combination of the contributing metabolites as described in the following equation:
| (1) |
where Ai represents the contribution from each metabolite. Using least square fitting that minimizes the square of D (see equation 2) where D represents the square of the difference between theoretical and measured values, will identify the values of Ai and therefore the contribution of each metabolite to the final BaP spectrum:
| (2) |
j represents the number of images collected between 399 and 600 nm with 10.7 nm bandwidth and providing the spectral data and i represents the number of metabolites measured per experiment. The maximum number of metabolites that can be measured simultaneously with the Zeiss 510 linear unmixing process is 8.
The linear equations are usually solved using the singular value decomposition method and values of Ai that results in best separations of metabolites are determined (Zimmermann 2005).
Additional Validation of Multiphoton Microscopy for Analysis of BaP and Metabolites
To validate the utility of multiphoton microscopy spectral analysis of BaP and metabolites in situ, the effects of brief, high intensity UV light exposure on BaP and its metabolites were evaluated. Cells treated with 2 μM BaP for 24 hr were washed and scanned before and after a 10 sec UV light exposure from an X-Cite 120Q 120 Watt mercury lamp (Photonic Solutions Inc., Mississauga, Ontario, Canada). Images were then analyzed using the reference spectra stored in the database and generated using the same scanning conditions described above. All images were collected with a C-APO 40X/1.2 NA water immersion objective designed for viewing specimens in an aqueous medium. Eight images per treatment were recorded and at least 3 experiments were performed, each on a different day.
To demonstrate the presence of the major carcinogenic metabolite of BaP (BPDE), and its precursor t7,8, as well as the sensitivity of the multiphoton microscopy unmixing approach to detect experimentally altered BaP biotransformation, the effects of both sEH and mEH on BaP metabolism were evaluated. Clone 9 cells were cultured for 8 hr, and then treated with one of the EHs for 18 hr, followed by exposure to 2 μM of BaP for 3 hr. Cells were then washed and cultured for another 18 hr in the presence of EH inhibitors.
Another confirmation of the presence of BPDE and t7,8 was performed by treating Clone 9 cells with the t7,8 standard and directly measuring the generation of BPDE and subsequent generation of a pyrene-like signal that would result from BPDE adduct formation. Similarly, direct conversion of BPDE to a pyrene adduct was monitored directly upon addition of the BPDE standard to Clone 9 cells.
To further verify the identity of metabolites generated in Clone 9 cells, triclosan was used as a substrate and inhibitor of glucuronidation and sulfonation (Wang et al. 2004) which in turn, should decrease the sulfonation and glucuronidation of the hydroxy metabolites of BaP. In addition, β-glucuronidase was used as a catalyst for the hydrolysis of the water-soluble glucuronide conjugates of BaP generated in phase II detoxification reactions which would increase the hydroxy metabolites of BaP (Kinoshita and Gelboin 1978). To conduct the triclosan experiments, Clone 9 cells were treated with 2 μM BaP, a combination of 2 μM BaP and 20–40 μM triclosan, 2 μM 3OH or the combination of 2 μM 3OH and 20 μM triclosan for 24 hr. Cells were then washed and scanned under the same experimental conditions. To conduct the β-glucuronidase experiments, cells were treated with 2 μM BaP, a combination of 2 μM BaP and 500 units/ml β-glucuronidase, 9OH, or a combination of 2 μM 9OH and 500 units/ml β-glucuronidase) for 24 hr, washed and then scanned. Spectra were then generated using the reference spectra as described above.
To test the ability of the unmixing method to decipher phase II products from their phase I precursors, a mixture of three closely related standards in suspension: 9OH (2 μM), 9-S (2 μM) and 9-G (2 μM) was scanned and a theoretical value for the amount of each standard was determined from the unmixing process and compared to the amount obtained with the standard alone at the same concentration.
Statistical Analysis
Data from the Biotek Synergy 4 plate reader for measuring EROD activity are presented as mean fluorescence intensities ± S.E. of 8 wells per concentration and were analyzed statistically by ANOVA followed by Dunnett’s multiple comparison test at P < 0.05. Data collected by multiphoton microscopy are reported as mean fluorescence intensities ± S.E. of at least 8 images per treatment and were analyzed by two-way ANOVA followed by Bonferroni test at P < 0.05. Data from the mEH inhibitors are reported as mean fluorescence intensity normalized to the mean of the corresponding control.
Results
Single Cell Multiphoton Spectral Analysis of BaP and Metabolites
Multiphoton microscopy spectral analysis of BaP fluorescence in Clone 9 cells treated with 2 μM BaP for 3 hr at an excitation wavelength of 740 nm and emission of 399–600 nm provided a sequence of images (Figure 1A). Each image in the sequence represents the fluorescence intensity generated in Clone 9 cells at a specific channel wavelength (400, 411, 422, 433, 444, 455, 477, 488, 499, 510, 521, 532, 543, 554, 565, 576, 587, 598 nm) with a bandwidth of 10.7 nm. Figure 1B represents the spectra generated from a similar sequence of images collected from Clone cells treated with 2 μM BaP for different time intervals (0.25, 1.0, 1.5, 3.0 hr). These spectral changes over time were indicative of metabolite formation.
Figure 1.

Multiphoton microscopy of Clone 9 cells exposed to BaP. (A). Typical spectral images collected at given wavelengths from 399 to 600 nm with 10.7 nm bandwidth in cells treated with 2 μM BaP for 3 h. The images shown include fluorescence emission contributions from BaP along with BaP metabolites generated within cells. (B) Multiphoton emission spectra recorded between 399 nm and 600 nm from cells loaded with 2 μM BaP for different time intervals (0.25, 1.0, 1.5 and 3 h).
