

Abstract
The copper-mediated and non-basic oxidative cross-coupling of organotrifluoroborates with phenols was applied to elaboration of the structures of thiirane-based inhibitors of matrix metalloproteinases. By revision of the synthetic sequence to allow this cross-coupling as the final step, and taking advantage of the neutral nature of organotrifluoroborate cross-coupling, a focussed series of inhibitors showing aryloxy and alkenyloxy replacement of the phenoxy substitutent was prepared. This reaction shows exceptional promise as an alternative to the classic copper-mediated but strongly basic Ullmann reaction, for the diversification of ether segments within base-labile lead structures.
Keywords: Thiirane, Ullmann reaction, Organotrifluoroborate salt, Diaryl ether synthesis, Cross coupling
There is a high demand for new synthetic methods for the preparation of diaryl ethers and alkenyl aryl ethers, owing to their importance as structural motifs in a wide range of molecules with various applications. The classical reaction for preparation of diaryl ethers is the copper-catalyzed Ullmann reaction, but this reaction is typically carried out at high temperatures in the presence of strong base, which significantly limits its use. 1 Recently, Batey and Quach have reported a novel protocol for ether formation from alcohols under essentially neutral conditions, using alkenyl and aryl trifluoroborate salts in the presence of copper.2 This method is of interest because of its mildness, which allows application to valuable and highly elaborated synthetics substrates.
In recent years, potassium organotrifluoroborates have become a promising alternative to the use of organoboronic acids. Organotrifluoroborates are crystalline solids that show exceptional stability toward oxygen and moisture, and they are easily prepared. 3 Many are now commercially available. Moreover, in many organic reactions potassium organotrifluoroborates have shown comparable and very often better yields compared to organoboronic acids. In addition, the tetracoordinated nature of these species allows their functionalization, increasing the molecular complexity of the organoboron reagent, providing a unique access to valuable organic synthons.4
We have previously described the synthesis, inhibitory activity, 5, 6 mechanism 7, 8 and metabolism studies 9, 10 with a valuable class of thiirane-containing highly selective gelatinase inhibitors, of which 1 (also known as SB-3CT) is a prototype. This inhibitor potently inhibits gelatinases (MMP-2 and MMP-9) in the nanomolar range, to the virtual exclusion of the other members of the MMP family. Compound 1 is a mechanism-based inhibitor, which operates by a distinct mechanism that gives rise to a stable complex with the gelatinases involving thiolate coordination to the active-site zinc atom. An important feature of the inhibitor is the biphenyl ether moiety that fits into the S1′ pocket of these gelatinases, and a thiirane ring that undergoes a gelatinase-catalyzed ring opening to give the stable zinc-thiol interaction of the inhibited enzyme.11
Despite its rapid metabolism in mice and other species,10 compound 1 is highly effective in animal models of several diseases. 12,13,14,15,16,17,18,19,20 This efficacy is in large measure due to the high gelatinase activity of one of its major metabolites.10 Although more potently active than compound 1, it has certain undesirable properties such as poor aqueous solubility, difficulty in formulation, and multiple routes of metabolism that all present obstacles to the progression of this compound in pharmacological evaluation.
SAR studies of variants of compound 1 carried out in our lab to date have shown that the structural template can be modified to improve solubility and metabolic stability of the compound class, concomitant with retention of selectivity and potency in inhibition of gelatinases. Furthermore, in some cases, it was possible to tweak the chemical structure to inhibit only MMP-2. Yet, in others the modifications gave rise to a widening of the spectrum of inhibited MMPs. These results reveal the richness of the structural space for the thiirane inhibitors, which is worthy of further exploration in generating libraries of structurally diverse molecules.
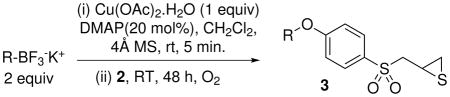
In order to aggressively develop this SAR, we have focused on the syntheses of structurally diverse libraries for biological evaluation. Here, we report our efforts toward the diversification of the phenoxy segment of 1. This ambition required a synthetic route wherein the diversification occurs at the final step of the synthesis, from a common intermediate, under conditions that are compatible with the other functionalities present in the molecule.
