

Abstract
Background
The interaction between small molecules and proteins is one of the major concerns for structure-based drug design since the principles of protein-ligand interactions and molecular recognition are not thoroughly understood. Fortunately, the analysis of protein-ligand complexes in the Protein Data Bank (PDB) enables unprecedented possibilities for new insights. Herein, we applied molecule-fragmentation algorithms to split the ligands extracted from PDB crystal structures into small fragments. Subsequently, we have developed a ligand fragment and residue preference mapping (LigFrag-RPM) algorithm to map the profiles of the interactions between these fragments and the 20 proteinogenic amino acid residues.
Results
A total of 4,032 fragments were generated from 71,798 PDB ligands by a ring cleavage (RC) algorithm. Among these ligand fragments, 315 unique fragments were characterized with the corresponding fragment-residues interaction profiles by counting residues close to these fragments. The interaction profiles revealed that these fragments have specific preferences for certain types of residues. The applications of these interaction profiles were also explored and evaluated in case studies, showing great potential for the study of protein-ligand interactions and drug design.
Conclusions
Our studies demonstrated that the fragment-residues interaction profiles generated from the PDB ligand fragments can be used to detect whether these fragments are in their favorable or unfavorable environments. The algorithm for a ligand fragment and residue preference mapping (LigFrag-RPM) developed here also has the potential to guide lead chemistry modifications as well as binding residues predictions.
Background
The Protein Data Bank (PDB)1, the largest protein structure database, is widely used in structure-based drug design. Several techniques have been developed for analyzing the interactions of protein-ligand complexes. For example, LIGPLOT 2 automatically analyzes and plots different intermolecular interactions, such as hydrogen bonds, ionic and aromatic interactions. Various sophisticated computer graphical interfaces, such as InsightII, Sybyl, and Maestro are available for analyzing 3D structure of proteins and their complexes.
Large scale systematical analyses of the interactions between proteins and their ligands at the fragment level are rare. SuperStar 3 and Relibase 4 are two examples. These two computational tools analyze the interaction of functional groups between ligands and proteins and can superimpose these functional groups to create scatterplots of the specific interactions. Relibase is also a database search, retrieval, and analysis program for protein-ligand complex structures. Another similar tool is SIF (Structural Interaction Fingerprint) 5, which generates a 1D binary fingerprint representations of the intermolecular interactions in a 3D protein-inhibitor complex.
Recently, fragment-based approaches have been recognized as valuable tools in drug design. Dissecting a drug or small organic molecule into smaller fragment pieces and into even smaller discrete functional groups simplifies the computational analysis of ligand binding and identifies different pharmacophore elements required for high-affinity binding 6. The method is based on the principle that the proper optimization of each unique interaction in the binding site and subsequent incorporation into a single molecular entity should produce a compound with a binding affinity that is the sum of the individual interactions 6.
Although successful applications of fragment-based approaches for drug discovery by NMR 7 and X-ray Crystallography 8 have been reported earlier, these methods are quite time-consuming and expensive. Furthermore, the patterns of interactions between ligands and protein residues can be useful for the prediction of binding modes and structural adaptations 9. A statistical analysis of the binding sites of proteins would contribute greatly to the understanding of the major determinants of ligand binding and specificity. Previous strategies or software tools have been limited in their ability to enumerate all of the fragments in PDB ligands and analyze the general interactions between the ligand fragments and protein residues.
We have therefore developed LigFrag-RPM, a novel ligand fragment and residue preference mapping algorithm. This approach splits the ligands in PDB complexes into fragments, then analyzes the fragment-residues interactions, and subsequently derives the fragment-residues interaction profiles from multiple individual interactions. Developing the LigFrag-RPM algorithm offers a promising technique for the systematic benchmarking of protein-ligand interactions. The resulting profiles can be used to detect disfavored ligand fragments and develop a catalogue of preferred fragments for residues in the protein binding pockets as a tool to guide lead modification.
Methods
Ligand data retrieval and filtering
In order to explore and map the interactions between ligand fragments and protein residues, the following protocols were used to extract protein-ligand complexes after cleanup of ligand binding sites in the PDB.
First, Ligand Expo 10 (http://ligand-expo.rcsb.org/index.html, accessed Dec 7, 2010) was used as the ligand resource in which ligands with their original coordinates were extracted from PDB crystal structure files. An advantage of the ligands compiled by Ligand Expo is that the PDB IDs are also encoded in the ligand names. Accordingly, the corresponding crystal structures can be readily identified. An example of a ligand name is ‘3gcs_BAX_1_A_401__B__’. In the Ligand Expo naming system, the first four characters, i.e. ‘3gcs’, represent the PDB ID, and BAX is the code for the co-crystallized ligand. A total of 456,642 ligand files were downloaded from Ligand Expo and were subjected to the subsequent filtering process, which removed ligands with less than six heavy atoms (such as solvents) and inorganic molecules (such as Mg2+, Na+ and Mn2+). Ligands without rings were also filtered out since the fragmentation algorithm requires at least one ring (see below). This filtering process was done by the SELECTOR module of Tripos Sybyl8.0. In addition, we also filtered out the PDB ligands files that contain either DNA or RNA by using the PDB advance search engine (http://www.rcsb.org/pdb/search/advSearch.do, accessed Dec 7, 2010). Finally, the PDB IDs were extracted from names of these qualified ligands. The corresponding PDB files were then downloaded from the PDB website. Only ligands from these complexes were considered in further analyses.