In order to identify these metabolites (Figure 1B) in real time within viable Clone 9 cells, database reference spectra with 2 μM concentrations for each of the standards (i.e., BaP, 3OH, 9OH, t7,8, BPDE, Pyr, 3-S, 3,6 BPQ) and the background were generated in solution (Figure 2) using the same experimental conditions for generation of spectra in Clone 9 cells treated with BaP (Figure 1B). These spectra were saved in the database and were used as reference spectra in order to identify the presence and amount of each metabolite detected in Clone 9 cells following BaP treatments for specific times. Then, using the linear unmixing process described above and the reference spectra saved in the database, each of the metabolites (for which reference spectra were collected) can be identified. Figure 3 represents the results of the unmixing process in Clone 9 cells treated with 2 μM BaP for 24 hr, identifying the presence of seven metabolites (BaP, 3OH, 9OH, t7,8, BPDE, Pyr, 3,6BPQ) and the background signal. Each image represents the fluorescence intensity of the metabolite detected. The last panel in figure 3 represents the overlay of all metabolites including the background.
Figure 2.

Multiphoton spectra of BaP and several metabolites. Multiphoton spectra of BaP, 3OH, 9OH, t7,8, BPDE, 3-S, 3,6BPQ, and Pyr obtained as reference standards along with background fluorescence (Bkg) were generated in solution and stored in the database for later analysis of BaP metabolism in cells. No spectra were identified for the two tetrol standards (c-tetrol and t-tetrol) examined.
Figure 3.
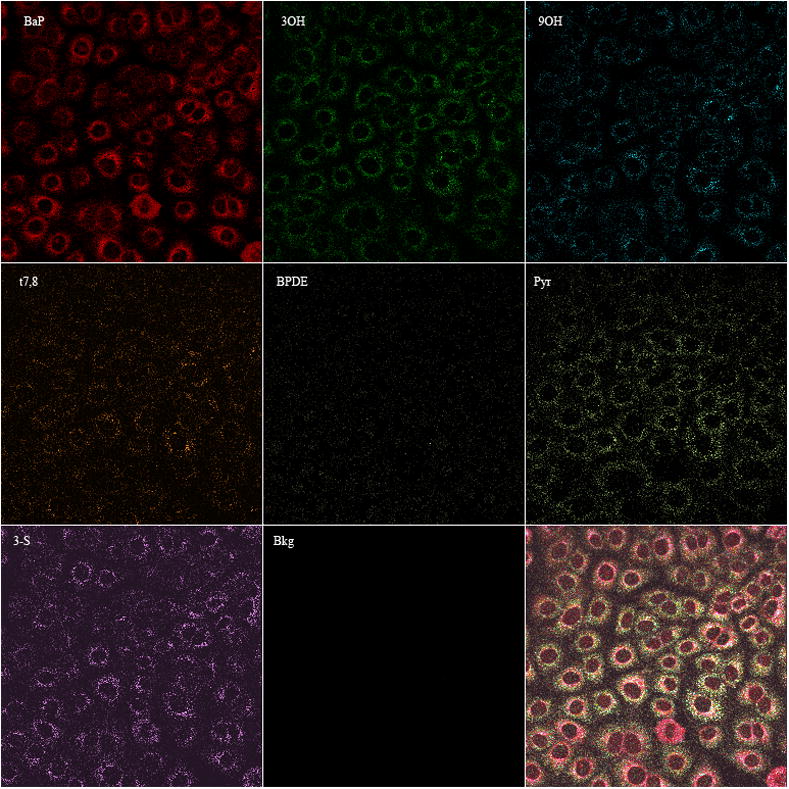
Images of BaP and seven metabolites. Images of BaP, 3OH, 9OH, t7,8, BPDE, Pyr, 3-S, and background were derived from linear unmixing process (equations 1&2) as described in methods that were present in viable Clone 9 cells directly after 24 h treatment with 2 μM BaP. The last panel represents the overlay of all 8 metabolites measured.
Time- and Concentration-Dependent Metabolism of BaP
Previous studies show that BaP is metabolized in Clone 9 cells (Barhoumi et al., 2000) and that BaP induces significant Cyp1A1-dependent EROD activity at concentrations = 0.2 μM (see supplemental figure S2). Minimal to non-detectable induction responses were observed for BaP metabolites (3OH, 9OH, t7,8, BPDE, 3-S, 9-S, 9-G, 3,6BPQ, Pyr) at concentrations up to 2 μM (data not shown) indicating that Cyp1A1, a key enzyme involved in BaP metabolism is induced only by the parent hydrocarbon at these concentrations. The relative distribution of BaP and several metabolites was investigated in Clone 9 cells treated with 2 or 10 μM BaP for 3 hr and analyzed for metabolite levels by multiphoton spectral analysis. Relative levels of 3OH, 9OH, t7,8, BPDE, 3-S and 3,6BPQ are similar between the 2 and 10 μM treatment levels (p > 0.05) whereas BaP levels were significantly higher in cells treated with 10 μM BaP (Figure 4A). A similar protocol was used to examine the time-dependent metabolism of BaP. Treatment of Clone 9 cells with 2 μM BaP for 3 hr followed by removal of BaP from the media showed that 6 hr after removal of BaP, the parent compound and the majority of metabolites decreased significantly with the exception of BPDE when compared to the corresponding metabolites at 0 hr (directly after removal of BaP). Twenty four hours after removal of BaP, the parent compound continued to decrease significantly while all other metabolites with the exception of 9OH were increased (Figure 4B). The time dependent effects of BaP metabolism were also examined during the treatment of Clone 9 cells with 2 μM BaP for up to 3 h. The fluorescence intensities for BaP and major metabolites (3OH, 9OH, and BPDE) increased over the initial treatment period and then reached saturation independently (Figure 4C). These results are in contrast to the time-dependent changes in BaP and metabolite profiles observed in Figure 4B. Thus 2 μM BaP in the extracellular media acts as a reservoir for intracellular BaP which is associated with lipid-rich organelles and maintains steady state levels of both BaP and the BaP metabolites over the treatment period.
Figure 4.

