

Abstract
The design and synthesis of a novel series of c-jun N-terminal kinase (JNK) inhibitors is described. The development of the 4-(pyrazol-3-yl)-pyridine series was discovered from an earlier pyrimidine series of JNK inhibitors. Through the optimization of the scaffold 2, several potent compounds with good in vivo profiles were discovered.
Protein kinases catalyse the phosphorylation of tyrosine and serine/threonine residues in proteins involved in the regulation of diverse cellular functions. Aberrant kinase activity is implicated in many diseases, which makes the inhibition of kinases an attractive target for the pharmaceutical industry [1]. As a member of the mitogen-activated protein kinase (MAPK) family, the c-Jun N-terminal kinases (JNKs) regulate the serine/threonine phosphorylation of several transcription factors when they are activated in response of a variety of stress-based stimuli such as cytokines, ultraviolet irradiation, heat shock, fatty acids and osmotic shock [2, 3]. Three distinct genes (jnk1, jnk2 and jnk3) encoding the ten splice variants of JNK (JNK1α1, JNK1β1, JNK1α2, JNK1β2; JNK2α1, JNK2β1, JNK2α2, JNK2β2; JNK3α1, JNK3α2) have been identified [4]. These isoforms differ in their tissue distribution profile and functions, with JNK1 and JNK2 being ubiquitously expressed, whereas JNK3 is expressed predominantly in the brain and at lower levels in the heart and testis [5, 6].
In recent studies, JNK-1, often in concert with JNK-2, has been suggested to play a central role in the development of obesity-induced insulin resistance which implies therapeutic inhibition of JNK1 may provide a potential solution in type-2 diabetes mellitus [7, 8]. JNK2 has been described in the pathology of autoimmune disorders such as rheumatoid arthritis and asthma, and it also has been implicated to play a role in cancer, as well as in a broad range of diseases with an inflammatory component [9–13]. JNK3 has been shown to play important roles in the brain to mediate neurodegeneration, such as beta amyloid processing, Tau phosphorylation and neuronal apoptosis in Alzheimer’s disease, as well as the mediation of neurotoxicity in a rodent model of Parkinson’s disease [14–16]. JNK3 is almost exclusively found in the brain. Identifying potent inhibitors of JNK3, with selectivity within the wider MAPK family (one of which is p38), may contribute towards neuroprotection therapies with reduced side effect profiles.
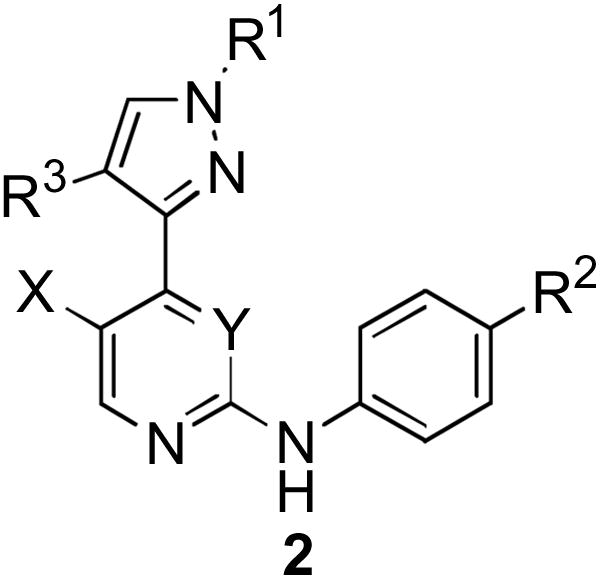
Therefore, developing JNK inhibitors as therapeutics has gained considerable interest over the past few years [17–32]. As part of our medicinal chemistry research program, we initiated a JNK3 project with a key objective of identifying brain penetrant compounds with good JNK3 potency and selectivity over p38. We previously reported on the synthesis and SAR of 4-phenyl substituted pyrimidines [31]. Compounds in this class had good in vitro potency, but only modest in vivo profiles (rodent pk and brain penetration). In the continuous development and optimization of JNK3 inhibitors, we found 4-pyrazole substituted pyrimidines were also potent inhibitors [33]. The first synthesized 4-(pyrazol-3-yl)-pyrimidine, compound 1, inhibited JNK3 with an IC50 = 0.63 μM with no detectable inhibition of p38 (> 20 μM). Encouraged by the JNK3 potency and selectivity against p38, we initiated a structure-activity relationships (SAR) study [34]. Our strategy was based on scaffold 2 and more precisely on the maintenance of the core structure of 1 wherein we could vary either X, Y, R1, R2 and R3 groups by modification or introduction of substituents.