Using these criteria, a total of 71,798 ligands from 21,198 PDB complexes (as of Sep 28, 2009) passed the filters for further studies.
Ligand fragmenting and encoding
Ligands were split into small fragments to explore the individual interactions between these fragments and the 20 proteinogenic amino acid residues. We adopted the ring-cleavage fragmentation algorithm and the fragment encoding method established by Lameijer et al. 11. Molecules were split into ring systems, substituents and various types of linkers, and these fragments were then encoded into strings containing the information of atoms, bonds and connectivity. All of the unique fragment strings were then numbered by continuous integers starting with 0.
The algorithms used in our studies were developed with the open source software chemistry development kit (CDK) 12, and Marvin 13 was used for displaying structures.
Generating interaction profiles
For each fragment, the corresponding interacting residues from protein-ligand complexes were recorded to generate the residue preference profile. An interacting residue of a fragment was originally defined as a residue containing at least one heavy atom located within 5 Å from any heavy atoms of the fragment. However, a threshold of 5 Å may include many residues that do not really interact with the fragment. We therefore restricted the threshold to require at least two heavy atoms of the residue to be located within less than 5 Å of the fragment’s heavy atoms. A residue close to a fragment was considered as an interacting residue, and was counted in the statistical analysis, only if it satisfied this criterion, which we named “2-pairs-in-5Å”. The interactions of the ions and other small species with these ligand fragments are ignored as we only analyze the interactions between ligand fragments and protein residues.
We assumed that if a fragment prefers one or more particular residues, the probability of these residues close to and interacting with this fragment should be higher than for those unfavorable residues in protein-ligand complexes. The occurrences of these 20 standard residues close to a fragment were counted as its interaction profile. However, many of these fragments may be overrepresented because of multiple occurrences in similar or even identical proteins in the PDB database. For example, querying with keywords “HIV-1 PROTEASE” retrieved 426 structures from the PDB database. Among them, 197 ligands were co-crystallized with HIV-1 protease. Especially, a ligand named 1UN or 2-[2-hydroxy-3-(3-hydroxy-2-methyl-benzoylamino)-4-phenyl sulfanyl-butyl]-decahydro-isoquinoline-3-carboxylic acid tert-butylamide, occurs in 10 PDB structures with HIV-1 protease. Even though these PDB structures are in different resolutions or with different protein mutants, for most of the fragments of 1UN, the ligand environments or the nearby residues are similar or identical. Considering the fragments from 1UN, the sum of the interactive residues would overrepresent the interacting residues in the binding pocket of HIV-1 protease. To remedy this problem, no-redundancy PDB (nr-PDB) IDs (ftp://resources.rcsb.org/sequence/clusters/clusters70.txt, accessed Dec 7, 2010) were used as a reference list to remove similar or identical proteins that interact with the same fragment. The nr-PDB IDs were compiled according to the sequence similarity of the proteins. Similar proteins were grouped into a cluster according to a similarity threshold of 70%. When multiple proteins from the same protein cluster interacted with the same fragment, we selected interacting residues only from a representative protein of this cluster. Because the focus of our study is about the interactions between residues and fragments, a protein with the highest number of interacting residues close to this fragment was selected as the representative protein. Other proteins in the same cluster with fewer interacting residues were discarded. An example of this selection process is described later.
After removing redundant proteins for each individual fragment, all the interactive residues of a fragment were counted according to residue names. The original counts were then ranked in descending order. It should be pointed out that the frequencies of 20 amino acids occurring in natural proteins are different, so normalization is necessary to calibrate these profiles. We took the residue frequencies from a published paper14, which was calculated from 36,498 unique eukaryotic proteins15. Each residue count in the original profile was then normalized by dividing with the correspondent residue frequency in all natural proteins.
Results and Discussion
Fragmentation results of PDB ligands
Figure 1 is an illustration of the fragmentation result of the ligand 1AW or 1-[1-(3-aminophenyl)-3-tert-butyl-1H-pyrazol- 5-yl]-3-phenylurea in PDB 3F3U. The ring-cleavage (RC) algorithm splits off the ring substituents (fragment 240 and fragment 35 in Figure 1), while the ring systems (fragment 322 and fragment 1) are intact. A linker (fragment 79) between fragment 322 and fragment 1 is also generated by the RC algorithm. Through this process, the ring cleavage algorithm yielded 4,032 fragments from these 71,798 PDB ligands, including 1,404 rings, 1,711 substituents and 917 linkers.
