

Abstract
Blind sterile 2 (bs2) is a spontaneous autosomal recessive mouse mutation exhibiting cataracts and male sterility. Detailed clinical and histological evaluation revealed that bs2 mice have cataracts resulting from severely disrupted lens fiber cells. Analysis of bs2 testes revealed the absence of mature sperm and the presence of large multinucleate cells within the lumens of seminiferous tubules. Linkage analysis mapped the bs2 locus to mouse chromosome 2, approximately 45cM distal from the centromere. Fine mapping established a 3.1Mb bs2 critical region containing 19 candidate genes. Sequence analysis of alkylglycerone-phosphate synthase (Agps), a gene within the bs2 critical region, revealed a G to A substitution at the +5 position of intron 14. This mutation results in two abundantly expressed aberrantly spliced Agps transcripts: AgpsΔexon14 lacking exon 14 or AgpsexonΔ13–14 lacking both exons 13 and 14 as well as full-length Agps transcript. Agps is a peroxisomal enzyme which catalyzes the formation of the ether bond during the synthesis of ether lipids. Both aberrantly spliced AgpsΔexon14 and AgpsexonΔ13–14 transcripts led to frame shift, premature stop and putative proteins lacking the enzymatic FAD domain. We present evidence that bs2 mice have significantly decreased levels of ether lipids. Human mutations in Agps result in rhizomelic chondrodysplasia punctata type 3 (RCDP3), a disease for which bs2 is the only genetic model. Thus, bs2 is a hypomorphic mutation in Agps, and represents a useful model for investigation of the tissue specificity of ether lipid requirements which will be particularly valuable for elucidating the mechanism of disease phenotypes resulting from ether lipid depletion.
Keywords: Agps, ether lipids, peroxisome, RCDP3, cataract, mouse model
INTRODUCTION
Peroxisomes are ER-derived organelles present in most mammalian cells [1–6]. They are simple in structure and are comprised of a single membrane and about 50 proteins. All peroxisomal proteins are encoded by nuclear genes, synthesized on free ribosomes and are postranslationally imported. The molecular mechanisms of peroxisomal protein import have been recently reviewed [7], indicating a complex mechanism capable of transporting folded and oligomeric proteins across the membrane [8, 9]. The vast majority of peroxisomal proteins contain peroxisome targeting signal 1 (PTS1)[6], a C-terminal Ser-Lys-Leu consensus sequence [10] that binds to the PTS1 receptor which mediates import into the peroxisome [11]. However, a small subset of peroxisomal proteins are imported via an alternative mechanism involving the N-terminal Arg/Lys-Leu-X5-Gln/His-Leu peroxisomal targeting signal 2 (PTS2) [6] and corresponding PTS2 receptor [12–15]. Peroxisomal proteins play a role in peroxisomal biogenesis and at least eight independent metabolic pathways including α- and β-oxidation of fatty acids, catalytic decomposition of hydrogen peroxide, and ether lipid synthesis [2, 6, 16]. The essential roles of peroxisomal function in mammals are reflected by a number of hereditary peroxisomal disorders in humans. Clinical phenotypes in these disorders may result from mutations in genes essential for peroxisomal formation, fusion, protein import, or functional defects of individual peroxisomal proteins.
Peroxisomal disorders are typically classified into two groups: peroxisomal biogenesis disorders (PBD), and isolated peroxisomal enzyme deficiencies [17–19]. The PBD group includes Zellweger spectrum disorders in which affected patients exhibit variable clinical phenotypes, most commonly including craniofacial dysmorphism, profound neurological abnormalities, severe psychomotor retardation, hypotonia, neonatal seizures, glaucoma, retinal degeneration and impaired hearing [17–19]. Mutations in at least 11 peroxisomal genes (PEX1,2,3,5,6,10,12,13,14,16,19, and 26) have been identified as being associated with the Zellweger spectrum disorders[19, 20]. These genes play an essential role in peroxisome biogenesis, and/or peroxisomal PTS1 protein import [17–19]. In addition to Zellweger spectrum disorders, rhizomelic chondrodysplasia punctata type 1 (RCDP1) is also classified within the PBD group of disorders. Mutations in PEX7, the gene encoding the PTS2 receptor, cause RCDP1 [12–14]. Clinical phenotypes of RCDP1 patients differ greatly from patients affected with Zellweger spectrum disorders. RCDP1 patients exhibit severe shortening of rhizomelic bones, bilateral cataracts, severe growth and motor delays, dysmorphic facial characteristics, stippled epiphyses, and vertebral coronal cleft [17, 21, 22]. Unlike RCDP1, rhizomelic chondrodysplasia punctata type 2 (RCDP2) and rhizomelic chondrodysplasia punctata type 3 (RCDP3) are classified as isolated peroxisomal enzyme deficiency disorders, resulting from mutations in glyceronephosphate O-acyltransferase (GNPAT) and alkylglycerone phosphate synthase (AGPS) respectively [23, 24]. Interestingly, while only a single enzyme is deficient, RCDP2 and RCDP3 patients exhibit clinical phenotypes identical to those observed in RCDP1 patients. AGPS and GNPAT are peroxisomal enzymes required for the synthesis of plasmalogens, a class of ether lipid species containing a vinyl ether bond at the sn-1 position of the glycerol backbone [24]. AGPS is one of the few peroxisomal proteins that is imported via the PTS2 signal/PEX7 receptor mechanism [2]. Although in RDCP1 all PTS2-mediated protein import is compromised, it has been shown that the RCDP1 phenotype is primarily determined by a loss of AGPS function [25]. Thus, disruption of the plasmalogen synthesis pathways has been established as the primary defect associated with clinical outcomes for all three forms of RCDP.
To better understand the molecular etiology of RCDP disorders, as well as the role of plasmalogens in-vivo, mouse models carrying null alleles of Pex7 and Gnpat were previously generated as models for RCDP1 and RCDP2 [26, 27]. Both Pex7−/− and Gnpat−/− mice exhibit cataracts and male sterility phenotypes [26, 27]. To our knowledge, Agps null mice have not yet been described. In this study we show that blind sterile 2 (bs2), a spontaneous mouse mutation identified to exhibit cataracts and male sterility [28], is a hypomorphic mutation in Agps resulting in severe plasmalogen deficiency. We also show that bs2 is not allelic with another spontaneous mouse mutation called blind sterile (bs) which like bs2 exhibits phenotypes of cataracts and male sterility and maps to chromosome 2 [29, 30]. As such, bs2 represents the first genetic model of RCDP3, further providing an opportunity for evaluation of the role of Agps in-vivo. In addition, the availability of the bs2 mouse mutant allows for comparative analysis between mouse and human phenotypes associated with all forms of RCDP.
MATERIALS AND METHODS
Mice, clinical evaluation, and histology
CAST/EiJ, C57BL/6, bs and bs2 mice were all obtained from The Jackson Laboratory (Bar Harbor, ME). All mice showed normal life expectancy and breeding patterns with the exception of bs and bs2 homozygote males which, consistent with previous results, were unable to produce litters [28, 30]. Mouse eyes were examined with a Topcon SL-D8Z slit lamp biomicroscope with a Nikon SLR-based Photo Slit Lamp imaging system following mydriasis with 1% Atropine Sulfate (Bausch & Lomb). For WT and bs2 histology, tissues were fixed in Zinc-formalin, or Davidson’s solution, embedded in paraffin and sectioned to 4 microns thickness as previously described [31]. Following H&E staining, sections were mounted and photographed with a Nikon DS-Fi1 camera on a Nikon Eclipse 80i microscope.
