

Abstract
This Letter describes a chemical lead optimization campaign directed at VU0108370, a weak M1 PAM hit with a novel chemical scaffold from a functional HTS screen within the MLPCN. An iterative parallel synthesis approach rapidly established SAR for this series and afforded VU0405652 (ML169), a potent, selective and brain penetrant M1 PAM with an in vitro profile comparable to the prototypical M1 PAM, BQCA, but with an improved brain to plasma ratio.
The muscarinic acetylcholine receptors (mAChRs) are members of the family A G-protein-coupled receptors (GPCRs) and include five subtypes denoted M1-M5. All five of the mAChRs are known to play critical roles in multiple basic physiological processes and represent attractive therapeutic targets for a number of peripheral and CNS pathologies.1-3 Within the mAChRs, a major challenge has been a lack of subtype selective ligands to study the specific contribution of discrete mAChRs in various disease states.3,4 To address this limitation, we have focused on targeting allosteric sites on mAChRs as a means to develop subtype selective small molecules, both allosteric agonists and positive allosteric modulators (PAMs).5-9 Moreover, the emerging phenomenon of ligand-biased signaling requires the development of diverse chemical scaffolds of M1 ligands to successfully dissect of the roles of M1 activation through multiple, discrete ligand-biased signaling pathways.10,11
As members of the Molecular Libraries Production Center Network (MLPCN),12 we performed a real-time cell-based calcium-mobilization assay employing a rat M1/CHO cell line (Z′ averaged 0.7) and screened a 63,656 member MLPCN compound library following a triple-add protocol to simultaneously identify M1 antagonists, agonists (both orthosteric and allosteric) and positive allosteric modualtors (PAMs). This screen proved to be a major success providing viable leads that were optimized into potent and highly selective M1 ligands (Fig. 1): an M1 antagonist (1, VU0255035, ML012),13 an M1 allosteric agonist (2, VU0357017, ML071),14 and both an M1 PAM (3, VU0366369, ML137)15 and the first M5 PAM (4, VU0238429, ML129)16 derived from a pan-M1,M3,M5-PAM (5, VU0119498).17,18 However, the brain penetration (brainAUC/plasmaAUC = 0.1) and efficacy (60% ACh Max) of 3 were poor, as was the brain penetration of the prototypical M1 PAM, BQCA (6, brainAUC/plasmaAUC = 0.1)19-21; therefore, M1 PAM ligands with improved physicohemical properties for in vivo studies and novel scaffolds to address ligand-biased signaling are required. In this Letter, we describe the development of VU0405652 (ML169), a highly selective M1 PAM MLPCN probe, with a novel chemical scaffold and improved brain penetration.
Figure 1.

Structures of selective M1 and M5 MLPCN probes developed from hits from a triple-add functional M1 HTS MLPCN screen (1-5) and BQCA (6).
Perusal of the HTS data, which also yielded the non-selective hit 5, identified a second weak M1 PAM hit 7, VU0108370, with an EC50 of ∼13 μM. Confirmation of 7 from fresh powder and counter-screening against M2-M5 increased our enthusiasm for this highly M1 mAChR selective hit (Fig. 2); however, the CRC did not plateau, suggesting the M1 EC50 was actually >13 μM. Despite the weak potency, the confirmation of a novel M1 PAM scaffold with high M1 selectivity initiated a lead optimization campaign to improve M1 potency while maintaining the high M2-M5 selectivity.
Figure 2.

Concentration response curves (CRCs) for M1-M5 for HTS hit VU0108370. M1 EC50 ∼13 μM (does not plateau) and M2-M5 EC50 >30 μM.
Our initial optimization strategy is outlined in Figure 3, and as SAR with allosteric ligands is often shallow, we employed an iterative parallel synthesis approach, along with targeted syntheses for structures encompassing more speculative modifications. Attempted modifications of the Eastern oxazole-amide, although not extensive, met with no success, returning only compounds with undetectable activity. In a straightforward attempt to reduce molecular weight the benzyl group attached to the indole nitrogen was omitted, but met with a similar lack of success (EC50 > 10 μM) as did the sulfide and sulfoxide congeners.
