

Abstract
A series of analogs of the amamistatin natural products was designed and synthesized to facilitate additional anticancer structure-activity-relationships. The results indicate that the anticancer activity is relatively independent of stereochemistry, ester or amide linkage and replacement of the oxazoline/oxazole based iron-binding group with a catechol.
Amamistatins A (1) and B (2) are natural products isolated from the actinomycete Nocardia asteroides (Figure 1).1–3 Amamistatin A (1) exhibits anti-proliferative activity against a variety of cancer cell lines, including MCF-7 breast, A549 lung, and MKN45 stomach cancer. Amamistatins A and B also show cytotoxicity against mouse lymphocytic leukemia cells P388 (IC50 15 and 16 ng/mL, respectively).1 In addition, these compounds structurally resemble mycobactins,4 analogs of which exhibit anti-tuberculosis activity and have other therapeutic potential due to the chelation of iron (III),4–7 and other more recently reported siderophores including the nocardimicins,8,9 nocobactin NA,10,11 formobactin,12 and brasilibactin A.13–15 Iron chelation by these similar siderophores arises from three coordination sites, a hydroxyphenyl oxazoline or oxazole and two hydroxamic acids. The origin of the anticancer activity of amamistatins is not yet known, but because of they are natural metal ligands, they are capable of chelating metals essential for enzymatic activities. It was previously proposed that the anticancer activity of amamistatins was due to HDAC inhibition by the N-formyl hydroxylamine ligand.5 However, recent studies showed that the synthetic amamistatin B and an analog lacking the N-hydroxy group on the cyclic hydroxamate displayed no HDAC activity at the concentration of 100 µM.6
Figure 1.

Amamistatins, analogs and their retrosynthetic analysis.
Herein, we wish to report the design and syntheses of new analogs of amamistatins and their activity against MCF-7 and PC-3 human cancer cell lines. The first series is represented by analogs 3–6, with variation of the oxazole moiety and the ester linkage as well as controlled stereochemical modification. The second compound type involves change of the hydroxyphenyl oxazole to a catechol (7) since catechols are frequent ligands in other natural siderophores.16 (Figure1).
The retro-synthetic analysis of amamistatin B analogs is shown in Figure 1. Disconnection of the ester reveals the constituent mycobactic acid and cobactin and further simplification gives four fragments: a hydroxyphenyl oxazole or its analog (A), a linear hydroxamic acid (B), a β-hydroxy or β-amino acid (C), and a lysine-based cyclic hydroxamic acid, N-hydroxycaprolactam (D). Syntheses of fragments A (compounds 8 to 10), B (compound 11), and D (compound 12) (Scheme 2) were carried out according to published procedures.5–7 The oxazole, and oxazoline were prepared from salicylic acid and the corresponding amino acid, serine (R1 = H) or threonine (R1 = Me). Both the linear and cyclic hydroxamic acid were prepared from Cbz-protected lysine.
Scheme 2.

The β-hydroxy carboxylic acid 16 (fragment C) was derived from Meldrum’s acid (13) and octanoyl chloride using an acylation, decarboxylation, and asymmetric reduction protocol (Scheme 1). Thus, treatment of Meldrum’s acid with octanoyl chloride in the presence of pyridine followed by refluxing in MeOH afforded β-keto ester 14 in 77% yield.17 Asymmetric hydrogenation of 14 with a catalyst generated in situ from (cod)Ru(2-methylallyl)2 and (R)- or (S)-BINAP worked very efficiently under one atmosphere of hydrogen to provide the (R)- or (S)-β-hydroxy ester 15R/15S in excellent optical (ee) and chemical yields.18 Saponification of these β-hydroxy esters with LiOH and subsequent acidification gave the corresponding β-hydroxy carboxylic acids 16R/16S.
Scheme 1.