Linkage Analysis
The bs2 locus has been maintained on the congenic C57BL/6 background. For linkage studies, given that bs2/bs2 male mice are sterile, bs2/bs2 female mice were outcrossed to CAST/EiJ; the resulting F1 progeny were subsequently intercrossed to generate 262 F2 progeny. At four weeks of age F2 progeny were clinically evaluated for the presence of cataract as described above. F2 progeny were euthanized and tissues were collected. Genomic DNA was purified from collected tissues and initially genotyped with D2Mit1, D2Mit465, D2Mit64, D2Mit7, D2Mit205, D2Mit247, D2Mit389, D2Mit459, D2Mit408, D2Mit260, D2Mit51, D2Mit457 as previously described [32]. Subsequent fine mapping analysis was performed on F2 progeny utilizing D2Mit327, D2Mit245, D2Mit37, D2Mit219, D2Mit435, D2Mit93, and D2Mit183 as previously described [32]. Microsatellite alleles were scored following electrophoresis on a 2.5% agarose gel and EtBr staining. Linkage data was analyzed with MapManager QTX (http://www.mapmanager.org/mmQTX.html).
Genomic and cDNA sequence analysis
For sequence evaluation of exons and intron/exon junctions, primers (Table 1) were designed to anneal in introns about 50 bp from intron/exon junctions as previously described [32]. For cDNA analysis RNA was isolated from mouse tissues or tissue cultures, reverse transcribed, and PCR amplified using primers in Table 1. All generated PCR products were electrophoresed, gel-purified and sequenced as previously described [31]. Comparative sequence analysis was performed using DNAStar software (Madison, WI). For semi-quantitative analysis of Agps transcript levels, RT-PCR products were generated while in the exponential phase of PCR amplification using Gapdh as an internal control (Table 1). PCR band intensities were quantified using ImageJ software (http://rsbweb.nih.gov/ij/) and are expressed relative to Gapdh. The results represent at least three independent experiments performed in triplicates. Comparison between WT and bs2 groups was analyzed by the Student’s t-test and the data is expressed as mean ± SEM. A value of p<0.05 was designated as being statistically significant. The IVS 14+5G>A substitution in Agps identified as being responsible for the bs2 phenotype introduces an AluI restriction site which allowed us to develop a genotyping assay. Primers were designed to amplify a 496 bp Agps genomic fragment that spans exon 14 and a portion of intron 14. The generated PCR products were digested with AluI (NEB) following the manufacturer’s protocol, separated on a 1.5% agarose gel, and scored. Following AluI digestion, the 496 bp PCR product containing the WT Agps IVS14+5G allele results in 3 fragments: 394 bp, 68 bp and 34 bp; the PCR product containing the bs2 Agps IVS14+5A results in 4 fragments: 248 bp, 146 bp, 68 bp and 34 bp.
Mass spectrometry
Mouse brains from age-matched pairs (ranging from 4–10 weeks) of bs2 and WT siblings were collected and homogenized in a Dounce homogenizer in 1.0 ml of PBS. Lipids were extracted with 1:1:0.5 chloroform: methanol: water. The combined organic phases were washed once with 0.5 ml of KCl (1 M) followed by 0.5 ml of water, dried at 40°C under N2 gas, and stored at −80°C. For phospholipid profiling, an electrospray ionization-tandem mass spectrometry approach was used, as previously described [33].
Cell culture, western blot, and immunocytochemistry analysis
Mouse embryonic fibroblasts (mEFs) were isolated from E13.5 mouse embryos and maintained as previously described [31]. For western blot analysis, mEFs were lysed with 0.5 ml RIPA buffer (Sigma) containing protease inhibitor cocktail (Sigma). Cell lysates were boiled for 5 min in Laemmli buffer containing β-ME, and separated on a 10% SDS-PAGE gel (all from Bio-Rad). Proteins were transferred at 100V to a PVDF membrane and blocked in 10% milk in 1xPBST for 1 hour. The blots were incubated overnight in 1x PBST containing either 1:2500 rabbit polyclonal catalase (Abcam) or 1:500 rabbit polyclonal Pmp70 (Abcam) primary antibodies. 1:5000 HRP-conjugated β-actin primary antibody (Abcam) was added to each primary incubation as a loading control. After three 10 minute washes in 1x PBST, 1:5000 Peroxidase-conjugated AffiniPure Donkey Anti-Rabbit IgG (H+L) (Jackson ImmunoResearch) was incubated in 1x PBST with the blot for 1 hour at 4°C. The detection was performed using the ECL Western Blot Analysis System (Amersham) following the manufacturer’s instructions. In immunocytochemical studies, mEFs were grown on coverslips until reaching 50–70% confluency. The cells were fixed for 10 min in 4% PFA, washed 3x for 5 min with 1x PBS, and incubated overnight at 4°C in 1x PBST containing 1:1000 rabbit polyclonal catalase or 1:250 rabbit polyclonal PMP70 primary antibody (both Abcam). Cells were again washed 3x for 5 min with 1xPBS and incubated with 1:1000 Alexa 488-conjugated Goat-anti-rabbit secondary (Invitrogen) for 1hr at room temperature. Slides were mounted using Vectashield w/DAPI (Vector Labs), and imaged using a Nikon DS-Fi1 camera on a Nikon Eclipse 80i microscope.
Complementation studies
To determine if bs and bs2 are allelic loci, bs/bs females were outcrossed to bs2/+ males. 34 progeny from 6 litters were generated. Progeny were clinically evaluated for cataracts between 6 and 8 weeks of age as described above. For genotyping, 1 mm tail clippings were collected and boiled in 300 μl of 50 mM NaOH for 15 min, neutralized with 26 μl of 1M Tris pH8.0, and spun at 12,000g for 6 min. 2ul of the supernatant was used for PCR genotyping of the bs2 allele as described above.
RESULTS
bs2 phenotypes
Analysis with slit-lamp biomicroscopy, following mydriasis, confirmed the previously reported bs2 cataract phenotype (Figure 1B) [28]. Histological analysis of bs2 eyes identified smaller globes and severely disrupted lenses (Figure 1D). Although lens epithelial cells at the anterior of the lens appeared normal, fiber cells within the bow region of the lens were smaller in size and were detached from the epithelium (Figure 1F). Bladder cells and liquefied lens material were present within the cortex; morgagnian globules were noted at the posterior poles (Figure 1F). Except for the lens, no ocular structural defects were observed in bs2 histological sections. Consistent with the initial report [28], breeding of bs2 homozygote males did not produce any litters. Evaluation of bs2 testes revealed that the testes are smaller in size when compared to WT controls (Figure 2A). Histological analysis in bs2 sections identified severely disrupted seminiferous tubules (Figure 2D) and the absence of mature spermatozoa and elongating spermatids (Figure 2E). In addition, multinucleate cells were present within the lumen of the bs2 seminiferous tubules (Figure 2E).
Figure 1.