We have prepared compound 2 in five synthetic steps from 4-hydroxythiophenol in the context of a focused series of sulfonate derivatives of the thiirane inhibitors.21 The thiirane ring of 2 is readily opened by base, by a mechanism identical to that which occurs in matrix metalloproteinase inhibition. Consequently, 2 is incompatible as a reagent for traditional Ullmann ether formation. Indeed, in our hands even the milder Ullmann conditions given by Chan and Evans22,23 (2 equiv boronic acid, 1 equiv Cu(OAc)2, 2 equiv Et3N, molecular sieves, RT) were unsuccessful.
As a final effort to enable Ullmann cross-coupling diversification from 2 as an intermediate, we explored the use of aryl trifluoroborates for phenol O-arylation as described by Batey and Quach. The result from the initial reaction (Table 1, entry 1). was encouraging. Given the modest yield for 1, and the intrinsic complexity of this reaction—key variables include the choice of copper salt catalyst, catalyst loading, catalyst ligand, reagent stoichiometry, solvent, temperature, oxidant, and nature of the molecular sieves and its source—we explored many of these variables as a prelude to library diversification. Cu(OAc)2 was chosen as the Cu(II) source. The addition of 1,2-dimethylimidazole 24 and N-methylimidazole 25 monodentate copper ligands, as were recently used in similar copper-catalyzed cross-couplings, gave a small increase in the reaction yield. 1,2-Dimethylimidazole was the better ligand of the two. DMSO and MeCN as sole solvents, or as co-solvents (in different ratios) with dichloromethane, were compared to dichloromethane alone. None of these changes improved the yields. The use of 5 equiv (instead of 2 equiv) of the potassium phenyltrifluorborate actually reduced the yield. This outcome may be attributed to phenyltrifluorborate homocoupling.24 Furthermore, we discovered that different sources for the molecular sieves, all exhaustively activated at 200 °C under vacuum prior to use, gave different yields. These differences may be attributed to the variability of the constituents and the mixtures of ingredient minerals in different molecular sieves from different origins.
Table 1.
Optimization experiment for Cu(II)-mediated etherification of the base-labile phenol 2 to produce compound 1
 | ||||
|---|---|---|---|---|
| entry | Equiv Cu(OAc)2 | Equiv DMAP | Temperature | % yielda |
| 1 | 0.1 | 0.2 | rt | 26 (61) |
| 2 | 0.1 | 2 | rt | 0 (14) |
| 3 | 1 | 0.2 | reflux | 24 (29) |
| 4 | 1 | 0.2 | rt | 72(0) |
Isolated yield; data in parentheses are the recovered starting materials.
As is shown in Table 1, we decided to increase the amount copper using DMAP as ligand. We found that 1 equiv of Cu(OAc)2, 0.2 equiv of DMAP, 2 equiv of potassium phenyltrifluoroborate in the presence of 4-Å molecular sieves gave the best results for this funtionalization (Table 1, entry 4). As Batey and Quach observed previously, under these conditions, phenylboronic acid also undergoes cross-coupling with phenol 2, although in lower yield (with 15% of cross-coupling product and 49% of starting material recovered).
We realize that when the number of components for the reaction is many (as is required for this reaction), many different conditions could be tried. Regardless, the conditions given in Table 1, entry 4, were satisfactory and we were poised to prepare a series of analogs of compound 1 by this method. The results of these efforts are given in Table 2.
Table 2.
The Cu(II)-mediated etherification of phenol 2 with different RBF3−K+ salts
 | |||
|---|---|---|---|
| entry | trifluoroborate | product | % yielda |
| 1 |  |
 |
44 (33) |
| 2 |  |
 |
65 (0) |
| 3 |  |
 |
36 (0) |
| 4 |  |
 |
39 (0) |
| 5 |  |
 |
74 (0) |
| 6 |  |
 |
16 (27) |
| 7 |  |
 |
53 (15) |
| 8 |  |
 |
14 (57) |
| 9 |  |
 |
24 (33) |
| 10 |  |
14 (41) | |
| 11 |  |
– | 0 (12) |
| 12 |  |
– | 0 (78) |
Isolated yield; data in parentheses are the recovered starting materials.