Analysis of BaP metabolism in Clone 9 cells. (A). Concentration-dependent metabolism of BaP. A comparison of metabolites fluorescence present in Clone 9 cells after three hour treatment with two different concentrations of BaP (2 and 10 μM). * represent significant difference from the corresponding metabolite at p < 0.05. (B). Time-dependent metabolism of BaP. Parent compound and metabolites generated in Clone 9 cells treated with 2 μM BaP for 24 h, washed and examined directly (0 h, open bar), 6 h (hatched bar) and 24 h (black bar) following removal of BaP. Data represent mean fluorescence intensity ± SEM of each metabolite collected from at least 8 images per treatment. * represents significant difference from the corresponding metabolite at p < 0.05. (C). Changes in fluorescence intensity of 2 μM BaP and selected metabolites ( 3OH, 9OH, and BPDE ) within Clone 9 cells treated with 2 μM BaP at different time points (up to 3 h) and in the continuous presence of BaP. Fluorescence intensity scales are adjusted for each metabolite signal in order to better illustrate the changes over time. The solid line in each graph of figure C represents the exponential curve fitting of data that will identify the plateau value for each metabolite measured.
Formation of BaP Metabolite Conjugates
Hydroxylated BaP metabolites form both glucuronide and sulfate conjugates (Zhu et al. 2008). The multiphoton spectral analysis assay was used to investigate formation of these conjugates in Clone 9 cells. Incubation of Clone 9 cells with 2 μM BaP alone or in combination with 500 units of β-glucuronidase for 24 hr resulted in a significant increase in the free 3OH and 9OH metabolites in the latter treatment group (Figure 5A). A significant decrease in BPDE formation was also observed. In a parallel experiment, Clone 9 cells were treated with 2 μM 9OH for 24 h in the presence or absence of 500 units of β-glucuronidase (Figure 5B). In the absence of β-glucuronidase, levels of 9OH were minimal due to rapid intracellular glucuronidation. However, the extracellular presence of glucuronidase resulted in significant increase in 9OH levels.
Figure 5.

Manipulation of BaP metabolite conjugates using inhibitors of glucuronidation and sulfation. (A). Effects of β-glucuronidase on BaP metabolism. Clone 9 cells were treated with 2 μM BaP alone (white bar) or in combination with 500 units of β-glucuronidase (black bar) for 24 h. (B). Effects of β-glucuronidase on 9OH metabolism. Clone 9 cells were treated with 2 μM 9OH alone (white bar) or in combination with 500 units of β-glucuronidase (black bar) for 24 h. Metabolites were identified and quantified by multiphoton spectral analysis as described in the Materials and methods. (C). Effects of triclosan on BaP metabolism. Cells were treated with 2 μM BaP (white bar), 2 μM BaP and 20 μM triclosan (hatched bar), and 2 μM BaP and 40 μM triclosan (black bar). (D). Effects of triclosan on 3OH metabolism. Cells were treated with 2 μM 3OH (white bar), or 2 μM 3OH and 20 μM triclosan for 24 h. Data represent mean fluorescence intensity ± SEM normalized to the corresponding control for each metabolite measured and collected from at least 8 images per treatment. * Indicates significant difference from the corresponding metabolite at p < 0.05.
The role of sulfate conjugation of BaP metabolites was also investigated in Clone 9 cells treated for 24 h with 2 μM BaP alone or in the presence of 20 or 40 μM triclosan, a competitive inhibitor of sulfation and to a lesser extent glucuronidation. After treatment, there were significant increases in both the 3OH and 9OH metabolites in cells treated BaP plus 40 μM triclosan; and this is consistent with decreased formation of their corresponding sulfate and glucuronidated conjugates (Figure 5C). There was also an increase in t7,8 metabolite but this was not accompanied by a corresponding increase in BPDE, an oxidative metabolite of t7,8. Clone 9 cells were also treated with 2 μM 3OH for 24 h in the presence or absence of 20 μM triclosan (Figure 5D) and multiphoton spectral analysis demonstrated that inhibition of 3OH sulfation and glucuronidation resulted in a significant increase in the free 3OH metabolite (see supplemental figure 3S). It is important to note that triclosan treatment alone did not alter the background spectrum of the cells (data not shown).
Analysis of t7,8 and BPDE Metabolites in Clone 9 Cells
t7,8 is a metabolic precursor of BPDE which has been identified as a carcinogenic BaP metabolite that alkylates DNA (Gammon et al. 2004). Formation of t7,8 is dependent on metabolism of BaP into the 7,8 oxide which in turn is hydrolyzed to t7,8; therefore we used real time multiphoton spectral analysis to follow this critical metabolic pathway in Clone 9 cells (Figure 6). Treatment of cells with the sEH inhibitors 1709 and 1728 had no significant effect on BaP metabolites generated within cells (data not shown). However both mEH inhibitors (mEH#16 and mEH#29) significantly decreased metabolism of BaP into t7,8 and BPDE. This was also accompanied by decreased levels of the 3OH, 9OH, Pyr and 3,6BPQ metabolites (Figure 6A and 6B).
Figure 6.

Analysis of t7,8 and BPDE metabolism. Effects of microsomal epoxide hydrolase inhibitors mEH#16 (A) and mEH#29 (B) on BaP metabolism. Clone 9 cells were treated with 2 μM BaP alone or in combination with 20 μM mEH#16 or mEH#29 and metabolites were identified and quantified by multiphoton spectral analysis as described in the Materials and Methods. Data for each metabolite represent fluorescence intensity ± SEM normalized to mean fluorescence intensity of the parent compound collected from at least 8 images per treatment. * indicates significant difference from the corresponding metabolite control at p < 0.05. (C). Analysis of t7,8 metabolism in situ. Clone 9 cells were treated with 2 μM t7,8 reference standard. The formation of BPDE and “pyrene-like metabolites” were determined over a period of 60 min by multiphoton spectral analysis. Note that BPDE formation decreases and pyrene formation increases within 6 min of addtition of t7,8 to the culture. (D). Analysis of BPDE metabolism in situ. Clone 9 cells were treated with 2 μM BPDE reference standard and levels of BPDE and “pyrene-like” metabolites were determined over a period of 60 min by multiphoton spectral analysis.