The analogues 7–9 were synthesized from compound 3 or 5 as described in Scheme 1 and data is shown in Table 1 [35]. The 2- and 4-positions of pyrimidine 3 were subsequently replaced with a substituted aniline and a chlorine atom to afford intermediate 4. The pyrazole ring was introduced via a Suzuki coupling to give compounds 7 with R1 = H. N-alkylation with alkyl halides gave 7 with R1 = alkyl. The analogues 8 were synthesized from compound 5 in 5 steps. Stille coupling of trichloropyrimidine 5 followed by hydrolysis afforded ketone 6. Introduction of the aniline followed by pyrazole ring formation via a two-step procedure gave compounds 8–9.
Scheme 1.

Reagents: a) R2PhNH2, EtO(CH2)2OH, 75°C, 16h; b) conc. HCl, 90°C, overnight; POCl3, 100°C; c) 3-pyrazoleboronic acid, Pd(PPh3)4, K2CO3, DME/H2O, 140°C, 1h, microwave; d) MeI, K2CO3, DMF, 50°C, overnight; e) Tributyl(1-ethoxyvinyl)tin, Pd(PPh3)4, toluene, 125°C, 1.5h, microwave; f) conc. HCl, THF, 2h, rt; g) R2PhNH2, EtO(CH2)2OH/H2O, 120°C, 15h; h) N,N-dimethylformamide dimethylacetal, 110°C, overnight; i) MeNHNH2, EtOH, rt, 55h.
Table 1.
JNK3 inhibition by analogues of 7–9.
| Compds | X | R1 | R2 | JNK3 IC50 (μM)a | p38 IC50 (μM)a |
|---|---|---|---|---|---|
| 1 | H | H | A | 0.63 | > 20 |
| 7 | H | Me | A | 1.45 | nt |
| 8 | Cl | Me | A | 0.86 | nt |
| 9 | Cl | Me | B | 0.73 | nt |
Values are means of three experiments, nt = not tested, All standard deviation ≤ 20%
Unfortunately, compounds containing pyrrazoles bearing free NH’s had poor brain penetration. To decrease the polarity of our lead compounds, and hopefully improve brain penetration, the nitrogen atom of the pyrazole ring was alkylated with a methyl group. This led to a decrease in JNK3 inhibition to 1.45μM for 7 (Table 1). The introduction of a 5-chloro group on the pyrimidine ring recovered some of the potency (8) lost by N-alkylation. We also found that modification of the R2 substituent did little in the way of improving inhibition (ie: 9), but did play a role in modulating in vivo parameters.
In the literature, kinase inhibitors with a central pyrimidine core are ubiquitous whereas those containing pyridine cores are far less common [33, 36–41]. With intermediates in hand, we decided to investigate if 4-pyrazole substituted pyridines had any activity against JNK3. Additionally, removing one heteroatom from the pyrimidine core would further reduce polarity. Pyridine analogues were synthesized as described in Scheme 2.
Scheme 2.

Reagents: a) 3-pyrazoleboronic acid, Pd(PPh3)4, K2CO3, DMF, 100°C, 3h; b) MeI, K2CO3, DMF, 50°C, overnight; or Ar1X, CuI, K2CO3, trans-N,N′-dimethylcyclohexane-1,2-diamine, dioxane, 120°C, overnight; c) R2PhNH2, Pd(OAc)2, xantphos, Cs2CO3, dioxane, 130°C, 1h, microwave.
The halogenated pyridine 10 was coupled with pyrazole boronic acid to give 11. Anilines were introduced into the 2-position via Buchwald-Hartwig conditions to afford compounds 12–13 with a hydrogen atom as the R1 substituent [42]. Compounds 14–23 were synthesized by N-alkylation (R1 = alkyl) or by a variation of the Ullmann reaction (R1 = aryl), followed by a Buchwald-Hartwig reaction. The rapid synthesis of these compounds allowed us to evaluate a series of analogues with a variety of substituents (not all data shown). Of all R2 substituents examined, only a few (A-F) are presented here (Figure 2).