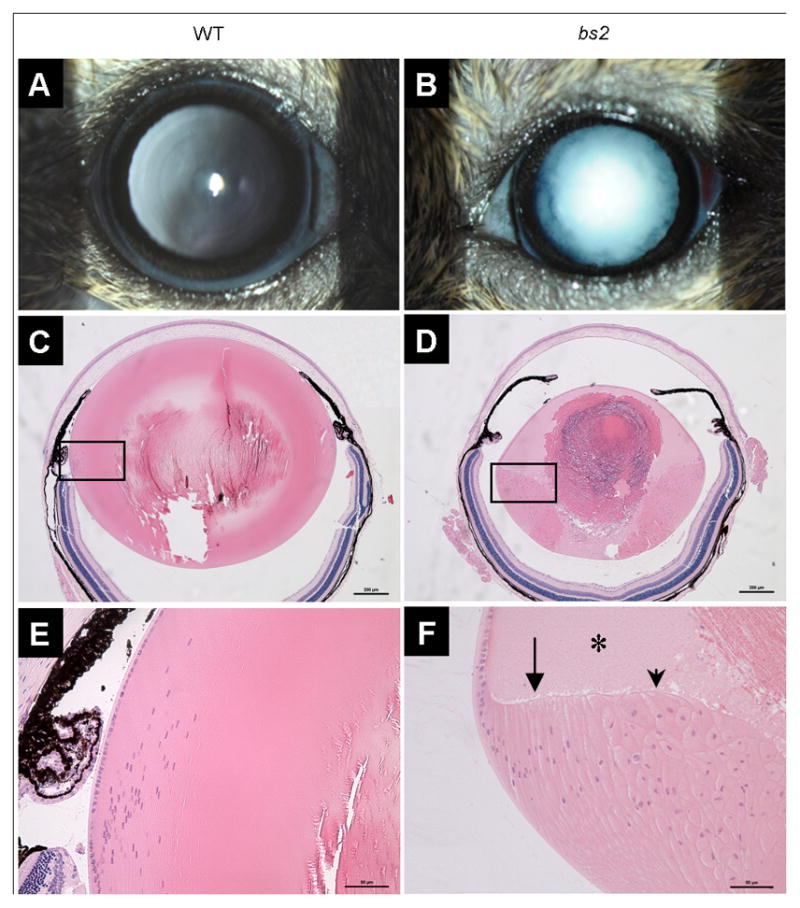
bs2 eye phenotype. Adult bs2 mice (B) exhibit nuclear cataracts whereas WT littermate controls have transparent lenses (A). Examination of bs2 sections, 20x magnification, P28 (D), reveals micropthalmia and a smaller lens as well as a large nuclear cataract compared to the normal WT control (C) (scale bar: 200μm). Boxes indicate regions of 40x magnification in E, F (scale bar: 50μm). bs2 mice exhibit improper differentiation of lens epithelial cells to fiber cells (F) including vacuolation, morgagnian globules (asterisk), bladder cells (arrowhead), and detachment of the apical-apical junctions of epithelial and fiber cells (arrow).
Figure 2.
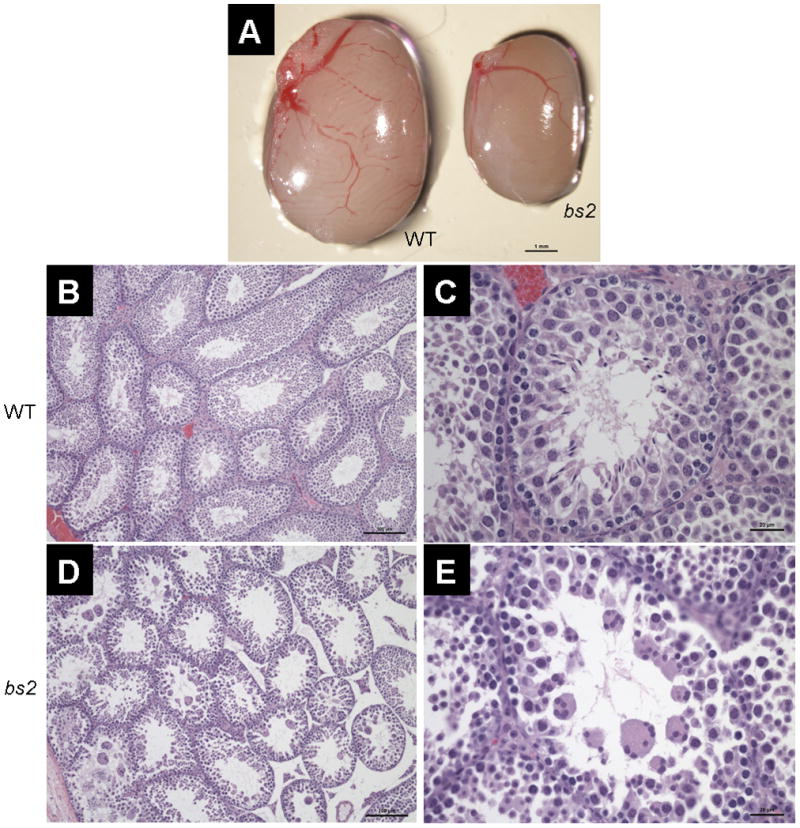
bs2 testis phenotype. Adult testes from bs2 mice (A, right) are smaller than WT siblings (A, left, scale bar: 1mm). Histological analysis of adult testis reveals that in bs2 mice (D, 10x) the seminiferous tubules are severely disrupted (scale bar: 100μm). Higher magnification (C, E, 40x) reveals that bs2 tubules contain no mature spermatozoa but do contain large multinucleate cells (scale bar: 20μm)
Positional cloning of the bs2 locus
The initial report established linkage of the bs2 locus to mouse chromosome 2, but did not implicate a specific chromosomal location [28]. To further map the bs2 locus, we utilized an intercross breeding strategy to generate 262 F2 progeny. At four weeks of age 15% (39/262) of F2 progeny were identified as affected, thus deviating from the expected 25% Mendelian ratio for a recessive locus as evaluated by chi square (p<0.05). F2 progeny were genotyped with Chr. 2 microsatellites spaced approximately 10 cM apart. The results confirmed linkage of bs2 to chromosome 2. The bs2 locus was assigned as closest to D2Mit247 in a region between D2Mit205 and D2Mit239 (Figure 3A). To further refine the genetic position of bs2, we genotyped the F2 progeny with microsatellites between D2Mit205 and D2Mit239. This analysis established the bs2 critical region as between D2Mit219 and D2Mit159, encompassing a 3.1 Mb physical region (Figure 3B). Analysis of the mouse genome sequence (July 2007 build) revealed that the bs2 critical region encompasses 19 previously established genes and one hypothetical gene (Table 2). In order to prioritize sequencing, we evaluated the NCBI (http://www.ncbi.nlm.nih.gov/gene/) and MGI databases (http://www.informatics.jax.org/) to identify if known mutations in any genes within the bs2 critical region were reported to lead to cataracts or male sterility in humans or mice. This analysis led to identification of cytochrome c, testis (Cyct; NM_009989) a gene exclusively expressed in the testes; Cyct−/− null mice exhibit reduced sperm counts, impaired fertilization, and testicular atrophy [34]. The second candidate gene selected was alkylglycerone phosphate synthase (Agps; NM_172666) a ubiquitously expressed peroxisomal protein present in all mammalian cells; mutations in AGPS result in RCDP3 where affected patients exhibit congenital cataracts as a part of their syndromic phenotype [35]. Thus, we proceeded to evaluate both Cyct and Agps for mutations in bs2 mice. Sequence analysis of Cyct exons, intron/exon junctions, and cDNA did not identify any sequence differences between WT and bs2 mice (data not shown). Sequence analysis of Agps cDNA from WT mEFs identified a single Agps transcript that matched the full-length Agps sequence (NM_172666.2). In contrast, three Agps transcripts were identified in bs2 mEFs (Figure 4A): full-length Agps transcript, AgpsΔexon14 which lacks exon 14, and AgpsexonΔ13–14 which lacks both exons 13 and 14 (Figures 4B and 4C). Full-length Agps transcript in bs2 mEFs comprised only 7% of total Agps transcript (Figure 4C). Both AgpsexonΔ14 and AgpsexonΔ13–14 are predicted to result in a frameshift followed by a premature stop codon and truncated AgpsGlu467fsX474 and AgpsPhe441fsX456 proteins, respectively (Figure 4E). The same aberrantly spliced transcripts as well as relative abundances of full-length Agps were also observed in bs2 lens, testes, spleen, and liver whereas only the single full-length transcript was observed in the same tissues of WT controls (not shown).