Figure 1. Ring cleavage (RC) algorithm for ligand fragmentation.

This figure shows the ligand 1AW or 1-[1-(3-aminophenyl)-3-tert-butyl-1H-pyrazol-5-yl]-3-phenylurea and the fragments generated by the ring cleavage (RC) algorithm.
The frequencies of these fragments in PDB structures were analyzed next, as shown in Figure 2. Among the 4,032 fragments, 2,111 fragments appeared only once in the PDB structure files. For example, the fragment 3003 (an acetonitrile group) only appeared once in PDB 3CY3. In addition, 636 fragments appeared twice and 319 fragments appeared 3 times in PDB structures (Figure 2). These 3,066 fragments (2,111+636+319) were deleted and were not included in our studies. The remaining 1,346 fragments appeared at least four times in crystal structures and were selected to generate the interaction profiles. The rationale was that the interaction profiles generated from fragments occurring in a limited number of co-crystal studies may be less meaningful.
Figure 2. The histogram of the occurrences of 4,032 ligand fragments from PDB structures.

2,111 fragments occur only once in PDB structures (For example, the fragment 3303 colored with red, occurs only in PDB 3CY3), 636 fragments occur twice, 319 fragments occur 3 times, and a total of 1,346 fragments occur in at least four crystal structures (For example, fragment 18 or hydroxyl attached to carbon, occurs in 13505 structures of our selected PDB complexes dataset.).
Interaction profiles of PDB ligand fragments
Fragment 112 (a trifluoromethyl group) serves as an example to illustrate how a fragment-residues interaction profile was generated (Figure 3A). The fragment 112 occurs in 238 co-crystallized ligands of 340 PDB complexes. According to the non-redundant PDB data (nr-PDB), the proteins in these 340 structures are from 77 clusters that were classified with 70% as the similarity threshold. For each co-crystal complex, the interactive residues of fragment 112 were identified by the 2-pairs-in-5Å criterion. In each cluster, the protein that had the highest number of interacting residues with fragment 112 was selected as the representative protein for this cluster. A total of 77 representative proteins from these 77 clusters then were identified as a refined subset. All interactive residues of fragment 112 from the refined protein subset were counted according to residue names. As shown in Figure 3A, in ligand 3gcs_BAX_1_A_401__B (or the ligand BAX from PDB 3GCS), fragment 112 interacts with chain A of 3GCS (3GCS:A), which is in the cluster 3645 of the nr-PDB list. In this case, three ILEs (ILE84, ILE141 and ILE166), 1 HIS (HIS148), 1 LEU (LEU74), 1 MET (MET78), 1 VAL (VAL83) and 1 ASP (ASP168) were identified as interacting residues of fragment 112 (Figure 3A). While in the ligand 3gz9_D32_1_A_1__C (the ligand D32 from PDB 3GZ9), the fragment 112 interacts with chain A of 3GZ9 (3GZ9:A), which is in the cluster 6994 of nr-PDB; 1 ILE (ILE249), 2 LEU (LEU255 and LEU353), 2 VALs (VAL281 and VAL348), 1 ARG (ARG284), 1 PHE (PHE352) and 1 TRP (TRP264) were identified as the interacting residues of fragment 112. If similar co-crystal complexes had limited interactive residues for a fragment, the interacting residues from those protein–ligand complexes were not considered in our analysis. For example, chain A of PDB 2ZNP (2ZNP:A) interacts with the fragment 112 of the ligand K55 (2znp_K55_1_A_922__D__), and 2ZNP:A belongs to the same cluster (6994) of protein 3GZ9:A. However, fragment 112 in the protein 2ZNP:A has fewer interacting residues than 3GZ9:A. The interacting residues of fragment 112 from 2ZNP:A were therefore not counted but interacting residues of fragment 112 from 3GZ9:A were considered when generating the fragment-residues interaction profiles.
Figure 3. An interaction profile is mapped out between the fragment 112 and 20 amino acid residues.

A: The interactive residues for fragment 112 (trifluoromethyl) in the non-redundant PDB structures are counted according to residues names, and the residue counts are then normalized by the correspondent residue frequencies in natural proteins. B. The graph plot of the interaction profile (normalized residues counts) for fragment 112. This interaction profile shows that the top five residues (MET, PHE, TRP, TYR and LEU) are the most favored five residues whereas THR, ASP, GLU, ASN and LYS are the least favored five residues for fragment 112.