Figure 2.

R2 substituents
Surprisingly, pyridine containing compounds were more potent than their corresponding pyrimidine analogues (Table 2). Compound 12 with no substitution at the 1-position on the pyrazole ring or at the 5-position on the pyridine ring had an IC50 value 160 nM for the inhibition of JNK3 with no detectable activity against p38. The introduction of a chlorine atom at C-5 (X substituent in 13) provided a 2-fold boost in activity. Unfortunately, the N-alkylation of the pyrazole nitrogen (14) led to a slight decrease in potency which is a tradeoff as it also serves to reduce compound polarity. Once again, modification of the aniline substituent provided no advantage with regards to in vitro potency. A 5-fluoro substituent (18) was equipotent to the 5-chloro (14) substituted compound. Substitution at C-5 with a methyl group (23) led to a 4-fold drop in potency (23 versus 16 and 22). Fortunately, all analogs tested showed no inhibition against p38.
Table 2.
Inhibition of JNK3 by compounds 12–23.
| Compds | X | R1 | R2 | JNK3 IC50 (μM)a | p38 IC50 (μM)a |
|---|---|---|---|---|---|
| 12 | H | H | E | 0.16 | >20 |
| 13 | Cl | H | E | 0.07 | >20 |
| 14 | Cl | Me | E | 0.13 | >20 |
| 15 | Cl | Me | D | 0.16 | >20 |
| 16 | Cl | Me | B | 0.20 | >20 |
| 17 | Cl | Me | C | 0.60 | >20 |
| 18 | F | Me | E | 0.16 | >20 |
| 19 | F | Me | D | 0.34 | >20 |
| 20 | F | Me | C | 0.20 | >20 |
| 21 | F | Me | F | 0.48 | >20 |
| 22 | F | Me | B | 0.16 | >20 |
| 23 | Me | Me | B | 0.75 | >20 |
Values are means of three experiments, All standard deviation ≤ 20%
Surprisingly, the phenyl ring in the aniline did not tolerate substitution, as both fluoro-substituted analogues were inactive against JNK3 (> 20 μM) (Figure 3, compounds 24 and 25). Additionally, 1-methyl-5-pyrazole isomers 26–28 (JNK3 IC50 1.6 μM, 6.0 μM and 6.2 μM, respectively) were considerably less potent JNK inhibitors than their corresponding 1-methyl 3-pyrazole isomers (14 and 18).
Figure 3.

Additional pyrazole JNK inhibitors.
We then looked to optimize pyrazole substitution (R1) as a means to improve JNK3 inhibition (Table 3). Compound 29 with an N-benzyl group was slightly more potent than 18 (R1 = Me). Phenethyl substitution (30) led to a slight decrease in potency. The morpholino-ethyl group was also less active (compound 31). The introduction of substituted aromatic rings (32–35) also led to a drop in JNK potency depending on substitution.
Table 3.
Inhibition of JNK3 by compounds 29–35.
| Compds | X | R1 | R2 | JNK3 IC50 (μM)a | p38 IC50 (μM)a |
|---|---|---|---|---|---|
| 29 | F | Bn | E | 0.10 | >20 |
| 30 | F | (CH2)2Ph | E | 0.17 | > 20 |
| 31 | F | 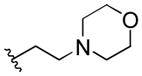 |
E | 0.32 | >20 |
| 32 | H | 3-MeOPh | D | 0.65 | > 20 |
| 33 | H | 3-CNPh | D | 0.24 | >20 |
| 34 | H | 4-MeSPh | D | 1.33 | >20 |
| 35 | H | 2-Py | D | 0.34 | >20 |
Values are means of three experiments, All standard deviation ≤ 20%
We next investigated substitution at the 4-position on the pyrazole ring (Table 4, R3). To facilitate SAR of the R3 substituent, we chose to use an N-methyl as the R1 substituent, and dimethyltriazole or morpholine (respectively noted D and E) as the R2 substituent.