Figure 3.
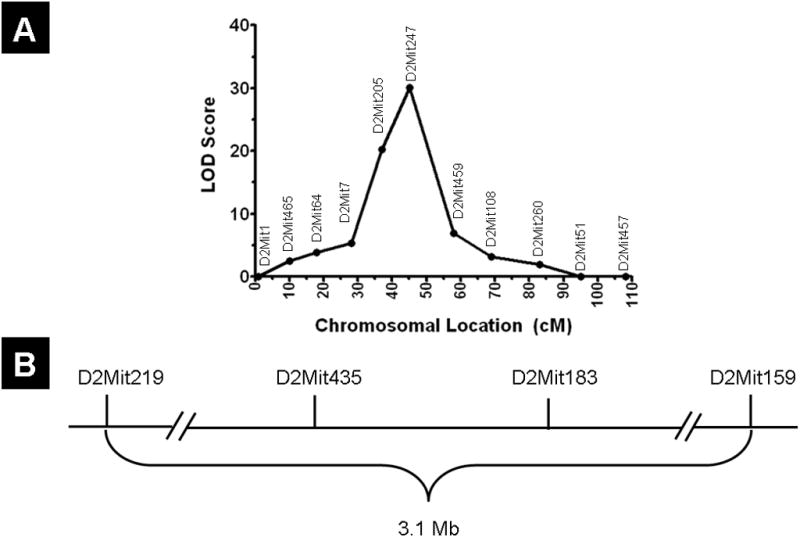
Linkage mapping and mutation identification. (A) Microsatellite markers were used to identify linkage to mouse chromosome 2, approximately 45 cM distal from the centromere. (B) Fine mapping narrowed the critical region to 3.1 Mb, between D2Mit219 and D2Mit159.
Figure 4.

Alternative splicing of Agps transcript in bs2 mice. (A) RT-PCR from mEF cDNA amplified three products in bs2 mice: 564bp, 451bp, and 371bp. In contrast, only the WT 564bp band was observed in WT controls. (B) Sequencing of the DNA contained in these bands identified the 564bp band as the full-length transcript, the 451bp band as skipping exon 14 (Agps Δ14), and the 371bp band as skipping exons 13 and 14 (Agps Δ13–14). (C) Semi-quantitative RT-PCR shows that 7% of Agps transcript was full-length, 55% Agps Δ14, and 38% Agps Δ13–14. (D) Semi-quantitative PCR also reveals that full length Agps transcript was present at 15% the level of that seen in WT controls. (p<0.05 for C, D) (E) AgpsGlu467fsX474 and AgpsPhe441fsX456, putative proteins formed from Agps Δ14 and Agps Δ13–14 transcripts, would contain an immediate frameshift at the beginning of exon 15, followed by 7 or 15 amino acids of nonsense, as well as a premature stop codon.
To further investigate the molecular basis for the aberrant Agps splicing observed in bs2, we sequenced the 12.3kb genomic region between exon 12 and exon 15. In bs2, sequence analysis identified a G to A substitution in Agps IVS14+5 (Figure 5A). The G nucleotide in Agps IVS14+5 is a part of the consensus 5′ splice sequence (Figure 5B). All affected intercross animals genotyped A/A at Agps IVS14+5, while none of the unaffected mice genotyped A/A, indicating co-segregation of Agps IVS14+5A with the bs2 phenotype. Evaluation of the SNP database did not reveal any previously reported polymorphisms for Agps intron 14 (http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?locusId=228061). Furthermore, we evaluated the Agps IVS14+5 locus in C57BL6/J, CAST/EiJ, AKR/J, C3H, RIIIS/J, DBA1/J, and DBA2/J strains and identified only G/G genotypes (not shown). These findings collectively suggest that the identified Agps IVS14+5G->A substitution is most likely responsible for the aberrant Agps splicing observed in bs2.
Figure 5.
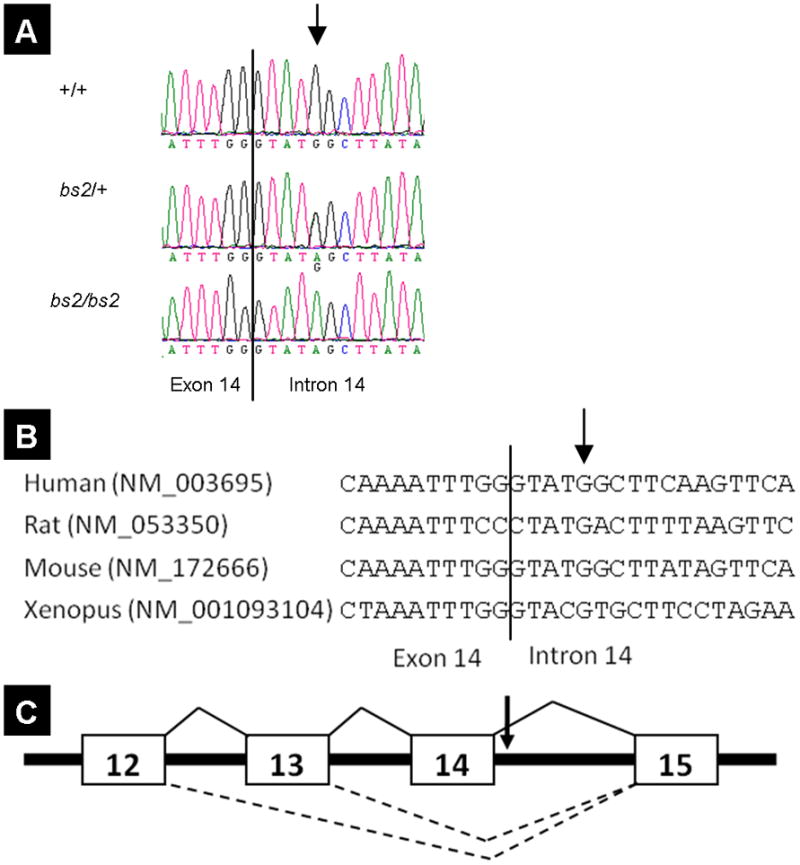
Identification of a single-base pair substitution in Agps intron 14. (A) A single base pair substitution, IVS14+5G>A, was identified in bs2 mice. All affected F2 mice genotyped A/A while unaffected mice genotyped G/A or G/G. (B) IVS14+5G is a key nucleotide within the 5′ splice donor site, which is conserved in human, rat, mouse, and xenopus. (C) IVS14+5 G>A disrupts a conserved splice-donor sequence (arrow). In the absence of this nucleotide, splice donor sites from intron 12 or 13 compete for the intron 14 splice acceptor, resulting in the Agps Δ13–14 and Agps Δ14 products.