An original (or non-normalized) interaction profile for fragment 112 was mapped by counting the interacting residues from these no-redundant proteins. The finial interaction profile was then generated by normalizing the original profile with background residues frequencies, which demonstrates that the fragment 112 prefers MET, PHE, TRP, TYR and LEU most (Figure 3B). Moreover, the interaction profile not only revealed its main preference for those five residues but also indicated its lowest preference to be LYS (Figure 3B), which is attributed to that the fragment 112 favors more hydrophobic interaction with these residues than that with the polar residue LYS. An interesting observation is that TRP is located in a lower ranking position (15th) in the original profile, but it is located in a higher ranking position (3rd) after normalization. Such a normalization effect could be attributed to the fact that TRP appears more times near fragment 112 than it should according to its low occurrence in natural proteins.
The interaction profiles of other fragments were generated in the same way as for fragment 112. We ignored those interaction profiles where the maximum of their original residues counts are less than 10, since those profiles might have no statistical meaning. Only 315 interaction profiles remained after this filtration, and these corresponding 315 fragments were considered as the most popular fragments among the PDB ligands, including methoxy, benzene, piperidine, and pyridine, etc. The 315 fragments and their original fragment-residue interaction profiles together with the normalized profiles are listed in Part 1 and Part 2 of the supplemental material, respectively.
Residue preference of PDB ligand fragments
The ranking positions of the original residue-counts profiles reflect the residues preference in the protein binding pockets. By analyzing the 315 original (non-normalized) interaction profiles, LEU appears more often nearby most of these 315 popular fragments. Figure 4 shows that LEU occurred in the first ranking position in 89 of the 315 original profiles, and LEU never occurs at lowest ranking position in any of the 315 original profiles. This result is congruent with the publication that LEU is the most popular residue among the protein amino acids 16, and that the probability of its occurrence near the binding pocket is much higher than for any other residues. LEU, as a non-polar hydrophobic residue, has the proper size, is not too bulky compared to TRP, not too small compared to ALA, and with a flexible side chain that can exhibit London dispersion interactions and adapt to abroad range of ligands. Therefore, LEU is suitable to interact with benzene, naphthalene, indole, –F, -Cl, -Br and -NO2 fragments. In contrast, the CYS residue was found more often at the lower positions for most of these 315 original profiles. It appeared in the 20th ranking position in 56 original profiles, while it did not appear in the first ranking position in any of these 315 original profiles (Figure 4).
Figure 4. The histogram of ranking positions of LEU and CYS in the 315 original (non-normalized) fragment–residue interaction profiles.

The histogram shows that LEU is found in the first ranking position in 89 of the 315 original interaction profiles, but it is not found in the 19th or 20th ranking positions of any original profiles. The histogram also shows that CYS is found in the 20th ranking position in 56 of the 315 original interaction profiles but it is not found in the first ranking position in any original profile.
To further analyze the residues preference for each fragment, we defined the count ratio of a profile as the ratio of the highest residue count divided by the lowest residue count in the normalized profiles. A high count ratio usually means that this fragment has a high preference for certain types of residues. If a residue count in a non-normalized profile is zero, a corrected profile is generated by adding 0.5 as a pseudo-count to all the original residues counts in this profile to avoid division by zero. Such corrections were also made in other cheminformatics tools such as Molprint2D software 17. The corresponding count ratio was calculated based on the normalized corrected profile. A histogram (Figure 5) was generated from these 315 count ratios, showing that the count ratios range from 2.62 to 239.62 with a mean value of 36.78. Among these 315 fragment-residues interaction profiles, 262 profiles had count ratios larger than 10, and 197 profiles had count ratios larger than 20. Obviously, the data indicate that most of the 315 fragments had specific preferences for certain types of residues.
Figure 5. The histogram for count ratios of 315 fragment-residues interaction profiles generated from popular fragments.

The histogram shows that the count ratios of these 315 interaction profiles are in a range of a minimum of 2.62 to a maximum of 239.62 and a mean value of 36.78. 262 of these 315 profiles have the count ratios larger than 10 and 197 of these 315 profiles have the count ratios larger than 20. The count ratio represents the residues preference for each fragment and is defined as the ratio of the highest residue count divided by the lowest residue count in the normalized profile.
Potential application of the interaction profiles by case studies
We have shown how our LigFrag-RPM algorithm was used to generate the profiles of fragment-residues interactions. We further demonstrated in the studies shown below that these profiles can be used to guide the structure-based design or chemistry optimization of a lead compound by analyzing the residues close to each individual fragment of the lead based on the generated interaction profiles. Two examples of such studies are illustrated here and more are given in Part 3 of the supplemental material.