Figure 3.

Initial optimization strategy for VU0108370, 7.
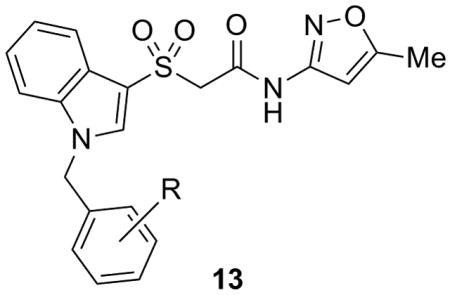
Thus, we planned to hold the northern portion of 7 constant, and survey diversity on the southern benzyl moiety employing a library synthesis approach. As shown in Scheme 1, the key library scaffold 12 was readily prepared in 3 steps from methyl thioglycolate 8. A PyClu-mediated microwave-assisted coupling between 9 and 10 provided 11 in 71% yield, which was then oxidized to the corresponding sulfone 12 with Oxone in 88% yield. An 18-membered library of analogs 13 was then prepared by treatment with NaH and 18 diverse benzyl halides.
Scheme 1.

Reagents and conditions: (a) i. indole, I2, KI, MeOH, H2O; ii. 2M LiOH, THF (38%); (b) PyClu, DCE, 110 °C, 20 min, mw (71%); (c) Oxone, MeOH, H2O (88%); NaH, DMF, BnX (50-90%).
As shown in Table 1, SAR, as with many allosteric ligands, was shallow affording only five active compounds from the eighteen synthesized. Upon resynthesis, HTS hit 7 (a 2-Cl congener) showed an EC50 of 9.7 μM with 83% ACh Max; however, the CRC once again did not plateau. The other four actives, all with substituents in the 3-position, did afford sigmoidal CRCs with up to 96% ACh Max and EC50s in the 3.8 to 6.5 μM range. The most potent analog was 13g, a 3-Br derivative (EC50 = 3.8 μM, 91% ACh Max) which provided a significant increase in potency, but at the cost of physiochemical properties (cLogP >4 and poor solubility). Replacement of the benzyl moiety with a pyridyl analog also led to an inactive compound (data not shown). However, this first generation library indicated that substitution at the 3-position of the benzyl moiety was preferred. This result then prompted us to employ 13g as a starting material for a small Suzuki coupling library (Scheme 2) to replace the lipohilic bromide with various aryl and heteroaryl moieties to introduce basicity and/or polarity.
Table 1.
Structures and activities of analogs 13.
 | |||
|---|---|---|---|
| Cmpd | R | M1EC50 (μM)a | %ACh Maxa |
| 7 | 2-Cl | 9.71 | 83 |
| 13a | 3-Cl | 5.82 | 96 |
| 13b | 4-Cl | >10 | - |
| 13c | 2-OMe | >10 | - |
| 13d | 3-OMe | 6.54 | 79 |
| 13e | 4-OMe | >10 | - |
| 13f | 3-F | 5.22 | 96 |
| 13g | 3-Br | 3.79 | 91 |
| 13h | 3-CF3 | >10 | - |
| 13i | 3-CN | >10 | - |
| 13j | 3,5-diBr | >10 | - |
| 13k | 3,4-diCl | >10 | - |
| 13l | 4-CF3 | >10 | - |
| 13m | 4-OCF3 | >10 | - |
| 13n | 2-F | >10 | - |
| 13o | 2,4-diF | >10 | - |
| 13p | 2-Br-4-F | >10 | - |
Average of at least three independent determinations.
Scheme 2.

Reagents and conditions: (a) Ar-B(OH)2 or Het-B(OH)2, 10 mol % Pd(t-Bu)2, 1.0 M aq Cs2CO3, THF, mw, 120 °C (65-90%).