Kinetic analysis of Clone 9 cells following direct addition of 2 μM t7,8 reference standard resulted in rapid formation of BPDE within the first 6 min, followed by a decrease in BPDE. Multiphoton spectral analysis detected a time-dependent increase in formation of “pyrene-ring” metabolites over the 60 min observation period (Figure 6C). Furthermore, kinetic analysis of Clone 9 cells following direct addition of 2 μM BPDE (Figure 6D) showed a rapid uptake and a slow decline of intracellular BPDE over the 60 min period and this was accompanied by a time-dependent increase in “pyrene-like” metabolites over the total treatment period.
Additional Validation of the Sensitivity of Multiphoton Spectral Analysis
To determine whether groups of closely related metabolites (phase II products and their phase I precursors) that exhibit subtle shift in spectra can be resolved, we evaluated individual spectra of 9OH, 9-S and 9-G and compared them to the spectrum of a mixture of 9OH, 9-S and 9-G. Figure 7 shows that each metabolite in the mixture was identified with a maximum error of < 5% when compared to the corresponding standard. Another confirmation of the utility of multiphoton microscopy for non-destructive, in situ evaluation of BaP and its metabolites within living cells, was also achieved by exposing Clone 9 cells treated with 2 μM BaP for 24 h to a 10 sec UV light (exposure with a 120W mercury lamp). Multiphoton microscopy analysis of these cells demonstrated that this brief UV exposure was sufficient to alter the BaP metabolites present in cells by a photocatalytic process (Figure 8) along with the sensitivity of the method to detect these photocatalytic events. Specifically, 3OH, t7,8, 3,6BPQ metabolites were increased whereas BaP, BPDE, and 3-S were decreased following UV exposure.
Figure 7.
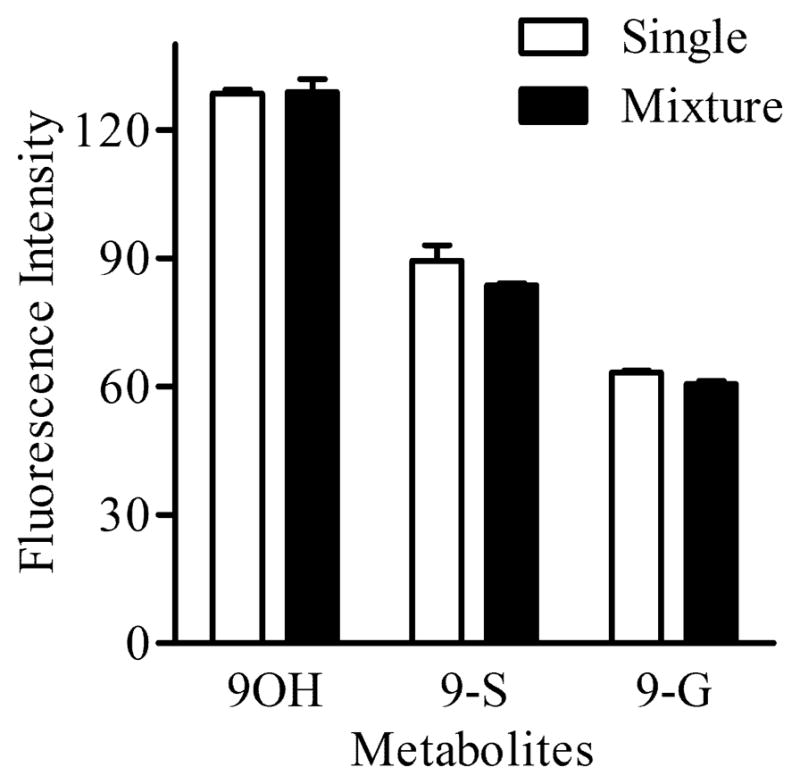
Multiphoton spectral analysis of closely related metabolites. Quantification of metabolites present in a mixture (black bar) of reference standards (2 μM 9OH, 2 μM 9-S and 2 μM 9-G) relative to a similar concentration of each standard alone (white bar) using the unmixing process. Note that each metabolite in the mixture was identified with a maximum error of <5% when compared to the corresponding standard alone.
Figure 8.
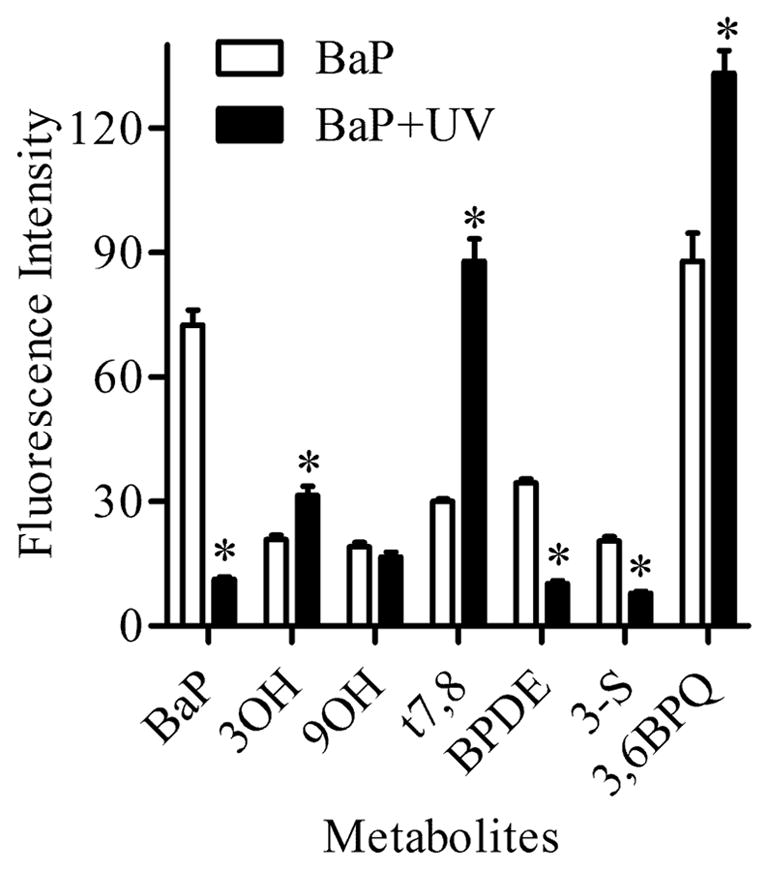
Analysis of photocatalytic oxidation of BaP. BaP and metabolites measured before (white bar) and after (black bar) 10 sec of UV exposure of Clone 9 cells treated with 2 μM BaP for 24 h. Note that UV exposure significantly decreased BaP, BPDE, and 3-S while other metabolites (3OH, t7,8, and 3,6BPQ) increase compared to the corresponding metabolites before UV exposure. * Indicates significant difference from the corresponding metabolite at p < 0.05.