Table 4.
4-substituted pyrazole SAR
| Compds | R2 | R3 | JNK3 IC50 (μM)a | p38 IC50 (μM)a |
|---|---|---|---|---|
| 37 | D | 4-FPh | 0.007 | 0.02 |
| 38 | D | 3-Py | 0.11 | 1.14 |
| 39 | E | 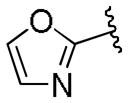 |
0.15 | 0.19 |
| 40 | E | 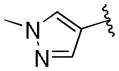 |
0.39 | > 20 |
| 41 | E | 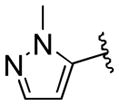 |
0.39 | > 20 |
| 42 | D | n-Pr | 1.40 | > 20 |
| 43 | D | Me | 0.40 | > 20 |
Values are means of three experiments, All standard deviation ≤ 20%
The synthesis of analogues (37–43) is outlined in Scheme 2. Compound 36 was obtained in two steps via a Suzuki coupling followed by N-alkylation. The regioselective bromination of the pyrazole ring at the 4-position, followed by another Suzuki coupling allowed for the introduction of R3-substituents [43]. The last step was the introduction of the substituted aniline under acidic conditions which provided the desired compounds.
Interestingly, 4-fluorophenyl substitution (37) resulted in a big boost in JNK3 potency concomitant with the loss of the selectivity between JNK3 and p38. This was true for several other phenyl substituted analogues (not shown). Heterocyclic rings did not afford the magnitude of potency enhancement that came with phenyl or substituted phenyl rings (38–41). However, it was interesting that pyrazole substitution, and smaller alkyl groups did not pick up p38 activity, although JNK activity also decreased.
Several compounds from this series were analyzed in vivo (Table 5) [44]. The compounds with morpholine as a R2 substituent (E) suffer from poor pharmacokinetics (high clearance, short half-life and low bioavailability). For the other R2 substituents, the compounds had a much improved in vivo profile (low clearance, good half-life and very good bioavailability). For most compounds tested in this series, the blood brain barrier penetration was also quite good (30–100%).
Table 5.
Key brain penetration and pharmacokinetic parameters.
| Cmpds | Drug concentration (μM)a Plasma/brain | Clp (mL/min/kg)b | t½b | Oral AUC (μM.h)b | %Fb |
|---|---|---|---|---|---|
| 15 | 10.4/5.3 | 9.0 | 1.3 | 7.1 | 73 |
| 16 | 6.6/3.5 | 0.9 | 6.5 | 45 | 75 |
| 20 | 9.0/10.3 | 4.2 | 2.3 | 10 | 69 |
| 21 | 3.3/0.74 | 4.0 | 3.4 | 14.9 | 73 |
| 37 | 9.0/1.4 | 6.4 | 1.5 | 6 | 51 |
mouse; 10mg/kg IP, 2h
rat; 1mg/kg IV; 2mg/kg PO
In summary, a novel series of pyrazole substituted pyrazoles has been developed. Optimization of this scaffold by modification of five substituents (X, Y R1, R2 and R3) afforded a series of potent JNK3 inhibitors free of p38 inhibition. Several compounds from this class have good pharmacokinetics and brain penetration. Evaluation of compounds of this type in vivo in CNS models of human disease (Parkinson’s and Alzheimer’s disease, stroke) is ongoing and will be reported in due course.
Figure 1.
Pyrazole pyrimidine JNK inhibitors
Scheme 3.

Reagents: a) 3-pyrazoleboronic acid, Pd(PPh3)4, K2CO3, DMF, 100°C, 3h; b) MeI, K2CO3, DMF, 50°C, overnight; c) NBS, DMF, 50°C, 4h; d) R3B(OH)2, Pd(PPh3)4, K2CO3, DME/H2O, 100°C, 1h, microwave; e) R2PhNH2, HCl(1M)/H2O/dioxane (ratio 2/4/1).