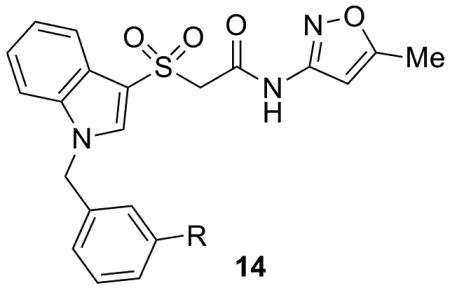
As shown in Table 2, SAR was again shallow, but we identified pyrazole as a preferred heterocyclic replacement for the bromide. N-Me pyrazole 14a possessed an M1 EC50 of 3.2 μM (102% ACh Max), while the unsubstituted pyrazole congener 14b was essentially equipotent (M1 EC50 = 2.2 μM, 90% ACh Max). Moreover, both pyrazole congeners lowered cLogP a full order of magnitude relative (cLogP = 3.1) to 13g. Additional steric bulk on the pyrazole, as in the sec-butyl derivative 14c, led to an inactive analog. Phenyl derivative 14d and basic amino pyridine analogs 14e-14h, were inactive (M1 EC50s >10 μM).
Table 2.
Structures and activities of analogs 14.
 | |||
|---|---|---|---|
| Cmpd | Ar/Het | M1 EC50 (μM)a | %ACh Maxa |
| 14a | 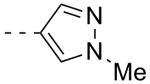 |
3.21 | 102 |
| 14b | 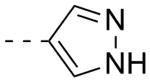 |
2.19 | 90 |
| 14c | 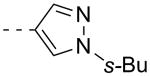 |
>10 | - |
| 14d | 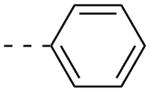 |
>10 | - |
| 14e | 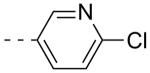 |
>10 | - |
| 14f | 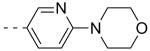 |
>10 | - |
| 14g | >10 | - | |
| 14h |  |
>10 | - |
Average of at least three independent determinations
To further the development of this novel series of M1 PAMs we focused on three of the more potent analogs (13a, 13g and 14b) and applied a fine-tuning process of introducing fluorine atoms at various locations to provide analogs 15 (Table 3), an approach we found successful for multiple allosteric ligands. Across the series, substitution at the 4-position was uniformly not tolerated (15a-c), consistent with the SAR appearing in Table 1. Bis-fluorination of the indole ring eroded activity in the context of the bromine analog 15d but conversely augmented the activity of the pyrazole congener 15e. This type of subtle/confounding SAR was similarly observed with respect to fluorination at the R2 position in analogs 15f-h. While the presence of a fluorine at R2 was neutral or slightly beneficial in the context of the chlorine analog (15f), its presence resulted in clearly decreased activity for both the bromine and pyrazole analogs, 15g and 15h. Lastly, the introduction of a single fluorine at the R6 position could either be moderately detrimental in the context of pyrazole 15i or decidedly beneficial with respect to bromine analog 15j, where an almost 3-fold improvement in potency was observed. In this manner, both VU0405652 (15j) and the related difluorindole analog VU0405645 (15e) were chosen for further evaluation.
Table 3.
Structures and activities of analogs 15.
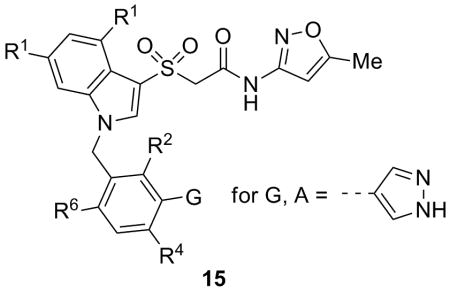 | |||||||
|---|---|---|---|---|---|---|---|
| Cmpd | G | R1 | R2 | R4 | R6 | M1 EC50 (μM)a | %ACh Maxa |
| 15a | A | H | H | F | H | >10 | - |
| 15b | Br | H | H | F | H | >10 | - |
| 15c | Cl | H | H | F | H | >10 | - |
| 15d | Br | F | H | H | H | 5.37 | 103 |
| 15e | A | F | H | H | H | 1.85 | 102 |
| 15f | Cl | H | F | H | H | 5.10 | 56 |
| 15g | Br | H | F | H | H | >10 | - |
| 15h | A | H | F | H | H | 4.40 | 103 |
| 15i | A | H | H | H | F | 3.07 | 96 |
| 15j | Br | H | H | H | F | 1.38 | 84 |
Average of at least three independent determinations.