Discussion
Metabolism of BaP is complex and involves biological activation through oxidative metabolism by cytochrome p450s and other enzymes responsible for Phase I reactions. The proposed ultimate carcinogen, benzo[a]pyrene 7,8, diol-9–10-epoxide (BPDE) results from metabolic activation by cytochrome P4501A1 and 1B1 enzymes and hydrolysis by epoxide hydrolase (Thakker et al. 1984). Numerous additional metabolites are also generated including epoxides, phenols, dihydrodiols, quinones, triols, tetrols and diol epoxides (Weeks et al., 1991; Kim et al., 1998) and these metabolic products can affect a wide variety of cellular responses. Many of these metabolites can be conjugated to glucuronic acid, sulfate and glutathione in Phase II reactions to become more water soluble facilitating their excretion (Zhu et al. 2008).
Quantitative analysis of BaP and BaP metabolites and their binding to macromolecules within cells and tissues has previously been performed using high-performance liquid chromatography (HPLC) methods with fluorescence detection (Stampfer et al. 1981); (Miles et al. 1996); (Ramesh et al. 2001) following tissue isolation and extraction. Time course studies of BaP metabolism in extracted tissues have also utilized spectrofluorimetry at excitation and emission wavelengths specific for each metabolite of BaP (Moore and Cohen 1978). However, quantitative analysis of BaP metabolism in real time within viable cells and tissues by fluorescence methods is more complicated due to: 1) the sensitivity of BaP to photocatalytic oxidation and photobleaching which complicate microscopic analysis using conventional fluorescence (UV light source) and confocal (continuous wavelength laser) microscopes, and 2) the overlap in excitation and/or emission spectra of metabolites. These two disadvantages limit opportunities to simultaneously identify all or many of the major metabolites. Multiphoton laser scanning microscopy has been used to identify the tissue distribution of BaP and some of its metabolites in medaka embryos and post-hatch larvae by taking advantage of differences in excitation spectral properties of the parent compound and metabolites (Hornung et al. 2004); (Hornung et al. 2007). An excitation below 830 nm and emission at 450/80 nm was used to identify the presence of the parent compound, whereas excitation at 840 nm or 860 nm indicated the presence of conjugated metabolites (3-G, 3-S) and excitation at 880 nm indicated the presence of 3OH only.
Recently, we described a different approach to investigate BaP uptake, partitioning and metabolism that involved multiphoton microscopy spectral analysis using a single excitation and multiple emission wavelength bands (Barhoumi et al. 2009). The approach identified 4 major emission bands that are due to quinones, BPDE, monohydroxybenzo[a]pyrene, along with the parent compound. Although this approach was an improvement when compared to available methods in live cells, it was not able to separate closely related metabolites (e.g., positional isomers of monohydroxybenzo[a]pyrene including 3OH and 9OH).
In the current study, we extended the previous single cell analysis study of BaP uptake and partitioning into live cells for the purpose of identifying additional reactive metabolites using a novel method. This method required generation of spectra for standards and the use of these reference spectra to identify metabolites present in cells following treatment with BaP for fixed intervals. The method uses the advanced linear unmixing process, originally designed for separation of strongly overlapping fluorescence signals, and is mainly considered as an important tool for colocalization and fluorescence resonance energy transfer studies (Zimmermann 2005). This method appears to be well suited to analyze mixed fluorescence contributions from different metabolites to individual pixels as in the case of BaP. Our purpose was to apply this unmixing process to the spectral information obtained with parallel detection channels from BaP-treated Clone 9 cells. Once the signal from multiple parallel channels is detected and the reference emission spectra are known, the contribution Ai (see Equation 1) of each metabolite present in the cell can be determined by calculating the contribution value that most closely matches the detected signal in the channels using the least square fitting approach as described (Equation 2).
Before applying the unmixing process, CYP1A1-dependent EROD activity was evaluated in BaP treated cells as well as in cells loaded with each standard used (BaP, 3OH, 9OH, t7,8, BPDE, Pyr, 3-S, 9-S, 9-G, and 3,6 BPQ) following a 3 h treatment. The reasoning behind this analysis was to identify the p450 activity and therefore to identify whether the metabolites generated by BaP treatment are due to CYP1A1 activity induced by BaP and/or by the BaP metabolites. The results indicated the ability of Clone 9 cells to metabolize BaP and that formation of most of BaP metabolites are due to CYP1A1 induced primarily by BaP (data not shown). Because of this metabolism, the BaP spectra (Figure 1B) generated in Clone 9 cells following BaP treatment and using multiphoton spectral analysis, with an excitation wavelength of 740 nm and emission wavelength between 399 and 600 nm, is logically assumed to be the sum of weighted spectra of different metabolites that can be identified. The approach taken to identify these metabolites depends on: 1) generating a reference spectrum for each standard (Figure 2), 2) predicting what metabolites are generated according to the possible chemical reactions (Ramesh et al. 2004), and 3) using the reference spectra of standards anticipated as metabolites generated by BaP in the unmixing process in order to quantify them (Figure 3). Based on this strategy, we were able to determine the presence and the relative amount of the following metabolites: BaP parent compound, 3OH, 9OH, t7,8, BPDE, 3-S and 3,6 BPQ in Clone 9 cells treated with BaP (2 μM or 10 μM) for 3 hr (Figure 4A). The same approach was also efficient in determining changes in some of these metabolites over time (Figure 4B and 4C).