Mass spectrometry of lipid species
Both AgpsexonΔ14 and AgpsexonΔ13–14 are predicted to result in truncated non-functional proteins. In bs2 mice, the remaining full-length Agps transcript represents only 15% of Agps levels observed in WT mice (Figure 4D). Thus, the bs2 mutation most likely results in severely reduced functional Agps protein levels that would in turn result in a severe reduction of plasmalogens. To test this hypothesis, we analyzed brain phospholipid levels by mass spectrometry in WT and bs2 mice. The bs2 brains contained only 15% of the levels of ether-phosphatidyl ethanolamine (ePE) detected in brains from WT mice (Figure 6B). Similarly, in bs2 samples, ether-phosphatidyl choline (ePC) levels were 35.1% of the levels of ePC observed in WT brains (Figure 6D). Interestingly, accumulation of phosphatidylethanolamine (PE), the precursor to ePE (Figure 6A), was detected in bs2 brains; no accumulation of phosphatidylcholine (PC), precursor to ePC, was observed (Figure 6C).
Figure 6.
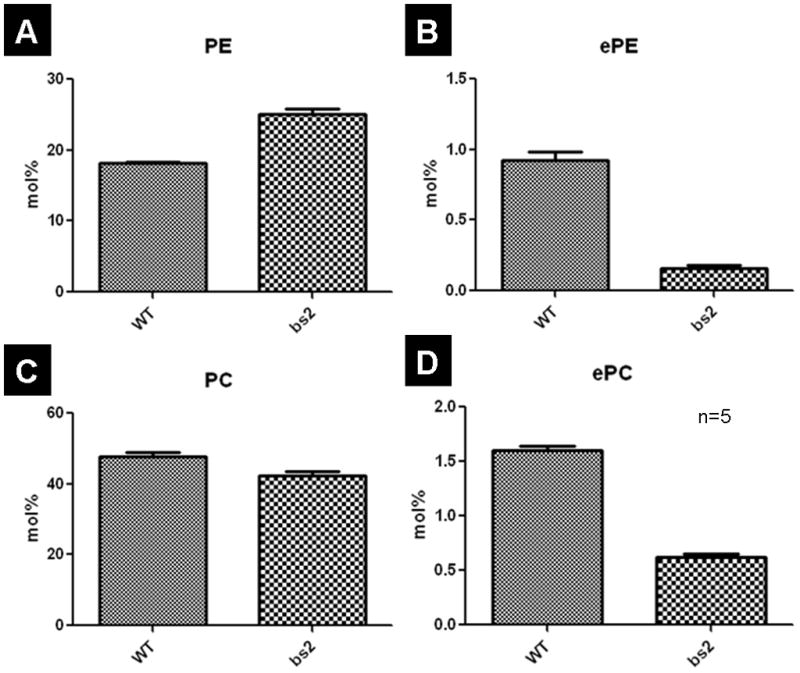
Mass spectrometry analysis of lipid species from brain lipid extracts. Ether-phosphatidylethanolamine (ePE; B) and ether phosphatidylcholine (ePC; D) were both significantly lower in bs2 mice (p<0.0001, both). Phosphatidylcholine (PC; C) levels are also lower in bs2 mice (p=0.0089), while phosphatidylethanolamine (PE; A) levels are higher (p<0.0001) in bs2 brains. Five replicates were performed for each genotype and significance was calculated via a t-test.
Peroxisomal assembly and PTS-mediated protein import
To determine if peroxisomal assembly and PTS1 or PTS2 mediated peroxisomal protein import were compromised in bs2, we evaluated bs2 mEFs. Immunofluorescence of WT and bs2 mEFs with Pmp70, catalase (not shown), and peroxisomal thiolase (not shown) antibodies revealed a typical peroxisomal punctate staining pattern (Figure 6B). Western blotting did not show any difference in protein levels of Pmp70 (Figure 7C), catalase (Figure 7D), and thiolase (not shown) between WT and bs2 mEFs. These findings suggest that peroxisome assembly as well as PTS1 and PTS2 mediated peroxisomal protein import did not differ between WT and bs2 mEFs.
Figure 7.
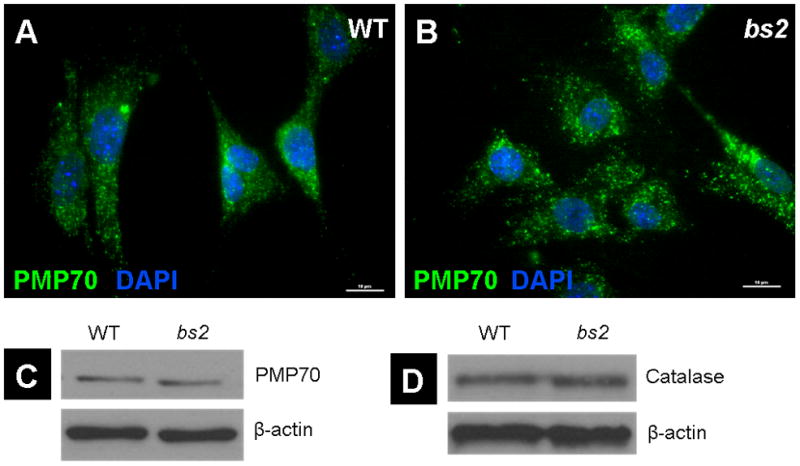
Peroxisomal assembly and PTS-mediated protein import. Immunofluorescent staining of Peroxisomal membrane protein 70 (PMP70, Green) counterstained with DAPI (Blue) confirms the presence of peroxisomes in bs2 mEFs (B) with no noted morphological deviations from those seen in WT controls (A) (scale bars: 10μm). Protein levels of PMP70 (C) and catalase (D) were not significantly different between WT and bs2 samples, as evaluated by western blot.
Examining possible allelism between bs2 and bs
The lens and testicular phenotypes in bs2 mice reported in this study resemble the phenotypes reported for the blind sterile (bs) mouse, another spontaneous mouse mutation on chromosome 2 resulting in cataracts and male sterility [29, 30]. The genetic position for bs was previously assigned to chromosome 2 between Hao1 and Emv-15 loci suggesting that bs and bs2 most likely are mutations in two separate genes on chromosome 2 resulting in similar cataract and male infertility phenotypes [29, 30]. To exclude the possibility that bs and bs2 are allelic, we set up a cross between bs/bs females and bs2/+ males, generating 34 F1 progeny. Genotyping revealed that 12 F1 progeny carried the Agps IVS14+5A allele, indicating 12/34 were compound bs and bs2 heterozygote mice. Clinical evaluation of all F1 progeny at >6 weeks of age did not identify any cataracts. Fertility in male bs/bs2 compound heterozygotes did not differ from WT mice.