With all fragments in hand, analogs 3, with either the (R) or (S) stereochemistry of the β-hydroxy amide and the unnatural (for amamistatins) L-(S)-configuration of the linear lysine components were first targeted for syntheses, because we had previously shown that the diastereomer of amamistatin B with the (S) configuration of the linear lysine had anticancer activity similar to amamistatin B, itself, which has a D-(R)-linear lysine component. Thus, additional changes of stereochemistry at other stereogenic centers including the β-hydroxy amide were anticipated to help elaborate structure-activity-relationship (SAR) studies. A convergent approach was designed to couple oxazole A with lysine derivative B to give the corresponding mycobactic acid and couple separate enantiomers of β-hydroxy acids C (Y = O) with cyclic hydroxamate D to generate diastereomeric cobactins. Final coupling of the mycobactic acid with the individual diastereomers of the cobactins would complete the syntheses.
As anticipated, DCC-mediated coupling of β-hydroxy carboxylic acids 16R and 16S with hydroxamate 12 provided diastereomerically pure cobactins 17R and 17S in high yields (Scheme 2). Interestingly, HOAt/EDC-mediated coupling of 16R and 12 was much less effective and gave only a 34% yield of 17R. However, use of HOAt/EDC was more effective than a DCC-mediated processes for coupling of oxazole 8 and hydroxamic acid 18 to give protected mycobactic acid 19, especially with addition of DMAP. LiOH-mediated saponification of methyl ester 19 gave mycobactic acid 20 cleanly.
The direct coupling of mycobactic acid 20 and cobactins 17R and 17S was anticipated to provide the dibenzyl-protected forms of diastereomeric amamistatin analogs 3. However, all attempts at esterification were unsuccessful, including use of EDC/4-PPY, HOAt/EDC or Yamaguchi coupling conditions. Since we had previously found that coupling of simple protected forms of linear lysine derivatives (fragment B) with other cobactins, including those containing a tertiary alcohol instead of the secondary alcohol of 18, was effective,5 we attempted the same method on separate substrates 17R/S. Again, to our surprise, these less hindered alcohols (17R/S) did not react with the linear lysine component 11 under the same conditions. A variety of other conditions, including 1-methylimidazole/TsCl mediated coupling,19,20 Mitsunobu reaction,21 EDC/4-PPY all failed to generate 21. Fortunately, in this case, Yamaguchi conditions provided the desired individual diastereomers of 21 in good yields.22 HOAt/EDC-mediated coupling of acid 8 and the individual diastereomers of dibenzyl protected amines 22R/S, derived from treatment of the Cbz-protected derivatives 21R/S with HBr/HOAc provided both isomers (23R/S) of the benzyl protected amamistatin analogs in approximately 80% yield. Separate hydrogenolysis of 23R and 23S generated analogs 3R and 3S, resepectively, in about 70% yield each (Scheme 3).
Scheme 3.

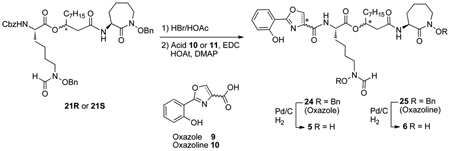
The des-methyl oxazole analogs 5R and 5S were also synthesized using a similar strategy. Starting from individual diastereomers of 21, removal of the Cbz-protecting groups with HBr/HOAc, followed by EDC/HOAt-mediated couplings with oxazole acid 9 afforded the protected analogs 24, which were debenzylated by hydrogenolysis to give analogs 5 (Table 1).
Table 1.
 | ||||
|---|---|---|---|---|
| Product | Acid | Compound 21 | % Yield of 24 or 25 | % Yield of 5 or 6 |
| 5R | 9 | 21R | 70 | 85 |
| 5S | 9 | 21S | 73 | 80 |
| 6SR | 10S | 21R | 36 | 94 |
| 6SS | 10S | 21S | 52 | 90 |
| 6RR | 10R | 21R | 59 | 93 |
| 6RS | 10R | 21S | 51 | 92 |
Because of the different biological activities exhibited by mycobactins S and macobactin T which differ in their growth promotion or inhibition of Mycobacterium smegmatis and Mycobacterium tuberculosis only on the basis of their diastereomeric relationship,23 individual amamistatin diastereomers 6, each with varied configuration at the stereogenic center of the oxazoline and the β-hydroxy ester components, were also prepared from 21R and 21S. The yields for the four isomers of 6 and 25 are also summarized in Table 1.
We next turned our attention to analogs 4R and 4S in which the normal ester was replaced by an amide. This required preparation of the corresponding diastereomeric amines, 25 which was accomplished by reaction of the individual diastereomeric alcohols 17R and 17S with diphenyl phosphoryl azide under Mitsunobu-like conditions and subsequent reduction of the resulting azides (Scheme 4). To complete the syntheses of the amides, we again used a sequential process in which compounds 26R/S were synthesized first before the final coupling to the oxazole. Diastereomers 26 were produced by coupling individual diastereomers of 25 with the NHS activated ester of 11. Removal of the Cbz-protecting groups followed by coupling with the NHS-activated ester 27 of oxazole 8 provided the benzyl-protected analogs 28R/S. Hydrogenolytic deprotection of diastereomeric forms of 28 afforded the desired amide-linked analogs 4 in excellent yields.
Scheme 4.

Alteration of iron binding moieties of amamistatins was also considered. Thus, in a second series of analogs, we replaced the oxazoline/oxazole group with a catechol to form the separate diastereomers 7R and 7S. The syntheses, summarized in Scheme 5, are similar to those used to prepare 4 and 6.
Scheme 5.