Both 15e and 15j were selective (Fig. 4A) for M1 (>30 μM vs. M2-M5) and both compounds demonstrated impressive left-ward shifts of the ACh CRC (98-fold and 49-fold, respectively) in fold-shift assays at 30 μM (Fig. 4B), values comparable to BQCA. However, a combination of calculated properties (TPSA, Hacc, etc…), ancillary pharmacology and in vivo PK suggested 15j was the more optimal MLPCN probe molecule compared to 15e.
Figure 4.
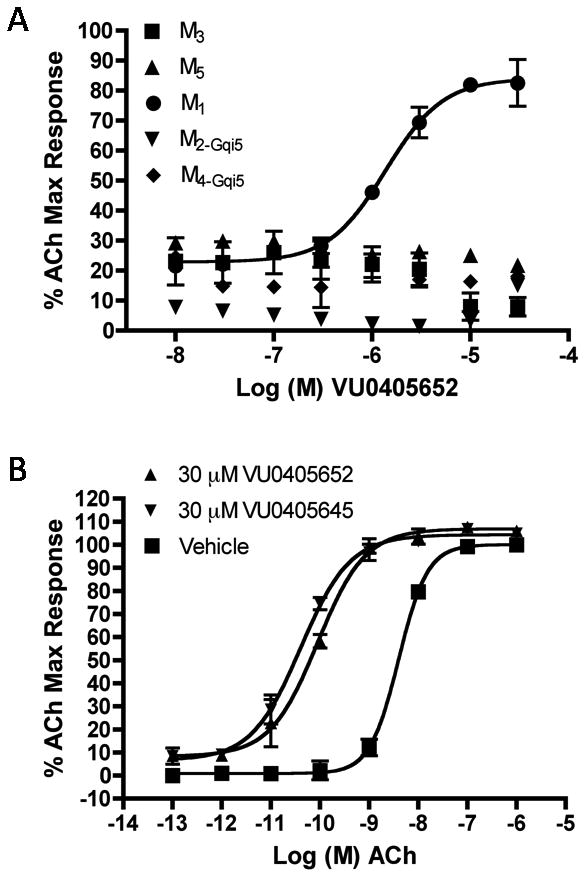
A) CRC for 15j, VU0405652 at M1-M5 (15e, VU0405645 looked similar); B) ACh fold-shift experiment at 30 μM for VU040652 (49-fold) and VU0405645 (98-fold).
In terms of ancillary pharmacology, the Lead Profile screen at Ricerca, evaluating 68 GPCRs, ion channels and transporters in radioligand binding assays, resulted in only two significant activities (DAT, 83% at 10 μM and sodium channel site 2, 83% at 10 μM); importantly, 15j was selective versus the biogenic amines (D2, H-HT2B, etc…) and displayed no orthosteric binding at M1-M5. Based on the exciting in vitro profile, we then evaluated 15j for brain penetration in rat. A 10 mg/kg IP dose of 15j afforded a brain AUC/plasmaAUC of 0.32 at 1 hour, providng an improvement over both 3 (ML137) and BQCA with brainAUC/plasmaAUC of ∼0.1. Based on this profile, 15j (VU0405652) was declared an MLPCN probe, ML169.
As we have previously demonstrated with both an M1 PAM (BQCA)20 and M1 allosteric agonists (2, ML071 and TBPB),23 ML169 also shifted APP processing towards a non-amyloidogenic pathway.24 As shown in Figure 5, 10 μM carbachol (CCh) affords a significant increase in soluble APP (APPsα), while 100 nM CCh provides a modest increase. VU0405652 (ML169) at a dose of 2 μM has no effect, but in combination with low dose CCh (100 nM), ML169 potentiates the CCh-mediated non-amyloidogenic APPsα release to the same degree as 10 μM CCh. These data once again suggest that selective activation of M1 may have a disease modifying role in Alzheimer's disease.14,20
Figure 5.