Discussion
In this study, our goal was to identify a gene and mutation responsible for the bs2 cataract and male sterility phenotypes. Utilizing a positional cloning approach, we identified an IVS14+5G→A substitution in the 5′ splice site of Agps intron 14 as the causative mutation. The +5G position of the 5′ splice site is part of a consensus splice-donor sequence conserved across species which base pairs with U1 snRNA to initiate splicing [36, 37]. Positions −1 and +5 of the 5′ splice site base pair strongly with U1 snRNA, thus mutations at these positions have detrimental effects on splicing [37]. It has been recently established that 19.7% of diseases resulting from mutations in the 5′ splice sites are due to a mutation of the +5 nucleotide [36, 38, 39]. In bs2 mice the Agps IVS14+5G→A substitution resulted in skipping of exon 14 or both exons 13 and 14 and the formation of the aberrantly spliced AgpsΔexon14 and AgpsexonΔ13–14 transcripts. In addition to the two aberrantly spliced transcripts, we also identified a full length Agps transcript expressed at very low levels in bs2 compared to WT mice. The putative mutant Agps proteins, AgpsGlu467fsX474 and AgpsPhe441fsX456, would have frameshifts following Thr467 and Phe441, respectively, resulting in a premature stop codon. Both putative proteins would be truncated near the start of the C-terminal catalytic FAD oxidase domain and thus, would be catalytically inactive. Agps is a member of the FAD binding oxidoreductase/transferase type 4 family of proteins. The role of Agps is to catalyze the second step of ether lipid biosynthesis in which acyl-dihydroxyacetonephosphate (DHAP) is converted to alkyl-DHAP [24]. Phospholipid profiling of bs2 brains identified severely reduced ether-lipid levels further confirming that the bs2 phenotypes are associated with a disruption in the ether-lipid synthesis pathway. Ether-lipids were observed in bs2 brains at low levels, most likely resulting from the Agps activity encoded by the small fraction of properly spliced Agps transcript identified in bs2 tissues. These findings collectively show that bs2 is a hypomorphic mutation in Agps resulting in disruption of the ether lipid synthesis pathway, leading to a severe reduction of ether lipid levels.
Mutations in AGPS have been previously reported in only three RCDP3 patients [23, 35, 40]. Biochemical evaluation of fibroblasts from RCDP3 patients identified deficient AGPS enzymatic function and severely reduced plasmalogen levels [40, 41]. RCDP3 fibroblasts, similarly to RCDP2 fibroblasts, exhibit ether lipid deficiencies due to mutations in enzymes essential for ether lipid synthesis without defects in peroxisomal assembly and PTS1 or PTS2 mediated protein import [40, 42]. A mutation in AGPS was also identified in a plasmalogen-deficient CHO cell line following a mutagenesis screen [43]. Analysis of the plasmalogen deficient CHO line identified significantly reduced AGPS enzymatic function with intact peroxisomal assembly and protein import [43]. Functional evaluation of bs2 mEFs is consistent with these findings, indicating that Agps IVS14+5G→A results in a severe reduction of Agps function. In addition, we did not observe any difference in PTS1 and PTS2 mediated protein import or peroxisomal assembly between WT and bs2. Therefore, the severe loss of ether lipids observed in bs2 mice is the result of a mutation in Agps in which the resulting enzymatic deficiency is independent of peroxisomal assembly or peroxisomal protein import. As such, the bs2 mouse is the first animal model of RCDP3, providing an excellent opportunity for evaluation of Agps deficiency in-vivo.
The mechanism by which an overall reduction of plasmalogen levels results in tissue-specific RCDP phenotypes in humans and mice is not well understood. Plasmalogens contribute to about 18% of total phospholipid mass, although there is a unique tissue-specific distribution: ePE levels are high in brain, heart, lung, kidney, spleen, and testis whereas high levels of ePC were noted in the cardiac and skeletal muscles [2, 24]. Originally, the bs2 mouse was identified during a phenotypic screen of mice with spontaneous mutations exhibiting vision defects [28]. The initial study reported cataracts in bs2 mice [28]. Results from this study show that bs2 cataracts are associated with defects in the elongation of lens fiber cells. Histological evaluation further showed that attenuated lens fiber cells were also detached from the lens epithelium, suggesting either a defect in lens fiber elongation or defect in cell-to-cell communication between lens epithelial and fiber cells. The cataract phenotype was also reported for Pex7−/− and Gnpat−/− mice [26, 27]. Similarly, all patients affected with RCDP are either born with cataracts or develop cataracts within the first six months of life [21, 25]. These findings suggest that plasmalogens are essential for lens development and homeostasis in both humans and mice. Recently, analysis of mouse lenses identified ePE as the major ether lipid class in the lens although ePC, phosphatidyl serine, and sphingomyelin were also present [44]. It has been proposed that plasmalogens may be protecting lens membranes against oxidative stress and may be essential for signaling [44]. Further evaluation is necessary for better understanding of the role of plasmalogens in the lens.
In addition to cataracts, all three RCDP mouse models, Pex7−/−, Gnpat−/−, and bs2, exhibit disorganized seminiferous tubules with abnormalities in spermatogenesis resulting in testicular atrophy and male sterility [26, 27]. Loss of plasmalogens in the testes of Gnpat−/− mice was associated with down-regulation and mis-targeting of claudin-3 and consequent impairment of the blood-testis-barrier leading to deficient sealing of the transient intermediate compartment [45]. In addition to cataracts and testicular phenotypes, bs2 mice exhibited some prenatal/perinatal lethality which was also observed in Pex7−/− and Gnpat−/− mice [26, 27]. The cause of prenatal/perinatal lethality associated with a decrease in ether lipids remains unclear. Interestingly, in bs2 we did not observe any obvious hypotonia, delayed growth, defects in neuronal migration, or bone ossification defects reported for Pex7−/− [26] or myelination defects reported for Gnpat−/− [27, 46]. These differences may be due to residual plasmalogen levels present in bs2 tissues, suggesting that the severity of the phenotype in mice may be associated with plasmalogen levels. This is consistent with findings in patients affected with all forms of RCDP in which disease severity is directly related to plasmalogen levels [25, 47].
Finally, as a part of this study we showed that bs2 is not allelic with bs, a mouse model also exhibiting cataracts and male sterility that was previously mapped to chromosome 2 [29, 30]. This finding confirms that bs2 is the only genetic model for RCDP3. Further analysis of pathways implicated in the bs phenotype may yield additional information on the mechanism of the tissue specificity observed in both blind-sterile mouse models. In combination with existing RCDP models, the bs2 mouse provides an exciting opportunity to facilitate a better understanding of the molecular consequences and mechanisms involved in ether-lipid deficiency.
Supplementary Material
Acknowledgments
This work was supported in part by National Institutes of Health grants EY15173 (D.J.S), and EY19943 (B.C). The lipid analyses described in this work were performed at the Kansas Lipidomics Research Center Analytical Laboratory. Kansas Lipidomics Research Center was supported by National Science Foundation (EPS 0236913, MCB 0455318, DBI 0521587), Kansas Technology Enterprise Corporation, K-IDeA Networks of Biomedical Research Excellence (INBRE) of National Institute of Health (P20RR16475), and Kansas State University.