The serine protease factor Xa has a unique role in the coagulation cascade and represents an interesting target for the development of novel anticoagulants. Zbinden et al. 18 reported the identification of novel 3-aminopyrrolidine factor Xa inhibitors and their co-crystallized structures in 2009. The reported inhibitor LZI was co-crystallized with factor Xa in PDB 2VWM. In our study, this ligand was fragmentized by the ring-cleavage algorithm and then the interacting residues of individual fragments were identified by the 2-pairs-in-5Å criterion. Among the generated fragments, the fragment 124 (cyclopropane) is surrounded by ARG143, GLU147, LYS148 and GLN192 (Figure 6B). However, these four residues are located at the lower ranking positions of the interaction profile of fragment 124 (Figure 6A), 18th, 19th, 16th and 13th, respectively. Such low ranking positions calculated by our program indicate that fragment 124 is not in its favorite environment, and also suggest that the fragment 124 may not be a required functional group for factor Xa inhibition. In our further study, we found that another ligand (H22 in PDB 2VVU) without the fragment 124 still shows a 10-fold higher affinity (Ki = 8 nM) than LZI (Ki = 93 nM). This result confirmed our prediction and is consistent with our analysis of the derived fragment-residues interaction profile.
Figure 6. The fragment-residues interaction profile of fragment 124 (A) and the surrounding residues of fragment 124 in the crystal structure of PDB 2VWM (B).

The fragment 124 (cyclopropane) is surrounded by ARG143, GLU147, LYS148, and GLN192 of factor Xa in PDB 2VWM. These four residues are located at the lower ranking positions of the interaction profile of fragment 124, 18th, 19th, 16th and 13th, respectively. It is therefore not in its most favorable environment according to its fragment-residues interaction profile.
Furthermore, we have explored the established LigFrag-RMP method in the study of the selectivity of a compound for guiding the lead chemistry optimization by analyzing the fragment-residues interaction profiles of this compound. As illustrated in Figure 7, ovalicin is an inhibitor of methionine amino peptidases, and the structures of human type 1 and type 2 methionine aminopeptidases (MetAP1 and MetAP2) in complex with ovalicin were reported as PDB 2GZ5 19 and PDB 1B59 20, respectively. It is known that ovalicin binds with lower affinity to MetAP1 than to MetAP2. In 2GZ5, the methoxy group (fragment 0) is surrounded by HIS303, HIS310 and CYS301 (Figure 7B), whereas HIS is in the 5th and CYS is in the 10th ranking position of the interaction profile of fragment 0. In 1B59, the methoxy group is surrounded by ANS329, GLY330, HIS331 and HIS339 (Figure 7C), whereas ASN, GLY and HIS are in the 7th, 12th, and 5th ranking positions, respectively. As shown in Figure 7C, GLY is not a favorite residue of fragment 0 whereas ASN in MetAP2 (1B59) is more preferred by the fragment 0 than CYS in MetAp1 (2GZ5). In addition, the residue ASN has water-bridged interaction with an –OH group in the sugar ring of ovalicin (not shown in Figure 7C). The methoxy group, therefore, is in a more preferred environment in MetAP2 than in MetAP1. This observation can partly explain the binding affinity difference between the inhibitor and the two target proteins.
Figure 7. The fragment-residues interaction profile of fragment 0 (methoxy group) (A) and the surrounding residues of fragment 0 in the crystal structures of PDB 2GZ5 (B) and PDB 1B59 (C).

The structure of PDB 2GZ5 (B) shows the interactions between ovalicin and human type 1 methionine aminopeptidase, and the methoxy group (fragment 0) is surrounded by HIS303, HIS310 and CYS301 where HIS is in the 5th ranking position and CYS is in the 10th ranking position of its interaction profile. The structure of PDB 1B59 (C) shows the interactions between ovalicin and human type 2 methionine aminopeptidase, and the methoxy (fragment 0) is surrounded by HIS339, HIS331, GLY330, and ASN329 where ASN, GLY and HIS are in the 7th, 12th and 5th ranking positions of its interaction profile, respectively.
In contrast, it should also be possible to modify this ligand in PDB 2GZ5 by substituting fragment 0 (methoxy group) with other fragments that are preferred by HIS or CYS residues. The question then is which fragments should be chosen to pursue the lead optimization chemistry? By analyzing the profiles of the 315 most popular fragments identified above, a few fragments that have HIS in the top ranking positions of their profiles are easily found. Table 1 summarized the structures of six identified putative fragments that have favored interactions with HIS, and the corresponding interactive profiles of these fragments. Accordingly, we believe that the established algorithm can be used as an alternative strategy to select optimal fragment or substituent for the lead chemistry modification. Of course, other factors such as fragment size and orientation should also be considered carefully when replacing a fragment. It also should be mentioned that as the positioning of a new fragment can be influenced by where and how its predecessors are placed, this kind of fragment replacement may not always be successful. However, our studies did provide options of introducing various fragments for lead modification of an active molecule in order to enhance the bioactivity.
Table 1.