Another validation for measuring BaP metabolites using this new technique involved use of β-glucuronidase to catalyze hydrolysis of glucuronidated metabolites of BaP (e.g., 9-G) (Kinoshita and Gelboin 1978). Exposure of Clone 9 cells to 9OH for 24 h in the presence of β-glucuronidase increased levels of 9OH (Figure 5B). Moreover, cells treated with BaP in the presence of β-glucuronidase exhibited a significant increase in 3OH and 9OH as expected (Figure 5A). This is in agreement with the fact that most of the hydroxylated metabolites of BaP including phenols, dihydrodiols and epoxides form glucuronide conjugates whereas the quinones do not exhibit any glucuronidated conjugates (Nemoto and Gelboin 1976).
Triclosan readily undergoes sulfation and glucuronidation and can be used as a competitive inhibitor of sulfation and glucuronidation of BaP metabolites (Wang et al. 2004). The inhibitory effects of triclosan were investigated in cells treated with one metabolite standard (e.g., 3OH) and triclosan (Figure 5D) and by measuring the metabolites generated using the same unmixing process. The data indicate that Clone 9 cells accumulated more 3OH and less 3-S when treated with 3OH and triclosan simultaneously compared to 3OH alone. This is consistent with the mechanism of action of triclosan and supports the validity of the unmixing method. In addition, analysis of the effects of triclosan on BaP metabolism (Figure 5C) similarly identified increased 3OH, 9OH, t7,8, and 3,6 BPQ, each of which is known to generate glucuronidated metabolites.
The most critical evaluation for the accuracy of the unmixing method was performed using selective inhibitors of soluble and microsomal epoxide hydrolases (sEH and mEH) as tools to evaluate the generation of the ultimate carcinogen BPDE. It is known that epoxide hydrolases play an important role in enhancing or reducing toxicity depending on the xenobiotics under study. In the case of BaP, diol formation leads to the generation of the stable epoxide BPDE, through its precursor t7,8. Due to the lipophilic nature of BaP and t7,8, and the sub-cellular partitioning of BaP into cellular membranes, the mEH were hypothesized to be more potent metabolic inhibitors than sEH since it is well known that sEH do not hydrate epoxide on cyclic systems (Morisseau and Hammock 2005). This hypothesis was verified when the sEHs had little effect on the metabolites generated while mEH #16 and #29 significantly reduced the amount of t7,8 as well as BPDE generated in Clone cells following 18 h of removal of BaP (Figure 6A and 6B). These inhibitors also significantly reduced other metabolites (3OH, 9OH and 3,6 BPQ) as well. While the basis for reduction of the latter metabolites has not yet been evaluated, several possibilities exist. The mEH inhibitors may affect MFO induction or activity, as they were added before the BaP treatment which resulted in fewer epoxides and therefore fewer phenols. Another possibility is that inhibition of EHs may be compensated for by an increase in the metabolizing enzymes such as GST (Morisseau et al., 2008) which would result in more glutathione conjugates. The observed reduction in quinones is a consequence of reduced phenol generation (Figure 6A and 6B). The role of the mEH inhibitors in other metabolic pathways is currently being investigated.
The metabolism of BaP into t7,8 and BPDE is critical for the carcinogenic effects of BaP (Ramesh et al. 2004) and further metabolism of BPDE to tetrol or DNA adducts results in formation of pyrene derivatives due to loss of aromatization of the D ring of BaP. To explore detection of the ultimate carcinogen of BaP, BPDE and its precursor t7,8, the t7,8 standard was added to the cells prior to multiphoton microscopy spectral analysis. Recording changes in t7,8 fluorescent signal as well as formation of the BPDE and a pyrene-like signal (resulting from BPDE-adduct formation) confirmed the generation of BPDE within 7 min of addition of t7,8 and that BPDE decreased within 7–15 min while the Pyr signal increased. These observations support the hypothesis that BPDE-adducts form within minutes of addition of t7,8 (Figure 6C). Furthermore, the BPDE adduct formation was also verified by adding the BPDE standard directly to cells. In this case, BPDE decreased within 10 min and a pyrene-like signal was detected within the same time frame (Figure 6D). These results demonstrate the potential for real time determination of BaP metabolism in cells using multiphoton spectral analysis and we are currently developing this methodology for investigating the fate of BaP and selected carcinogenic PAHs in other relevant biological systems.
Although this approach was based on solid assumptions (chemical reactions and mathematical solution with singular value decomposition), several additional controls support these findings. A mixture of closely related standards (e.g. 9OH, 9-S and 9-G) proved that data obtained with the spectral unmixing technique is reliable and accurate within < 5% maximum error (Figure 7).
PAHs are sensitive to photocatalytic oxidation and photobleaching which complicate microscopic analysis using conventional fluorescence (UV light source) and confocal (continuous wavelength laser) microscopes. Because of this, multiphoton analysis (pulsed femtosecond infrared laser) of BaP metabolism when confined to an optimal optical window, is an improved approach to prevent BaP from reacting with oxygen to form endoperoxides as intermediates and quinones as stable photoproducts of irradiation (Kot-Wasik 2004).
In summary, the two-photon microscopy imaging (740 nm) surpasses the conventional fluorescence imaging techniques (Figure 8) and the unmixing process is a mathematical technique that seems applicable to analysis of BaP metabolites in living cells especially for analysis of changes of the ultimate carcinogen BPDE in living cells where other methods are lacking. This technique in conjunction with assessment of alterations of cellular homeostasis will make it possible to provide important insights into the cellular and molecular mechanisms of injury. Future studies will concentrate on using this technique not only to identify specific metabolites in cells and their relative changes under different experimental conditions but also to calibrate the absolute concentrations of each metabolite.
Supplementary Material
Acknowledgments
Confocal and multiphoton microscopy was performed in the Texas A&M University College of Veterinary Medicine & Biomedical Sciences Image Analysis Laboratory, supported by NIH-NCRR (1S10RR22532-01), and NIH grants P30-ES09106, P42-ES04917, T32 ES07273 and R01 ES002710. BDH is a George and Judy Marcus Senior Fellow of the American Asthma Foundation. This research was performed in part using compounds provided by the National Cancer Institute’s Chemical Carcinogen Reference Standards Repository operated under contract by Midwest Research Institute, No. N02-CB-07008.