References and notes
- 1.Parang K, Sung GQ. Design strategies for protein kinase inhibitors. Current Opinion in Drug Discovery & Development. 2004;7(5):617–629. [PubMed] [Google Scholar]
- 2.Barr RK, Bogoyevitch MA. The c-Jun N-terminal protein kinase family of mitogen-activated protein kinases (JNK MAPKs) International Journal of Biochemistry & Cell Biology. 2001;33(11):1047–1063. doi: 10.1016/s1357-2725(01)00093-0. [DOI] [PubMed] [Google Scholar]
- 3.Weston CR, Davis RJ. The JNK signal transduction pathway. Current Opinion in Cell Biology. 2007;19(2):142–149. doi: 10.1016/j.ceb.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 4.Gupta S, et al. Selective interaction of JNK protein kinase isoforms with transcription factors. Embo Journal. 1996;15(11):2760–2770. [PMC free article] [PubMed] [Google Scholar]
- 5.Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiological Reviews. 2001;81(2):807–869. doi: 10.1152/physrev.2001.81.2.807. [DOI] [PubMed] [Google Scholar]
- 6.Morrison DK, Davis RJ. Regulation of map kinase signaling modules by scaffold proteins in mammals. Annual Review of Cell and Developmental Biology. 2003;19:91–118. doi: 10.1146/annurev.cellbio.19.111401.091942. [DOI] [PubMed] [Google Scholar]
- 7.Hirosumi J, et al. A central role for JNK in obesity and insulin resistance. Nature. 2002;420(6913):333–336. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- 8.Bennett BL, Satoh Y, Lewis AJ. JNK: a new therapeutic target for diabetes. Current Opinion in Pharmacology. 2003;3(4):420–425. doi: 10.1016/s1471-4892(03)00068-7. [DOI] [PubMed] [Google Scholar]
- 9.Han ZN, et al. Joint damage and inflammation in c-Jun N-terminal kinase 2 knockout mice with passive murine collagen-induced arthritis. Arthritis and Rheumatism. 2002;46(3):818–823. doi: 10.1002/art.10104. [DOI] [PubMed] [Google Scholar]
- 10.Wong WSF. Inhibitors of the tyrosine kinase signaling cascade for asthma. Current Opinion in Pharmacology. 2005;5(3):264–271. doi: 10.1016/j.coph.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 11.Pelaia G, et al. Mitogen-activated protein kinases and asthma. Journal of Cellular Physiology. 2005;202(3):642–653. doi: 10.1002/jcp.20169. [DOI] [PubMed] [Google Scholar]
- 12.Blease K, Lewis A, Raymon HK. Emerging treatments for asthma. Expert Opin Emerg Drugs. 2003;8(1):71–81. doi: 10.1517/14728214.8.1.71. [DOI] [PubMed] [Google Scholar]
- 13.Zhang H, et al. Nocodazole-induced p53-dependent c-Jun N-terminal kinase activation reduces apoptosis in human colon carcinoma HCT116 cells. Journal of Biological Chemistry. 2002;277(46):43648–58. doi: 10.1074/jbc.M203214200. [DOI] [PubMed] [Google Scholar]
- 14.Resnick L, Fennell M. Targeting JNK3 for the treatment of neurodegenerative disorders. Drug Discov Today. 2004;9(21):932–9. doi: 10.1016/S1359-6446(04)03251-9. [DOI] [PubMed] [Google Scholar]
- 15.Kimberly WT, et al. Physiological regulation of the beta-amyloid precursor protein signaling domain by c-Jun N-terminal kinase JNK3 during neuronal differentiation. J Neurosci. 2005;25(23):5533–43. doi: 10.1523/JNEUROSCI.4883-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pan J, et al. K252a prevents nigral dopaminergic cell death induced by 6-hydroxydopamine through inhibition of both mixed-lineage kinase 3/c-Jun NH2-terminal kinase 3 (JNK3) and apoptosis-inducing kinase 1/JNK3 signaling pathways. Mol Pharmacol. 2007;72(6):1607–18. doi: 10.1124/mol.107.038463. [DOI] [PubMed] [Google Scholar]
- 17.Bennett BL, et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci U S A. 2001;98(24):13681–6. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ruckle T, et al. Design, synthesis, and biological activity of novel, potent, and selective (benzoylaminomethyl)thiophene sulfonamide inhibitors of c-Jun-N-terminal kinase. Journal of Medicinal Chemistry. 2004;47(27):6921–34. doi: 10.1021/jm031112e. [DOI] [PubMed] [Google Scholar]