The interaction profiles of six fragments with HIS in the top ranking positions.
| Fragment ID |
135 | 180 | 246 | 260 | 369 | 402 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Structure |  |
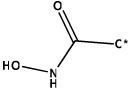 |
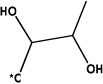 |
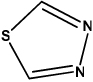 |
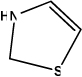 |
|||||||
| HIS | 1434 | HIS | 1173 | HIS | 2913 | TRP | 846 | HIS | 478 | HIS | 304 | |
| TRP | 923 | TRP | 153 | ARG | 903 | HIS | 521 | THR | 214 | MET | 217 | |
| THR | 321 | TYR | 151 | GLY | 513 | TYR | 272 | GLN | 125 | TYR | 151 | |
| VAL | 301 | PRO | 134 | TRP | 2692 | PHE | 238 | VAL | 111 | LEU | 134 | |
| SER | 219 | GLY | 125 | CYS | 1000 | GLN | 150 | LEU | 78 | ILE | 113 | |
| MET | 217 | GLU | 98 | GLN | 625 | GLU | 131 | TRP | 76 | GLY | 97 | |
| PHE | 214 | ARG | 96 | PHE | 571 | MET | 130 | ALA | 41 | VAL | 79 | |
| LEU | 146 | ASP | 80 | SER | 273 | ILE | 113 | GLU | 32 | TRP | 76 | |
| LYS | 125 | PHE | 71 | GLU | 311 | LEU | 112 | TYR | 30 | ASP | 60 | |
| ASN | 116 | ALA | 54 | TYR | 545 | SER | 109 | PHE | 23 | ALA | 41 | |
| ARG | 115 | GLN | 50 | ALA | 219 | ASP | 80 | ASP | 20 | CYS | 40 | |
| GLY | 97 | MET | 43 | ASP | 280 | CYS | 80 | LYS | 31 | |||
| ASP | 80 | CYS | 40 | MET | 521 | VAL | 79 | SER | 27 | |||
| PRO | 76 | LEU | 22 | LEU | 123 | ALA | 68 | PHE | 23 | |||
| GLN | 75 | ILE | 18 | ASN | 232 | PRO | 38 | THR | 17 | |||
| TYR | 60 | SER | 13 | VAL | 142 | THR | 35 | GLU | 16 | |||
| ILE | 56 | ILE | 113 | GLY | 27 | |||||||
| GLU | 49 | LYS | 93 | ASN | 23 | |||||||
| ALA | 41 | THR | 107 | ARG | 19 | |||||||
| PRO | 96 | |||||||||||
Limitations of the LigFrag-RPM
Our LigFrag-RPM method analyzes the residues close to a fragment and generated the fragment-residues interaction profile. However, the current version of the program also has some limitations that will need further improvements. For example, LigFrag-RPM still require users to manually analyze the relevant interactions, such as covalent bonds, hydrogen-bond interactions, charge-charge interactions, or hydrophobic interactions. An extreme case is that the profile of fragment 23 (Figure 8) has ASN (original count is 1,632 and the normalized count is 37,953) as its most favored residue, followed by TYR (original count is 457 and the normalized count is 21000), and ALA as the least favored residue (original count is 187, and the normalized count is 2,561), as shown in Figure 8. The count ratio for the fragment is 14.82 (i.e., 37,953/2,561=14.82). The result reveals that the fragment 23 may have specific interactions with the residue ASN. Actually, one approved drug molecule deposited in DrugBank 21, N-acetyl-D-glucosamine (NAG, DrugBank ID DB00141), has exactly this fragment. NAG is co-crystallized with its target protein SPP-40 (PDB ID 1XHG, Figure 8). The N atom of fragment 23 is 3.01 Å apart from the N atom of the amide of ASN39, because the C1 atom in the NAG ring is covalently bound to the N atom of amide in ASN39 (the distance is 1.42 Å). Thus, caution is needed when analyzing the residue preferences by the interaction profiles because the adjacent fragments often influence each other.
Figure 8. The fragment-residues interaction profiles of fragment 23 (A) and the fragment 23 in N-acetyl-D-glucosamine from PDB 1XHG (B).

A: The interaction profiles demonstrated that ASN is the most favorable residue and CYS is the least favorable residue for fragment 23. B: In PDB 1XHG, N-acetyl-D-glucosamine is co-crystallized with its target protein SPP-40. The N atom of fragment 23 is 3.01 Å from the N atom of amide of ASN39 because the C1 of NAG ring is covalently bound to the N atom of the amide in ASN39 (The distance is 1.42 Å).