Footnotes
Conflict of Interest: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barhoumi R, et al. Effects of benzo-a-pyrene on oxytocin-induced Ca2+ oscillations in myometrial cells. Toxicol Lett. 2006;165(2):133–141. doi: 10.1016/j.toxlet.2006.02.005. [DOI] [PubMed] [Google Scholar]
- Barhoumi R, et al. Multiphoton spectral analysis of benzo[a]pyrene uptake and metabolism in breast epithelial cell lines. J Toxicol Sci. 2009;34(1):13–25. doi: 10.2131/jts.34.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barhoumi R, et al. Characterization of calcium oscillations in normal and benzo[a]pyrene-treated clone 9 cells. Toxicol Sci. 2002;68(2):444–450. doi: 10.1093/toxsci/68.2.444. [DOI] [PubMed] [Google Scholar]
- Barhoumi R, et al. Analysis of benzo[a]pyrene partitioning and cellular homeostasis in a rat liver cell line. Toxicol Sci. 2000;53(2):264–270. doi: 10.1093/toxsci/53.2.264. [DOI] [PubMed] [Google Scholar]
- Bolton JL, et al. Role of quinones in toxicology. Chem Res Toxicol. 2000;13(3):135–160. doi: 10.1021/tx9902082. [DOI] [PubMed] [Google Scholar]
- Boysen G, Hecht SS. Analysis of DNA and protein adducts of benzo[a]pyrene in human tissues using structure-specific methods. Mutat Res. 2003;543(1):17–30. doi: 10.1016/s1383-5742(02)00068-6. [DOI] [PubMed] [Google Scholar]
- Dabestani RaINI. A Compilation of physical, spectroscopic and photophysical properties of polycyclic aromatic hydrocarbons. Photochem Photobiol. 1999;70:10–34. [Google Scholar]
- Donato MT, et al. A microassay for measuring cytochrome P450IA1 and P450IIB1 activities in intact human and rat hepatocytes cultured on 96-well plates. Anal Biochem. 1993;213(1):29–33. doi: 10.1006/abio.1993.1381. [DOI] [PubMed] [Google Scholar]
- Gammon MD, et al. Polycyclic aromatic hydrocarbon-DNA adducts and breast cancer: a pooled analysis. Arch Environ Health. 2004;59(12):640–649. doi: 10.1080/00039890409602948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gmur DJ, Varanasi U. Characterization of benzo[a]pyrene metabolites isolated from muscle, liver, and bile of a juvenile flatfish. Carcinogenesis. 1982;3(12):1397–1403. doi: 10.1093/carcin/3.12.1397. [DOI] [PubMed] [Google Scholar]
- Goryacheva IY, et al. Preconcentration and fluorimetric determination of polycyclic aromatic hydrocarbons based on the acid-induced cloud-point extraction with sodium dodecylsulfate. Anal Bioanal Chem. 2005;382(6):1413–1418. doi: 10.1007/s00216-005-3287-0. [DOI] [PubMed] [Google Scholar]
- Hecht SS. Cigarette smoking and lung cancer: chemical mechanisms and approaches to prevention. Lancet Oncol. 2002;3(8):461–469. doi: 10.1016/s1470-2045(02)00815-x. [DOI] [PubMed] [Google Scholar]
- Hornung MW, et al. Tissue distribution and metabolism of benzo[a]pyrene in embryonic and larval medaka (Oryzias latipes) Toxicol Sci. 2007;100(2):393–405. doi: 10.1093/toxsci/kfm231. [DOI] [PubMed] [Google Scholar]
- Hornung MW, et al. Use of multi-photon laser-scanning microscopy to describe the distribution of xenobiotic chemicals in fish early life stages. Aquat Toxicol. 2004;67(1):1–11. doi: 10.1016/j.aquatox.2003.11.009. [DOI] [PubMed] [Google Scholar]
- Hwang SH, et al. Orally bioavailable potent soluble epoxide hydrolase inhibitors. J Med Chem. 2007;50(16):3825–3840. doi: 10.1021/jm070270t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita N, Gelboin HV. beta-Glucuronidase catalyzed hydrolysis of benzo(a)pyrene-3-glucuronide and binding to DNA. Science. 1978;199(4326):307–309. doi: 10.1126/science.619459. [DOI] [PubMed] [Google Scholar]
- Kot-Wasik A, Dabrowska D, Namiesnik J. Photodegradation and biodegradation study of benzo[a]pyrene in different liquid media”. J Photochem Photobiol A. 2004;168:109–115. [Google Scholar]
- Lopp A, et al. Fluorescence method for the measurement of penetration and metabolism of carcinogens in mouse skin. Cancer Biochem Biophys. 1986;8(3):185–191. [PubMed] [Google Scholar]
- Miles PR, et al. Pulmonary microsomal metabolism of benzo[a]pyrene following exposure of rats to silica. J Toxicol Environ Health. 1996;48(5):501–514. doi: 10.1080/009841096161203. [DOI] [PubMed] [Google Scholar]
- Miller AG, Whitlock JP., Jr Heterogeneity in the rate of benzo[a]pyrene metabolism in single cells: quantitation using flow cytometry. Mol Cell Biol. 1982;2(6):625–632. doi: 10.1128/mcb.2.6.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore BP, Cohen GM. Metabolism of benzo(a)pyrene and its major metabolites to ethyl acetate-soluble and water-soluble metabolites by cultured rodent trachea. Cancer Res. 1978;38(9):3066–3075. [PubMed] [Google Scholar]
- Moore M, et al. Benzo[a]pyrene-resistant MCF-7 human breast cancer cells. A unique aryl hydrocarbon-nonresponsive clone. J Biol Chem. 1994;269(16):11751–11759. [PubMed] [Google Scholar]
- Morisseau C, Hammock BD. Epoxide hydrolases: mechanisms, inhibitor designs, and biological roles. Annu Rev Pharmacol Toxicol. 2005;45:311–333. doi: 10.1146/annurev.pharmtox.45.120403.095920. [DOI] [PubMed] [Google Scholar]