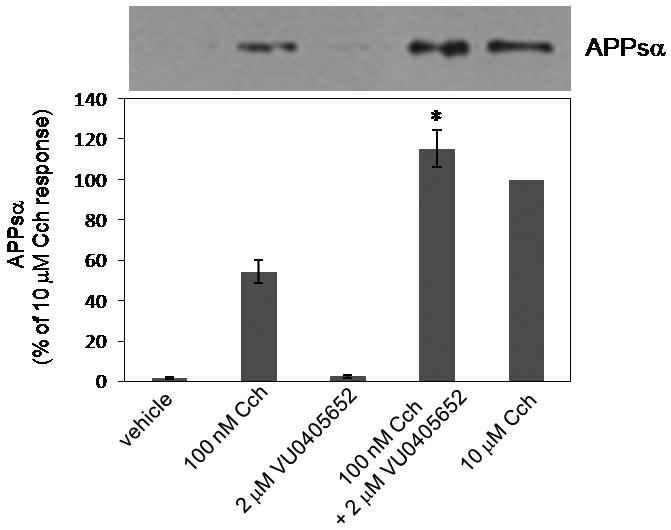
M1 PAM VU04505652 (ML169) potentiates the CCh-mediated non-amyloidogenic APPsα release in TREx293-hM1 cells.
In summary, we have developed a potent, selective and brain penetrant M1 PAM, ML169, based on a novel indole scaffold from an MLPCN functional HTS. Further in vivo evaluation of this probe is underway and results from preclinical models of Alzheimer's disease and schizophrenia will be reported in due course. ML169 is an MLPCN probe and freely available upon request.
Acknowledgments
The authors thank the MLPCN (1U54 MH084659), NIMH (1RO1 MH082867), NIH and the Alzheimer's Association (IIRG-07-57131) for support of our Program in the development of subtype selective allosteric ligands of mAChRs.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Bonner TI, Buckley NJ, Young AC, Brann MR. Science. 1987;237:527–532. doi: 10.1126/science.3037705. [DOI] [PubMed] [Google Scholar]
- 2.Bonner TI, Young AC, Brann MR, Buckley NJ. Neuron. 1988;1:403–410. doi: 10.1016/0896-6273(88)90190-0. [DOI] [PubMed] [Google Scholar]
- 3.Wess J. Annu Rev Pharmacol Toxicol. 2004;44:423–450. doi: 10.1146/annurev.pharmtox.44.101802.121622. [DOI] [PubMed] [Google Scholar]
- 4.Langmead CJ, Watson J, Reavill C. Pharmacol Ther. 2008;117:232–243. doi: 10.1016/j.pharmthera.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 5.Conn PJ, Jones C, Lindsley CW. Trends in Pharm Sci. 2009;30:25–31. doi: 10.1016/j.tips.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Conn PJ, Lindsley CW, Jones C. Trends in Pharm Sci. 2009;30:148–156. doi: 10.1016/j.tips.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Conn PJ, Christopoulos A, Lindsley CW. Nat Rev Durg Disc. 2009;8:41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bridges TM, LeBois EP, Hopkins CR, Wood MR, Jones JK, Conn PJ, Lindsley CW. Drug News & Perspect. 2010;23:229–240. doi: 10.1358/dnp.2010.23.4.1416977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lewis JA, Lebois EP, Lindsley CW. Curr Opin Chem Biol. 2008;12:269–279. doi: 10.1016/j.cbpa.2008.02.014. [DOI] [PubMed] [Google Scholar]
- 10.Digby GJ, Conn PJ, Lindsley CW. Curr Opin Drug Disc & Dev. 2010;13:577–586. [PMC free article] [PubMed] [Google Scholar]
- 11.Marlo JE, Niswender CM, Luo Q, Brady AE, Shirey JK, Rodriguez AL, Bridges TM, Williams R, Days E, Nalywajko NT, Austin C, Williams M, Xiang Y, Orton D, Brown HA, Kim K, Lindsley CW, Weaver CD, Conn PJ. Mol Pharm. 2009;75(3):577–588. doi: 10.1124/mol.108.052886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.For information on the MLPCN and information on how to request probe compounds, such as ML169, see: http://mli.nih.gov/mli/mlpcn/