In addition, the interactions can occur between residues and a molecular fragment composed of part of a ring+non-ring. Although we counted the residues that interact with two or more fragments simultaneously, other fragmentation algorithms should be considered for the analysis of the interaction between residues and ring+no-ring fragments. We would like to point it out that crystallization conditions can also influence the interaction between the fragments and the residues, but such information cannot be easily incorporated in our statistical analysis. For example, amino acid charges could be different and interactions involved would change significantly upon various pH conditions for crystallization. Fortunately, a study conducted by Kantardjieff and Rupp22 reported that “the frequency distribution for the reported crystallization pH of proteins is unimodal, with mean = 6.7, median = 6.9”. Their results show that most of the reported proteins were crystallized at nearly neutral (or physiological) pH values. Their result also shows that most of proteins were crystallized at nearly neutral pH values. However, a caution about pH influence still needs to be taken when using these profiles for individual structure analysis. Compared to traditional energy-based protein-ligand interaction analysis, the interaction profiles generated by LigFrag-RPM with statistical method are only qualitative and can be an alternative for data-mining protein-ligand interactions. It is obvious that not all profiles of the fragments in a known binding affinity complex can be acquired because some of these fragments are very rare in PDB structures. It thus is challenging to quantitatively link the contribution of each residue-fragment to the binding free energy for the current approach. However, as one can see from the case studies, the interaction profile directly links the fragment and its preferred residues. It therefore can be used as an alternative data-mining tool to guide fragment-based drug design.
Conclusions
We have presented a novel algorithm, LigFrag-RPM, for fragment-residues interaction analysis, and also validated it through the illustrated case studies. This algorithm fragmentizes the ligands in PDB and then counts the interactive residues to generate the fragment-residues interaction profiles. Our results indicate that most fragments have specific preferences for certain types of residues. We also demonstrated that the interaction profiles generated by the LigFrag-RPM algorithm can be used to identify whether a molecular fragment or functional group is in its favored or a rather unfavorable environment, and potentially to guide lead modification by replacing the residue-mismatched fragment with other more residue-matched fragments. The profile of each fragment derived from multiple protein-ligand complexes can help to estimate the probability of each amino acid residue around a fragment and thus may also be used potentially to guide site-directed mutagenesis studies of the protein binding pocket.
The current LigFrag-RPM algorithm is still at an evolutionary stage. The fragments from which we generated the LigFrag-RPM profiles represent only a small number of all possible fragments. For example, Lameijer et al. 11 reported their studies of the fragmentation of 250,251 compounds in the NCI library using a ring-cleavage algorithm yielding 65,612 fragments, which represents a much larger collection than those from the PDB. With increasingly available protein-ligand complexes in PDB, the diversity and number of fragments will help us to refine our algorithm. Furthermore, we are also exploring another potential application to use the developed profiles as a primary filtering process in our docking and virtual screening studies to remove ineligible compounds of a target protein, which will be reported elsewhere.
Supplementary Material
Acknowledgements
We acknowledge Dr. Lei Wang and Dr. Tom Jones at Tripos Company for technical support. We would like to thank Dr. Herbert Barry III for reading the manuscript. This project is supported by grants from NIH (NIGMS P50-GM067082 and R01 DA025612).
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
The research project was conducted by LR Wang. ZJ Xie and P Wipf participated and helped to draft the manuscript. XQ (Sean) Xie is the principal investigator. All authors read and approved the final manuscript.
Supporting Information
The Supporting Information is divided in three parts. The structures of the 315 fragments are given in Part 1. The fragment-residue interaction profiles of these 315 fragments are listed in Part 2. Part 3 provides four additional examples of PDB fragment-residue interaction analysis. This information is available free of charge via the Internet at http://pubs.acs.org.
Contributor Information
Lirong Wang, Email: liw30@pitt.edu.
Zhaojun Xie, Email: zhx10@pitt.edu.
Peter Wipf, Email: pwipf@pitt.edu.
Xiang-Qun Xie, Email: xix15@pitt.edu.
References
- 1.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28(1):235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wallace AC, Laskowski RA, Thornton JM. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995;8(2):127–134. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- 3.Verdonk ML, Cole JC, Taylor R. SuperStar: a knowledge-based approach for identifying interaction sites in proteins. J. Mol. Biol. 1999;289(4):1093–1108. doi: 10.1006/jmbi.1999.2809. [DOI] [PubMed] [Google Scholar]