- Morisseau C, et al. Development of metabolically stable inhibitors of Mammalian microsomal epoxide hydrolase. Chem Res Toxicol. 2008;21(4):951–957. doi: 10.1021/tx700446u. [DOI] [PubMed] [Google Scholar]
- Nemoto N, Gelboin HV. Enzymatic conjugation of benzo (a) pyrene oxides, phenols and dihydrodiols with UDP-glucuronic acid. Biochem Pharmacol. 1976;25(10):1221–1226. doi: 10.1016/0006-2952(76)90373-7. [DOI] [PubMed] [Google Scholar]
- Phillips DH. The Carcinogenic Effects of Polycyclic Aromatic Hydrocarbons. Imperial College Press; London: 2005. Macromolecular adducts as biomarkers of human exposure to polycyclic aromatic hydrocarbons; pp. 137–169. [Google Scholar]
- Plant AL, et al. Cellular uptake and intracellular localization of benzo(a)pyrene by digital fluorescence imaging microscopy. J Cell Biol. 1985;100(4):1295–1308. doi: 10.1083/jcb.100.4.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramesh A, et al. Comparative metabolism, bioavailability, and toxicokinetics of benzo[a]pyrene in rats after acute oral, inhalation, and intravenous administration. Polycyclic Aromat Compd. 2002;22:969–980. [Google Scholar]
- Ramesh A, et al. Metabolism, bioavailability, and toxicokinetics of benzo(alpha)pyrene in F-344 rats following oral administration. Exp Toxicol Pathol. 2001;53(4):275–290. doi: 10.1078/0940-2993-00192. [DOI] [PubMed] [Google Scholar]
- Ramesh A, et al. Modulation of adult rat benzo(a)pyrene (BaP) metabolism and DNA adduct formation by neonatal diethylstilbestrol (DES) exposure. Exp Toxicol Pathol. 2004;56(3):129–138. doi: 10.1016/j.etp.2004.08.005. [DOI] [PubMed] [Google Scholar]
- Ramesh A, et al. Bioavailability and risk assessment of orally ingested polycyclic aromatic hydrocarbons. Int J Toxicol. 2004;23(5):301–333. doi: 10.1080/10915810490517063. [DOI] [PubMed] [Google Scholar]
- Raspotnig G, et al. Colorimetric determination of cell numbers by Janus green staining. Anal Biochem. 1999;275(1):74–83. doi: 10.1006/abio.1999.4309. [DOI] [PubMed] [Google Scholar]
- Rose TE, et al. 1-Aryl-3-(1-acylpiperidin-4-yl)urea Inhibitors of Human and Murine Soluble Epoxide Hydrolase: Structure-Activity Relationships, Pharmacokinetics, and Reduction of Inflammatory Pain. J Med Chem. 2010 doi: 10.1021/jm100691c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samanta SK, et al. Polycyclic aromatic hydrocarbons: environmental pollution and bioremediation. Trends Biotechnol. 2002;20(6):243–248. doi: 10.1016/s0167-7799(02)01943-1. [DOI] [PubMed] [Google Scholar]
- Shimada T, et al. Arylhydrocarbon receptor-dependent induction of liver and lung cytochromes P450 1A1, 1A2, and 1B1 by polycyclic aromatic hydrocarbons and polychlorinated biphenyls in genetically engineered C57BL/6J mice. Carcinogenesis. 2002;23(7):1199–1207. doi: 10.1093/carcin/23.7.1199. [DOI] [PubMed] [Google Scholar]
- Stampfer MR, et al. Metabolism of benzo[a]pyrene by human mammary epithelial cells: toxicity and DNA adduct formation. Proc Natl Acad Sci U S A. 1981;78(10):6251–6255. doi: 10.1073/pnas.78.10.6251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakker DR, et al. Effects of a 6-fluoro substituent on the metabolism of benzo(a)pyrene 7,8-dihydrodiol to bay-region diol epoxides by rat liver enzymes. J Biol Chem. 1984;259(18):11249–11256. [PubMed] [Google Scholar]
- Wang LQ, et al. Triclosan as a substrate and inhibitor of 3’-phosphoadenosine 5’-phosphosulfate-sulfotransferase and UDP-glucuronosyl transferase in human liver fractions. Drug Metab Dispos. 2004;32(10):1162–1169. doi: 10.1124/dmd.104.000273. [DOI] [PubMed] [Google Scholar]
- Weston A, et al. Detection of metabolites of polycyclic aromatic hydrocarbons in human urine. Carcinogenesis. 1993;14(5):1053–1055. doi: 10.1093/carcin/14.5.1053. [DOI] [PubMed] [Google Scholar]
- Weyand EH, Bevan DR. Benzo(a)pyrene disposition and metabolism in rats following intratracheal instillation. Cancer Res. 1986;46(11):5655–5661. [PubMed] [Google Scholar]
- Wild E, et al. Use of two-photon excitation microscopy and autofluorescence for visualizing the fate and behavior of semivolatile organic chemicals within living vegetation. Environ Toxicol Chem. 2007;26(12):2486–2493. doi: 10.1897/06-279.1. [DOI] [PubMed] [Google Scholar]
- Xu XB, Jin ZL. High-performance liquid chromatographic studies of environmental carcinogens in China. J Chromatogr. 1984;317:545–555. doi: 10.1016/s0021-9673(01)91695-4. [DOI] [PubMed] [Google Scholar]
- Zhu S, et al. Simultaneous determination of benzo[a]pyrene and eight of its metabolites in Fundulus heteroclitus bile using ultra-performance liquid chromatography with mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2008;863(1):141–149. doi: 10.1016/j.jchromb.2008.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann T. Spectral imaging and linear unmixing in light microscopy. Adv Biochem Eng Biotechnol. 2005;95:245–265. doi: 10.1007/b102216. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.