As Batey and Quach noted, electron-rich aryl trifluoroborates give the best results (Table 2, entries 5 and 7), whereas electron-deficient aryl trifluoroborates with electron-withdrawing groups at the para position and alkyl organotrifluoroborates did not react, when treated with phenol 2 (Table 2, entries 11 and 12). However, the electron-deficient potassium 3-nitrophenyltrifluoroborate (Table 2, entry 4), with an electron-withdrawing group at the meta position, furnished the desired product in a moderate yield. With the exception of potassium trans-styryltrifluoroborate (Table 2, entry 2), alkenyl and heterocyclic trifluoroborates were modest cross-coupling partners (Table 2, entries 3 and 6).
The mechanism of this copper-mediated oxidative coupling with organotrifluoroborates is believed to go through a CuIII species. Stahl and co-workers 26 have suggested that after the initial transmetallation, the resulting aryl-CuII species is oxidized by another equivalent of CuII to yield an aryl-CuIII intermediate, which undergoes facile C-O reductive elimination.
Notwithstanding the modesty in yields, the procedure employed in the present study allows for a single-step rapid assembly of a series of analogs of 1 with variations in the ether moiety in the last step on a structurally sensitive precursor. We have expanded the mild and essentially neutral conditions developed by Batey and Quach for the syntheses of alkenyl-aryl and aryl-aryl ethers. The reaction was successfully applied to the base-labile thiirane 2 to generate a series of analogs of gelatinase inhibitor 1.
Supplementary Material
Figure 1.

SB-3CT (1), a potent and selective gelatinase inhibitor and intermediate base-labile phenol 2.
Acknowledgments
This research was supported by a grant from the National Institutes of Health (CA122417). The Mass Spectrometry & Proteomics Facility at the University of Notre Dame is supported by a grant from the National Science Foundation, (CHE-0741793).
Footnotes
Supplementary data associated with this article can be found, in the online version.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Ley SV, Thomas AW. Angew Chem Int Ed. 2003;42:5400. doi: 10.1002/anie.200300594. [DOI] [PubMed] [Google Scholar]
- 2.Quach TD, Batey RA. Org Lett. 2003;5:1381. doi: 10.1021/ol034454n. [DOI] [PubMed] [Google Scholar]
- 3.For reviews see: Molander GA, Figueroa R. Aldrichimica Acta. 2005;38:49.Stefani HA, Cella R, Vieira AS. Tetrahedron. 2007;63:3623.Molander GA, Ellis N. Acc Chem Res. 2007;40:275. doi: 10.1021/ar050199q.Darses S, Genet JP. Chem Rev. 2008;108:288. doi: 10.1021/cr0509758.