- 19.Graczyk PP, et al. The neuroprotective action of JNK3 inhibitors based on the 6,7-dihydro-5H-pyrrolo[1,2-a]imidazole scaffold. Bioorganic & Medicinal Chemistry Letters. 2005;15(21):4666–70. doi: 10.1016/j.bmcl.2005.07.076. [DOI] [PubMed] [Google Scholar]
- 20.Gaillard P, et al. Design and synthesis of the first generation of novel potent, selective, and in vivo active (benzothiazol-2-yl)acetonitrile inhibitors of the c-Jun N-terminal kinase. Journal of Medicinal Chemistry. 2005;48(14):4596–607. doi: 10.1021/jm0310986. [DOI] [PubMed] [Google Scholar]
- 21.Swahn BM, et al. Design and synthesis of 6-anilinoindazoles as selective inhibitors of c-Jun N-terminal kinase-3. Bioorganic & Medicinal Chemistry Letters. 2005;15(22):5095–9. doi: 10.1016/j.bmcl.2005.06.083. [DOI] [PubMed] [Google Scholar]
- 22.Stocks MJ, et al. Structure-driven HtL: design and synthesis of novel aminoindazole inhibitors of c-Jun N-terminal kinase activity. Bioorganic & Medicinal Chemistry Letters. 2005;15(14):3459–62. doi: 10.1016/j.bmcl.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 23.Swahn BM, et al. Design and synthesis of 2′-anilino-4,4′-bipyridines as selective inhibitors of c-Jun N-terminal kinase-3. Bioorganic & Medicinal Chemistry Letters. 2006;16(5):1397–401. doi: 10.1016/j.bmcl.2005.11.039. [DOI] [PubMed] [Google Scholar]
- 24.Angell RM, et al. N-(3-Cyano-4,5,6,7-tetrahydro-1-benzothien-2-yl)amides as potent, selective, inhibitors of JNK2 and JNK3. Bioorganic & Medicinal Chemistry Letters. 2007;17(5):1296–301. doi: 10.1016/j.bmcl.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 25.Alam M, et al. Synthesis and SAR of aminopyrimidines as novel c-Jun N-terminal kinase (JNK) inhibitors. Bioorganic & Medicinal Chemistry Letters. 2007;17(12):3463–7. doi: 10.1016/j.bmcl.2007.03.078. [DOI] [PubMed] [Google Scholar]
- 26.Christopher JA, et al. 1-Aryl-3,4-dihydroisoquinoline inhibitors of JNK3. Bioorganic & Medicinal Chemistry Letters. 2009;19(8):2230–4. doi: 10.1016/j.bmcl.2009.02.098. [DOI] [PubMed] [Google Scholar]
- 27.Cao J, et al. Structure-based design and parallel synthesis of N-benzyl isatin oximes as JNK3 MAP kinase inhibitors. Bioorganic & Medicinal Chemistry Letters. 2009;19(10):2891–5. doi: 10.1016/j.bmcl.2009.03.043. [DOI] [PubMed] [Google Scholar]
- 28.Jiang R, et al. 3,5-Disubstituted quinolines as novel c-Jun N-terminal kinase inhibitors. Bioorganic & Medicinal Chemistry Letters. 2007;17(22):6378–82. doi: 10.1016/j.bmcl.2007.08.054. [DOI] [PubMed] [Google Scholar]
- 29.Shin Y, et al. Synthesis and SAR of piperazine amides as novel c-jun N-terminal kinase (JNK) inhibitors. Bioorganic & Medicinal Chemistry Letters. 2009;19(12):3344–7. doi: 10.1016/j.bmcl.2009.03.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kamenecka T, et al. Structure-activity relationships and X-ray structures describing the selectivity of aminopyrazole inhibitors for c-Jun N-terminal kinase 3 (JNK3) over p38. Journal of Biological Chemistry. 2009;284(19):12853–61. doi: 10.1074/jbc.M809430200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kamenecka T, et al. Synthesis, biological evaluation, X-ray structure, and pharmacokinetics of aminopyrimidine c-jun-N-terminal kinase (JNK) inhibitors. Journal of Medicinal Chemistry. 2010;53(1):419–31. doi: 10.1021/jm901351f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.LoGrasso P, Kamenecka T. Inhibitors of c-jun-N-terminal kinase (JNK) Mini Rev Med Chem. 2008;8(8):755–66. doi: 10.2174/138955708784912120. [DOI] [PubMed] [Google Scholar]
- 33.0246184. Vertex Pharmaceuticals Incorporated; WO.