- 13.Sheffler DJ, Williams R, Bridges TM, Lewis LM, Xiang Z, Zheng F, Kane AS, Byum NE, Jadhav S, Mock MM, Zheng F, Lewis LM, Jones CK, Niswender CM, Weaver CD, Conn PJ, Lindsley CW, Conn PJ. Mol Pharmacol. 2009;76:356–368. doi: 10.1124/mol.109.056531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lebois EP, Bridges TM, Dawson ES, Kennedy Jp, Xiang Z, Jadhav SB, Yin H, Meiler J, Jones CK, Conn PJ, Weaver CD, Lindsley CW. ACS Chemical Neurosci. 2010;1:104–121. doi: 10.1021/cn900003h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bridges TM, Kennedy JP, Cho HP, Conn PJ, Lindsley CW. Bioorg Med Chem Lett. 2010;20:1972–1975. doi: 10.1016/j.bmcl.2010.01.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bridges TM, Marlo JE, Niswender CM, Jones JK, Jadhav SB, Gentry PR, Weaver CD, Conn PJ, Lindsley CW. J Med Chem. 2009;52:3445–3448. doi: 10.1021/jm900286j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bridges TM, Kennedy JP, Cho HP, Conn PJ, Lindsley CW. Bioorg Med Chem Lett. 2010;20:558–562. doi: 10.1016/j.bmcl.2009.11.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bridges TM, Kennedy JP, Hopkins CR, Conn PJ, Lindsley CW. Bioorg Med Chem Lett. 2010;20:5617–5622. doi: 10.1016/j.bmcl.2010.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ma L, Seager M, Wittman M, Bickel N, Burno M, Jones K, Graufelds VK, Xu G, Pearson M, McCampbell A, Gaspar R, Shughrue P, Danzinger A, Regan C, Garson S, Doran S, Kreatsoulas C, Veng L, Lindsley CW, Shipe W, Kuduk S, Jacobson M, Sur C, Kinney G, Seabrook GR, Ray WJ. Proc Natl Acad Sci USA. 2009;106:15950–15955. doi: 10.1073/pnas.0900903106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shirey JK, Brady AE, Jones PJ, Davis AA, Bridges TM, Jadhav SB, Menon U, Christain EP, Doherty JJ, Quirk MC, Snyder DH, Levey AI, Watson ML, Nicolle MM, Lindsley CW, Conn PJ. J Neurosci. 2009;29:14271–14286. doi: 10.1523/JNEUROSCI.3930-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang FV, Shipe WD, Bunda JL, Nolt MB, Wisnoski DD, Zhao Z, Barrow JC, Ray WJ, Ma L, Wittman M, Seager M, Koeplinger K, Hartman GD, Lindsley CW. Bioorg Med Chem Lett. 2010;20:531–536. doi: 10.1016/j.bmcl.2009.11.100. [DOI] [PubMed] [Google Scholar]
- 22.ML169, 2-((1-(5-bromo-2-fluorobenzyl)-1H-indol-3-yl)sulfonyl)-N-(5-methylisoxazol-3-yl)acetamide. To a solution of indole (3.00 g, 25.6 mmol) and methyl thioglycolate 8 (2.40 mL, 25.6 mmol) in methanol:water (80 mL: 20 mL) was added iodine (6.50 g, 25.6 mmol) and potassium iodide (4.25 g, 25.6 mmol). The reaction mixture was stirred at ambient temperature for 60 hours. Methanol was removed in vacuo and the aqueous layer diluted with a saturated solution of sodium bicarbonate and extracted with ethyl acetate. The organic layer was dried over magnesium sulfate, evaporated in vacuo and the resulting residue was purified on a silica gel column (0-100% ethyl acetate:hexanes over 33 min) to afford the ester as an oil (LCMS >98%). The ester was dissolved in a mixture of tetrahydrofuran (20 mL) and 2.0M aqueous LiOH (15 mL), then stirred vigorously at ambient temperature for 30 minutes. Tetrahydrofuran was removed in vacuo, the aqueous layer neutralized with 1.2 N HCl and extracted with CH2Cl2. The organic layer was dried over magnesium sulfate and removed in vacuo to produce an oily residue. Upon diluting the residue in dichloromethane a reddish-brown solid formed which