ABBREVIATIONS
- bs2
blind-sterile 2
- Agps
alkylglyceronephosphate synthase
- RCDP3
Rhizomelic chondrodysplasia punctata type 3
- PTS1
peroxisomal targeting signal 1
- PTS2
peroxisomal targeting signal 2
- FAD
flavin adenine dinucleotide
- PBD
peroxisomal biogenesis disorders
- RCDP1
rhizomelic chondrodysplasia punctata type 1
- RCDP2
rhizomelic chondrodysplasia punctata type 2
- GNPAT
glyceronephosphate O-acyltransferase
- PEX7
peroxisomal biogenesis factor 7
- Cyct
cytochrome c, testis
- mEFs
mouse embryonic fibroblasts
- PE
phosphatidyl ethanolamine
- ePE
ether phosphatidyl ethanolamine
- PC
phosphatidyl choline
- ePC
ether phosphatidyl choline
- U1snRNA
U1 spliceosomal RNA
- bs
blind-sterile
Footnotes
Conflict of Interest Statement:
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lazarow PB, Fujiki Y. Biogenesis of peroxisomes. Annu Rev Cell Biol. 1985;1:489–530. doi: 10.1146/annurev.cb.01.110185.002421. [DOI] [PubMed] [Google Scholar]
- 2.Wanders RJ, Waterham HR. Biochemistry of mammalian peroxisomes revisited. Annu Rev Biochem. 2006;75:295–332. doi: 10.1146/annurev.biochem.74.082803.133329. [DOI] [PubMed] [Google Scholar]
- 3.Hoepfner D, Schildknegt D, Braakman I, Philippsen P, Tabak HF. Contribution of the endoplasmic reticulum to peroxisome formation. Cell. 2005;122:85–95. doi: 10.1016/j.cell.2005.04.025. [DOI] [PubMed] [Google Scholar]
- 4.Kim PK, Mullen RT, Schumann U, Lippincott-Schwartz J. The origin and maintenance of mammalian peroxisomes involves a de novo PEX16-dependent pathway from the ER. J Cell Biol. 2006;173:521–532. doi: 10.1083/jcb.200601036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kunau WH. Peroxisome biogenesis: end of the debate. Curr Biol. 2005;15:R774–776. doi: 10.1016/j.cub.2005.08.056. [DOI] [PubMed] [Google Scholar]
- 6.Purdue PE, Lazarow PB. Peroxisomal biogenesis: multiple pathways of protein import. J Biol Chem. 1994;269:30065–30068. [PubMed] [Google Scholar]
- 7.Girzalsky W, Platta HW, Erdmann R. Protein transport across the peroxisomal membrane. Biol Chem. 2009;390:745–751. doi: 10.1515/BC.2009.104. [DOI] [PubMed] [Google Scholar]
- 8.McNew JA, Goodman JM. An oligomeric protein is imported into peroxisomes in vivo. J Cell Biol. 1994;127:1245–1257. doi: 10.1083/jcb.127.5.1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Glover JR, Andrews DW, Rachubinski RA. Saccharomyces cerevisiae peroxisomal thiolase is imported as a dimer. Proc Natl Acad Sci U S A. 1994;91:10541–10545. doi: 10.1073/pnas.91.22.10541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gould SJ, Keller GA, Hosken N, Wilkinson J, Subramani S. A conserved tripeptide sorts proteins to peroxisomes. J Cell Biol. 1989;108:1657–1664. doi: 10.1083/jcb.108.5.1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gatto GJ, Jr, Geisbrecht BV, Gould SJ, Berg JM. Peroxisomal targeting signal-1 recognition by the TPR domains of human PEX5. Nat Struct Biol. 2000;7:1091–1095. doi: 10.1038/81930. [DOI] [PubMed] [Google Scholar]
- 12.Motley AM, Hettema EH, Hogenhout EM, Brites P, ten Asbroek AL, Wijburg FA, Baas F, Heijmans HS, Tabak HF, Wanders RJ, Distel B. Rhizomelic chondrodysplasia punctata is a peroxisomal protein targeting disease caused by a non-functional PTS2 receptor. Nat Genet. 1997;15:377–380. doi: 10.1038/ng0497-377. [DOI] [PubMed] [Google Scholar]
- 13.Braverman N, Steel G, Obie C, Moser A, Moser H, Gould SJ, Valle D. Human PEX7 encodes the peroxisomal PTS2 receptor and is responsible for rhizomelic chondrodysplasia punctata. Nat Genet. 1997;15:369–376. doi: 10.1038/ng0497-369. [DOI] [PubMed] [Google Scholar]
- 14.Purdue PE, Zhang JW, Skoneczny M, Lazarow PB. Rhizomelic chondrodysplasia punctata is caused by deficiency of human PEX7, a homologue of the yeast PTS2 receptor. Nat Genet. 1997;15:381–384. doi: 10.1038/ng0497-381. [DOI] [PubMed] [Google Scholar]
- 15.Zhang JW, Lazarow PB. PEB1 (PAS7) in Saccharomyces cerevisiae encodes a hydrophilic, intra-peroxisomal protein that is a member of the WD repeat family and is essential for the import of thiolase into peroxisomes. J Cell Biol. 1995;129:65–80. doi: 10.1083/jcb.129.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van den Bosch H, Schutgens RB, Wanders RJ, Tager JM. Biochemistry of peroxisomes. Annu Rev Biochem. 1992;61:157–197. doi: 10.1146/annurev.bi.61.070192.001105. [DOI] [PubMed] [Google Scholar]
- 17.Steinberg SJ, Dodt G, Raymond GV, Braverman NE, Moser AB, Moser HW. Peroxisome biogenesis disorders. Biochim Biophys Acta. 2006;1763:1733–1748. doi: 10.1016/j.bbamcr.2006.09.010. [DOI] [PubMed] [Google Scholar]
- 18.Gould SJ, Valle D. Peroxisome biogenesis disorders: genetics and cell biology. Trends Genet. 2000;16:340–345. doi: 10.1016/s0168-9525(00)02056-4. [DOI] [PubMed] [Google Scholar]
- 19.Shimozawa N. Molecular and clinical aspects of peroxisomal diseases. J Inherit Metab Dis. 2007;30:193–197. doi: 10.1007/s10545-007-0516-z. [DOI] [PubMed] [Google Scholar]
- 20.Steinberg S, Chen L, Wei L, Moser A, Moser H, Cutting G, Braverman N. The PEX Gene Screen: molecular diagnosis of peroxisome biogenesis disorders in the Zellweger syndrome spectrum. Mol Genet Metab. 2004;83:252–263. doi: 10.1016/j.ymgme.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 21.White AL, Modaff P, Holland-Morris F, Pauli RM. Natural history of rhizomelic chondrodysplasia punctata. Am J Med Genet A. 2003;118A:332–342. doi: 10.1002/ajmg.a.20009. [DOI] [PubMed] [Google Scholar]
- 22.Wanders RJ, Waterham HR. Peroxisomal disorders I: biochemistry and genetics of peroxisome biogenesis disorders. Clin Genet. 2005;67:107–133. doi: 10.1111/j.1399-0004.2004.00329.x. [DOI] [PubMed] [Google Scholar]
- 23.Wanders RJ, Dekker C, Hovarth VA, Schutgens RB, Tager JM, Van Laer P, Lecoutere D. Human alkyldihydroxyacetonephosphate synthase deficiency: a new peroxisomal disorder. J Inherit Metab Dis. 1994;17:315–318. doi: 10.1007/BF00711817. [DOI] [PubMed] [Google Scholar]
- 24.Brites P, Waterham HR, Wanders RJ. Functions and biosynthesis of plasmalogens in health and disease. Biochim Biophys Acta. 2004;1636:219–231. doi: 10.1016/j.bbalip.2003.12.010. [DOI] [PubMed] [Google Scholar]