- 34.There is good selectivity when compounds show more than 100-fold selectivity for JNK3 over P38.
- 35.Biochemical Assays Measuring JNK Activity: Biochemical IC50s for JNK3 and p38 were determined using HTRF. Briefly, final assay concentrations of JNK3, biotinylated-ATF2 and ATP were 0.3nM, 0.4nM, and 1μM, respectively. Final assay concentrations of p38, biotinylated-ATF2, and ATP were 1nM, 0.4nM, and 11.5μM, respectively. In both assays, the phosphor- Thr71-ATF-2 product was detected by a Europiumcryptate-labeled anti-phospho-Thr71-ATF-2 antibody. Streptavidin-allophycocyanin -XL was used as the acceptor. A 10-point dose-response curve for each compound was generated in duplicate and data were fit to a four parameter logistic.
- 36.Burns CJ, et al. Phenylaminopyrimidines as inhibitors of Janus kinases (JAKs) Bioorganic & Medicinal Chemistry Letters. 2009;19(20):5887–92. doi: 10.1016/j.bmcl.2009.08.071. [DOI] [PubMed] [Google Scholar]
- 37.Aliagas-Martin I, et al. A class of 2,4-bisanilinopyrimidine Aurora A inhibitors with unusually high selectivity against Aurora B. Journal of Medicinal Chemistry. 2009;52(10):3300–7. doi: 10.1021/jm9000314. [DOI] [PubMed] [Google Scholar]
- 38.2008129071. Ingenium Pharmaceuticals G.m.b.H; Germany: WO.
- 39.2008099074. Sanofi-Aventis; Fr: WO.
- 40.2004084901. Signal Pharmaceuticals, LLC; USA: WO.
- 41.Paul R, et al. Preparation of substituted N-phenyl-4-aryl-2-pyrimidinamines as mediator release inhibitors. Journal of Medicinal Chemistry. 1993;36(19):2716–25. doi: 10.1021/jm00071a002. [DOI] [PubMed] [Google Scholar]
- 42.Janey JM. Buchwald-Hartwig Amination. Name Reactions for Functional Group Transformations. 2007:564–609. [Google Scholar]
- 43.Miyaura N, Suzuki A. Palladium-Catalyzed Cross-Coupling Reactions of Organoboron Compounds. Chemical Reviews. 1995;95(7):2457–2483. [Google Scholar]
- 44.Rat Pharmacokinetics and Mouse Brain Penetration: Pharmacokinetics of test compounds was assessed in Sprague-Dawley rats (n = 3). Compounds were dosed intravenously at 1 mg/kg and orally by gavage at 2 mg/kg. Blood was taken at eight time points (5, 15, 30 min, 1, 2, 4, 6, 8 h) and collected into EDTA containing tubes and plasma was generated using standard centrifugation techniques. Plasma proteins were precipitated with acetonitrile and drug concentrations were determined by LC-MS/MS. Data was fit by WinNonLin using a noncompartmental model and basic pharmacokinetic parameters including peak plasma concentration (Cmax), oral bioavailability, exposure (AUC), half-life (t1/2), clearance (CL), and volume of distribution (Vd) were calculated. CNS exposure was evaluated in C57Bl6 mice (n = 3). Compounds were dosed at 10 mg/kg intraperitoneally and after 2 h blood and brain were collected. Plasma was generated and the samples were frozen at −80°C. The plasma and brain were mixed with acetonitrile (1:5 v:v or 1:5 w:v, respectively). The brain sample was sonicated with a probe tip sonicator to break up the tissue, and samples were analyzed for drug levels by LCMS/MS. Plasma drug levels were determined against standards made in plasma and brain levels against standards made in blank brain matrix. All procedures were approved by the Scripps Florida IACUC.