- 4.Molander GA, Cooper DJ. J Org Chem. 2008;73:3885. doi: 10.1021/jo800383e. and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown S, Bernardo MM, Li ZH, Kotra LP, Tanaka Y, Fridman R, Mobashery S. J Am Chem Soc. 2000;122:6799. [Google Scholar]
- 6.Lee M, Bernardo MM, Meroueh SO, Brown S, Fridman R, Mobashery S. Org Lett. 2005;7:4463. doi: 10.1021/ol0517269. [DOI] [PubMed] [Google Scholar]
- 7.Forbes C, Shi Q, Fisher JF, Lee M, Hesek D, Llarrull LI, Toth M, Gossing M, Fridman R, Mobashery S. Chem Biol Drug Des. 2009;74:527. doi: 10.1111/j.1747-0285.2009.00881.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tao P, Fisher JF, Mobashery S, Schlegel HB. Org Lett. 2009;11:2559. doi: 10.1021/ol9008393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Celenza G, Villegas-Estrada A, Lee M, Boggess B, Forbes C, Wolter WR, Suckow MA, Mobashery S, Chang M. Chem Biol Drug Des. 2008;71:187. doi: 10.1111/j.1747-0285.2008.00632.x. [DOI] [PubMed] [Google Scholar]
- 10.Lee M, Villegas-Estrada A, Celenza G, Boggess B, Toth M, Kreitinger G, Forbes C, Fridman R, Mobashery S, Chang M. Chem Biol Drug Des. 2007;70:371. doi: 10.1111/j.1747-0285.2007.00577.x. [DOI] [PubMed] [Google Scholar]
- 11.Ikejiri M, Bernardo MM, Bonfil RD, Toth M, Chang M, Fridman R, Mobashery S. J Biol Chem. 2005;280:33992. doi: 10.1074/jbc.M504303200. [DOI] [PubMed] [Google Scholar]
- 12.Bonfil RD, Sabbota A, Nabha S, Bernardo MM, Dong Z, Meng H, Yamamoto H, Chinni SR, Lim IT, Chang M, Filetti LC, Mobashery S, Cher ML, Fridman R. Int J Cancer. 2006;118:2721. doi: 10.1002/ijc.21645. [DOI] [PubMed] [Google Scholar]
- 13.Kruger A, Arlt MJE, Gerg M, Kopitz C, Bernardo MM, Chang M, Mobashery S, Fridman R. Cancer Res. 2005;65:3523. doi: 10.1158/0008-5472.CAN-04-3570. [DOI] [PubMed] [Google Scholar]
- 14.Martin MD, Carter KJ, Jean-Philippe SR, Chang M, Mobashery S, Thiolloy S, Lynch CC, Matrisian LM, Fingleton B. Cancer Res. 2008;68:6251. doi: 10.1158/0008-5472.CAN-08-0537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gu Z, Cui J, Borwn S, Fridman R, Mobashery S, Strongin AY, Lipton SA. J Neurosci. 2005;25:6401. doi: 10.1523/JNEUROSCI.1563-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ota R, Kurihara C, Tsou TL, Young WL, Yeghiazarians Y, Chang M, Mobashery S, Sakamoto A, Hashimoto T. J Cereb Blood Flow Metab. 2009;29:1547. doi: 10.1038/jcbfm.2009.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luplertlop N, Misse D, Bray D, Deleuze V, Gonzalez JP, Leardkamolkarn V, Yssel H, Veas F. EMBO Rep. 2006;7:1176. doi: 10.1038/sj.embor.7400814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guo Z, Sun X, He Z, Jiang Y, Zhang X, Zhang JH. Neurol Res. 2010;32:715. doi: 10.1179/016164109X12478302362491. [DOI] [PubMed] [Google Scholar]
- 19.He ZJ, Huang ZT, Chen XT, Zou ZJ. Chin Med J (Engl) 2009;122:2346. [PubMed] [Google Scholar]
- 20.Yu F, Kamada H, Niizuma K, Endo H, Chan PH. J Neurotrauma. 2008;25:184. doi: 10.1089/neu.2007.0438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Testero SA, Lee M, Staran RT, Espahbodi M, Llarrull LI, Toth M, Mobashery S, Chang M. ACS Med Chem Lett. doi: 10.1021/ml100254e. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chan DMT, Monaco KL, Wang RP. Tetrahedron Lett. 1998;39:2933. [Google Scholar]
- 23.Evans DA, Katz JL, West TR. Tetrahedron Lett. 1998;39:2937. [Google Scholar]
- 24.Jouvin K, Couty F, Evano G. Org Lett. 2010;12:3272. doi: 10.1021/ol101322k. [DOI] [PubMed] [Google Scholar]
- 25.Bolshan Y, Batey RA. Tetrahedron. 2010;66:5283. [Google Scholar]
- 26.King AE, Brunold TC, Stahl SS. J Am Chem Soc. 2009;131:5044. doi: 10.1021/ja9006657. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.