- 25.Braverman N, Chen L, Lin P, Obie C, Steel G, Douglas P, Chakraborty PK, Clarke JT, Boneh A, Moser A, Moser H, Valle D. Mutation analysis of PEX7 in 60 probands with rhizomelic chondrodysplasia punctata and functional correlations of genotype with phenotype. Hum Mutat. 2002;20:284–297. doi: 10.1002/humu.10124. [DOI] [PubMed] [Google Scholar]
- 26.Brites P, Motley AM, Gressens P, Mooyer PA, Ploegaert I, Everts V, Evrard P, Carmeliet P, Dewerchin M, Schoonjans L, Duran M, Waterham HR, Wanders RJ, Baes M. Impaired neuronal migration and endochondral ossification in Pex7 knockout mice: a model for rhizomelic chondrodysplasia punctata. Hum Mol Genet. 2003;12:2255–2267. doi: 10.1093/hmg/ddg236. [DOI] [PubMed] [Google Scholar]
- 27.Rodemer C, Thai TP, Brugger B, Kaercher T, Werner H, Nave KA, Wieland F, Gorgas K, Just WW. Inactivation of ether lipid biosynthesis causes male infertility, defects in eye development and optic nerve hypoplasia in mice. Hum Mol Genet. 2003;12:1881–1895. doi: 10.1093/hmg/ddg191. [DOI] [PubMed] [Google Scholar]
- 28.Chang B, Hawes NL, Hurd RE, Wang J, Howell D, Davisson MT, Roderick TH, Nusinowitz S, Heckenlively JR. Mouse models of ocular diseases. Vis Neurosci. 2005;22:587–593. doi: 10.1017/S0952523805225075. [DOI] [PubMed] [Google Scholar]
- 29.Spence SE, Gilbert DJ, Harris BS, Davisson MT, Copeland NG, Jenkins NA. Genetic localization of Hao-1, blind-sterile (bs), and Emv-13 on mouse chromosome 2. Genomics. 1992;12:403–404. doi: 10.1016/0888-7543(92)90392-6. [DOI] [PubMed] [Google Scholar]
- 30.Varnum DS. Blind-sterile: a new mutation on chromosome 2 of the house mouse. J Hered. 1983;74:206–207. doi: 10.1093/oxfordjournals.jhered.a109768. [DOI] [PubMed] [Google Scholar]
- 31.Hassemer EL, Le Gall SM, Liegel R, McNally M, Chang B, Zeiss CJ, Dubielzig RD, Horiuchi K, Kimura T, Okada Y, Blobel CP, Sidjanin DJ. The waved with open eyelids (woe) locus is a hypomorphic mouse mutation in Adam17. Genetics. 185:245–255. doi: 10.1534/genetics.109.113167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Talamas E, Jackson L, Koeberl M, Jackson T, McElwee JL, Hawes NL, Chang B, Jablonski MM, Sidjanin DJ. Early transposable element insertion in intron 9 of the Hsf4 gene results in autosomal recessive cataracts in lop11 and ldis1 mice. Genomics. 2006;88:44–51. doi: 10.1016/j.ygeno.2006.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Devaiah SP, Roth MR, Baughman E, Li M, Tamura P, Jeannotte R, Welti R, Wang X. Quantitative profiling of polar glycerolipid species from organs of wild-type Arabidopsis and a phospholipase. Dalpha1 knockout mutant Phytochemistry. 2006;67:1907–1924. doi: 10.1016/j.phytochem.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 34.Narisawa S, Hecht NB, Goldberg E, Boatright KM, Reed JC, Millan JL. Testis-specific cytochrome c-null mice produce functional sperm but undergo early testicular atrophy. Mol Cell Biol. 2002;22:5554–5562. doi: 10.1128/MCB.22.15.5554-5562.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.de Vet EC, Ijlst L, Oostheim W, Wanders RJ, van den Bosch H. Alkyl-dihydroxyacetonephosphate synthase. Fate in peroxisome biogenesis disorders and identification of the point mutation underlying a single enzyme deficiency. J Biol Chem. 1998;273:10296–10301. doi: 10.1074/jbc.273.17.10296. [DOI] [PubMed] [Google Scholar]
- 36.Carmel I, Tal S, Vig I, Ast G. Comparative analysis detects dependencies among the 5′ splice-site positions. RNA. 2004;10:828–840. doi: 10.1261/rna.5196404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ast G. How did alternative splicing evolve? Nat Rev Genet. 2004;5:773–782. doi: 10.1038/nrg1451. [DOI] [PubMed] [Google Scholar]
- 38.Wang GS, Cooper TA. Splicing in disease: disruption of the splicing code and the decoding machinery. Nat Rev Genet. 2007;8:749–761. doi: 10.1038/nrg2164. [DOI] [PubMed] [Google Scholar]
- 39.Berget SM. Exon recognition in vertebrate splicing. J Biol Chem. 1995;270:2411–2414. doi: 10.1074/jbc.270.6.2411. [DOI] [PubMed] [Google Scholar]
- 40.Thai TP, Rodemer C, Jauch A, Hunziker A, Moser A, Gorgas K, Just WW. Impaired membrane traffic in defective ether lipid biosynthesis. Hum Mol Genet. 2001;10:127–136. doi: 10.1093/hmg/10.2.127. [DOI] [PubMed] [Google Scholar]
- 41.de Vet EC, Ijlst L, Oostheim W, Dekker C, Moser HW, van Den Bosch H, Wanders RJ. Ether lipid biosynthesis: alkyl-dihydroxyacetonephosphate synthase protein deficiency leads to reduced dihydroxyacetonephosphate acyltransferase activities. J Lipid Res. 1999;40:1998–2003. [PubMed] [Google Scholar]
- 42.Singh I, Lazo O, Contreras M, Stanley W, Hashimoto T. Rhizomelic chondrodysplasia punctata: biochemical studies of peroxisomes isolated from cultured skin fibroblasts. Arch Biochem Biophys. 1991;286:277–283. doi: 10.1016/0003-9861(91)90041-g. [DOI] [PubMed] [Google Scholar]
- 43.Nagan N, Hajra AK, Das AK, Moser HW, Moser A, Lazarow P, Purdue PE, Zoeller RA. A fibroblast cell line defective in alkyl-dihydroxyacetone phosphate synthase: a novel defect in plasmalogen biosynthesis. Proc Natl Acad Sci U S A. 1997;94:4475–4480. doi: 10.1073/pnas.94.9.4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thai TP, Rodemer C, Worsch J, Hunziker A, Gorgas K, Just WW. Synthesis of plasmalogens in eye lens epithelial cells. FEBS Lett. 1999;456:263–268. doi: 10.1016/s0014-5793(99)00968-0. [DOI] [PubMed] [Google Scholar]
- 45.Komljenovic D, Sandhoff R, Teigler A, Heid H, Just WW, Gorgas K. Disruption of blood-testis barrier dynamics in ether-lipid-deficient mice. Cell Tissue Res. 2009;337:281–299. doi: 10.1007/s00441-009-0809-7. [DOI] [PubMed] [Google Scholar]
- 46.Teigler A, Komljenovic D, Draguhn A, Gorgas K, Just WW. Defects in myelination, paranode organization and Purkinje cell innervation in the ether lipid-deficient mouse cerebellum. Hum Mol Genet. 2009;18:1897–1908. doi: 10.1093/hmg/ddp110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bams-Mengerink AM, Majoie CB, Duran M, Wanders RJ, Van Hove J, Scheurer CD, Barth PG, Poll-The BT. MRI of the brain and cervical spinal cord in rhizomelic chondrodysplasia punctata. Neurology. 2006;66:798–803. doi: 10.1212/01.wnl.0000205594.34647.d0. discussion 789. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.