was filtered and dried to yield compound 9 (2.00 grams, 9.65 mmol, 38% yield over 2 steps, LCMS >98%). Compound 10 (650 mg, 3.14 mmol), 3-amino-5-methyl-isoxazole (616 mg, 6.28 mmol), PyClU (2.00 g, 6.28 mmol), and DIEA (1.36 mL, 7.85 mmol) were added to dichloroethane (25 mL) and microwave irradiated at 110 °C for 20 minutes. After cooling, the solvent was removed in vacuo and the remaining residue purified on a silica gel column (0-70% ethyl acetate:hexanes over 33 min) to yield compound 11 (642 mg, 2.23 mmol, 71% yield, LCMS >98%). Compound 11 (502 mg, 1.77 mmol) was dissolved in 25 mL (9:1, methanol:water) and Oxone (10.0 g, 17.7 mmol) was added. Stirring at ambient temperature continued overnight. Water (20 mL) was added and the mixture extracted with ethyl acetate (3×20 mL). The organics were combined, dried over magnesium sulfate, and concentrated in vacuo to give an oily residue which was purified on silica gel (0-50% ethyl acetate:hexanes over 19 min) to yield compound 12 (500 mg, 1.57 mmol, 88% yield, LCMS >98%). In a 5 mL microwave vial, compound 12 (55.0 mg, 0.174 mmol) was dissolved in DMF (3 mL) and cooled to 0 °C. Sodium hydride (60% by weight, 14.0 mg, 0.348 mmol) was then added in one portion and the reaction mixture vigorously stirred at 0 °C for 15 minutes. 4-bromo-2-bromomethyl-1-flourobenzene (51.0 mg, 0.191 mmol) was added in one portion and the reaction mixture was stirred while being allowed to warm to ambient temperature over 3 hours. The reaction mixture was quenched with water (2 mL) and the solution was extracted with ethyl acetate (3×4 mL). The combined organics were dried over magnesium sulfate, concentrated in vacuo to give an oily residue which was purified on silica gel (0-70% ethyl acetate:hexanes over 19 min) to yield ML169 (45 mg, 0.088 mmol, 51% yield). LCMS >98% 214 nm, RT = 1.34 min, m/z = 506 ([79Br]m+1), 508 ([81Br]m+1). 1H NMR (400 MHz, DMSO-d6) 11.27 (s, 1H), 8.23 (s, 1H), 7.82 (d, J = 8.0 Hz, 1H), 7.63 (d, J = 8.0 Hz, 1H), 7.56-7.58 (m, 1H), 7.47 (dd, J = 2.4 Hz, 6.4 Hz, 1H), 7.35-7.23 (m, 3H), 6.54 (s, 1H), 5.62 (s, 2H), 4.43 (s, 2H), 2.37 (s, 3H), HRMS found: 506.0184; calculated for C21H17BrFN3O4S: 506.0185.
- 23.Jones CK, Brady AE, Davis AA, Xiang Z, Bubser M, Tantawy MN, Kane A, Bridges TM, Kennedy JP, Bradley SR, Peterson T, Baldwin RM, Kessler R, Deutch A, Lah JL, Levey AI, Lindsley CW, Conn PJ. J Neurosci. 2008;28(41):10422–10433. doi: 10.1523/JNEUROSCI.1850-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.APP processing. In order to test the effect of M1 PAM on M1-stimulated APPsα release, a human M1 overexpressing stable cell line was generated in TREx293 cells (Invitrogen). Cells were plated at 0.3×106 cells in 6-well plate 2 days prior to experiments. Cells were pretreated with 2μM VU0405652 or dimethylsulfoxide (DMSO) for 15 min. Immediately, 100nM or 10μM carbachol was added, and the medium was then conditioned for 1 h at 37°C. Western blot analysis of the endogenous APPsα in conditioned media was performed as described.23