The analogs of amamistatin B were submitted for biological studies using MCF-7 and PC-3 tumor cellular assays. All of the analogs were found to have moderate to good activity. As shown in Table 2, the anticancer activity varied only slightly among all of the analogs, including diastereomers, with the catechol (7), being only slightly less active overall.
Table 2.
| Compound | MCF-7 | PC-3 | ||
|---|---|---|---|---|
| %inhibition @ 20 µM | IC50 (µM) | %inhibition @ 20 µM | IC50 (µM) | |
| 3R | 94 | 0.2 | 72 | 4 |
| 3S | 91 | 0.32 | 62 | 8 |
| 4R | 96 | 0.55 | 72 | 9 |
| 4S | 95 | 2 | 68 | 14 |
| 5R | 91 | 0.45 | 85 | 3.75 |
| 5S | 93 | 0.8 | 60 | 9 |
| 6SR | 87 | 0.24 | 83 | 3 |
| 6SS | 89 | 0.5 | 79 | 3 |
| 6RR | 92 | 0.32 | 90 | 2 |
| 6RS | 90 | 3 | 79 | 7.5 |
| 7R | 86 | 6 | 40 | - |
| 7S | 81 | 1 | 63 | 7 |
Conclusion
The syntheses of a series of amamistatin analogs was accomplished and all of the analogs were found to be comparably active against MCF-7 and PC-3 human cancer cell lines indicating that metal chelation might be more important than stereochemistry or the specific metal ligand. Further studies to allow detailed elaboration of the mode of action of these interesting natural iron chelators as anticancer agents are under consideration.
Supplementary Material
Acknowledgments
We gratefully acknowledge support from the National Institutes of Health (AI 054193).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kokubo S, Suenaga K, Shinohara C, Tsuji T, Uemura D. Tetrahedron. 2000;56:6435. [Google Scholar]
- 2.Suenaga K, Kokubo S, Shinohara C, Tsuji T, Uemura D. Tetrahedron Lett. 1999;40:1945. [Google Scholar]
- 3.Yokokawa F, Izumi K, Omata J, et al. Tetrahedron. 200;56:3027. [Google Scholar]
- 4.Vergne AF, Walz AJ, Miller MJ. Nat. Prod. Rep. 2000;17:99. doi: 10.1039/a809397k. [DOI] [PubMed] [Google Scholar]
- 5.Fennell KA, Miller MJ. Org. Lett. 2007;9:1683. doi: 10.1021/ol070382e. [DOI] [PubMed] [Google Scholar]
- 6.Fennell KA, Moellmann U, Miller MJ. J. Org. Chem. 2008;73:1018. doi: 10.1021/jo7020532. [DOI] [PubMed] [Google Scholar]
- 7.Xu Y, Miller MJ. J. Org. Chem. 1998;63:4314. [Google Scholar]
- 8.Banks JC, Moody CJ. Tet. Lett. 2009;26:3371. [Google Scholar]
- 9.Ikeda Y, Nonaka H, Furumai T, et al. J. Nat. Prod. 2005;68:1061. doi: 10.1021/np050091j. [DOI] [PubMed] [Google Scholar]
- 10.Ratledge C, Snow GA. Biochem. J. 1974;139:407. doi: 10.1042/bj1390407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sakagami H, Ishihara M, Hoshino Y, et al. In Vivo. 2005;19:277. [PubMed] [Google Scholar]
- 12.Murakami Y, Kato S, Nakajima M, et al. J. Antibiotics. 1996;49:839. doi: 10.7164/antibiotics.49.839. [DOI] [PubMed] [Google Scholar]
- 13.Tsuda M, Yamakawa M, Oka S, et al. J. Natl Prod. 2005;68:462. doi: 10.1021/np0496385. [DOI] [PubMed] [Google Scholar]
- 14.Mitchell JM, Shaw JT. Org. Lett. 2007;9:1679. doi: 10.1021/ol070355o. [DOI] [PubMed] [Google Scholar]
- 15.Ying Y, Hong J. Tet. Lett. 2007;48:8104. [Google Scholar]
- 16.Hider RC, Kong X. Natl. Prod. Reports. 2010;27:637. doi: 10.1039/b906679a. [DOI] [PubMed] [Google Scholar]
- 17.Kocienski PJ, Pelotier B, Pons J-M, Prideaux H. J. Chem. Soc., Perkin Trans. 1998;1:1373. [Google Scholar]
- 18.Ratovelomanana-Vidal V, Girard C, Touati R, Tranchier JP, Ben Hassine B, Genet JP. Adv. Synth. Catal. 2003;345:261. [Google Scholar]
- 19.Wakasugi K, Iida A, Misaki T, Nishii Y, Tanabe Y. Adv. Synth. Catal. 2003;345:1209. [Google Scholar]
- 20.Nakatsuji H, Morimoto M, Misaki T, Tanabe Y. Tetrahedron. 2007;63:12071. [Google Scholar]
- 21.Dembinski R. Eur. J. Org. Chem. 2004:2763. [Google Scholar]
- 22.Inanaga J, Hirata K, Saeki H, Katsuki T, Yamaguchi M. Bull. Chem. Soc. Jpn. 1979;52:1989. [Google Scholar]
- 23.Hu J, Miller MJ. J. Am. Chem. Soc. 1997;119:3462. [Google Scholar]
- 24.Experimental details and characterization data for new compounds are provided in the Supporting Information.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.