- 4.Hendlich M, Bergner A, Gunther J, Klebe G. Relibase: design and development of a database for comprehensive analysis of protein-ligand interactions. J. Mol. Biol. 2003;326(2):607–620. doi: 10.1016/s0022-2836(02)01408-0. [DOI] [PubMed] [Google Scholar]
- 5.Deng Z, Chuaqui C, Singh J. Structural interaction fingerprint (SIFt): a novel method for analyzing three-dimensional protein-ligand binding interactions. J. Med. Chem. 2004;47(2):337–344. doi: 10.1021/jm030331x. [DOI] [PubMed] [Google Scholar]
- 6.Hajduk PJ, Greer J. A decade of fragment-based drug design: strategic advances and lessons learned. Nat. Rev. Drug Discovery. 2007;6(3):211–219. doi: 10.1038/nrd2220. [DOI] [PubMed] [Google Scholar]
- 7.Shuker SB, Hajduk PJ, Meadows RP, Fesik SW. Discovering high-affinity ligands for proteins: SAR by NMR. Science. 1996;274(5292):1531–1534. doi: 10.1126/science.274.5292.1531. [DOI] [PubMed] [Google Scholar]
- 8.Nienaber VL, Richardson PL, Klighofer V, Bouska JJ, Giranda VL, Greer J. Discovering novel ligands for macromolecules using X-ray crystallographic screening. Nat. Biotechnol. 2000;18(10):1105–1108. doi: 10.1038/80319. [DOI] [PubMed] [Google Scholar]
- 9.Gunther J, Bergner A, Hendlich M, Klebe G. Utilising structural knowledge in drug design strategies: applications using Relibase. J. Mol. Biol. 2003;326(2):621–636. doi: 10.1016/s0022-2836(02)01409-2. [DOI] [PubMed] [Google Scholar]
- 10.Feng Z, Chen L, Maddula H, Akcan O, Oughtred R, Berman HM, Westbrook J. Ligand Depot: a data warehouse for ligands bound to macromolecules. Bioinformatics. 2004;20(13):2153–2155. doi: 10.1093/bioinformatics/bth214. [DOI] [PubMed] [Google Scholar]
- 11.Lameijer EW, Kok JN, Back T, Ijzerman AP. Mining a chemical database for fragment co-occurrence: discovery of "chemical cliches". J. Chem. Inf. Model. 2006;46(2):553–562. doi: 10.1021/ci050370c. [DOI] [PubMed] [Google Scholar]
- 12.(a) Steinbeck C, Han Y, Kuhn S, Horlacher O, Luttmann E, Willighagen E. The Chemistry Development Kit (CDK): an open-source Java library for Chemo- and Bioinformatics. J. Chem. Inf. Comput. Sci. 2003;43(2):493–500. doi: 10.1021/ci025584y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Steinbeck C, Hoppe C, Kuhn S, Floris M, Guha R, Willighagen EL. Recent developments of the chemistry development kit (CDK) - an open-source java library for chemo- and bioinformatics. Curr. Pharm. Des. 2006;12(17):2111–2120. doi: 10.2174/138161206777585274. [DOI] [PubMed] [Google Scholar]
- 13.Marvin5.0.2.1. [accessed Dec 7, 2010];ChemAxon. ( http://www.chemaxon.com.
- 14.Halabi N, Rivoire O, Leibler S, Ranganathan R. Protein sectors: evolutionary units of three-dimensional structure. Cell. 2009;138(4):774–786. doi: 10.1016/j.cell.2009.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lockless SW, Ranganathan R. Evolutionarily conserved pathways of energetic connectivity in protein families. Science. 1999;286(5438):295. doi: 10.1126/science.286.5438.295. [DOI] [PubMed] [Google Scholar]
- 16.Carlson HA, Smith RD, Khazanov NA, Kirchhoff PD, Dunbar JB, Jr, Benson ML. Differences between high-and low-affinity complexes of enzymes and nonenzymes. J. Med. Chem. 2008;51(20):6432. doi: 10.1021/jm8006504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bender A, Mussa HY, Glen RC, Reiling S. Molecular similarity searching using atom environments, information-based feature selection, and a naive bayesian classifier. J. Chem. Inf. Comput. Sci. 2004;44(1):170–178. doi: 10.1021/ci034207y. [DOI] [PubMed] [Google Scholar]
- 18.Zbinden KG, Anselm L, Banner DW, Benz J, Blasco F, Decoret G, Himber J, Kuhn B, Panday N, Ricklin F, Risch P, Schlatter D, Stahl M, Thomi S, Unger R, Haap W. Design of novel aminopyrrolidine factor Xa inhibitors from a screening hit. Eur. J. Med. Chem. 2009;44(7):2787–2795. doi: 10.1016/j.ejmech.2008.12.025. [DOI] [PubMed] [Google Scholar]
- 19.Addlagatta A, Matthews BW. Structure of the angiogenesis inhibitor ovalicin bound to its noncognate target, human Type 1 methionine aminopeptidase. Protein Sci. 2006;15(8):1842–1848. doi: 10.1110/ps.062278006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu S, Widom J, Kemp CW, Crews CM, Clardy J. Structure of human methionine aminopeptidase-2 complexed with fumagillin. Science. 1998;282(5392):1324. doi: 10.1126/science.282.5392.1324. [DOI] [PubMed] [Google Scholar]
- 21.(a) Wishart DS, Knox C, Guo AC, Cheng D, Shrivastava S, Tzur D, Gautam B, Hassanali M. DrugBank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2008;36:D901–D906. doi: 10.1093/nar/gkm958. (Database issue) [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wishart DS, Knox C, Guo AC, Shrivastava S, Hassanali M, Stothard P, Chang Z, Woolsey J. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006;34:D668–D672. doi: 10.1093/nar/gkj067. (Database issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kantardjieff KA, Rupp B. Protein isoelectric point as a predictor for increased crystallization screening efficiency. Bioinformatics. 2004;20(14):2162. doi: 10.1093/bioinformatics/bth066. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.