

Abstract
The authors discuss the case of a 7-year-old female who presented with exertional cyanosis and was found to have pulmonary arterial hypertension. Despite normal left-sided heart function, the patient developed pulmonary oedema in response to pulmonary vasodilator therapy, increasing suspicion for pathology in the pulmonary capillaries and veins. Lung biopsy confirmed a diagnosis of pulmonary capillary haemangiomatosis (PCH), a rare cause of pulmonary hypertension in both children and adults. The diagnosis requires lung biopsy and is often made postmortem. She was treated with interferon α-2a and doxycycline for their antiangiogenic properties and reports of disease regression. Although she initially demonstrated improvement in her pulmonary hypertension in response to these medications, she succumbed to the disease within the time frame previously reported for PCH.
Background
We discuss the case of a 7-year-old female who presented with exertional cyanosis and pulmonary hypertension and was found to have pulmonary capillary haemangiomatosis (PCH). The case is important because it highlights1 the limited differential diagnosis of paediatric pulmonary hypertension associated with parenchymal lung disease,2 the importance of early lung biopsy in such cases,3 the limited reliability of non-invasive estimates of right ventricular pressure and4 our experience with medications to treat this rare and fatal disease.
Case presentation
A previously well 7-year-old female presented with an episode of self-resolving exertional cyanosis, which was not associated with cough, wheeze, shortness of breath, retractions, pallor, syncope or mottling. She had experienced similar but milder events in the preceding months. On her family physician’s examination, oxygen saturation was in the mid-80s, improving with supplemental oxygen. An echocardiogram, performed by a local cardiologist, showed a dilated, hypertrophic right ventricle and an estimated right ventricular systolic pressure (RVSP) of 77 mm Hg (tricuspid regurgitant jet velocity, TRJV, 4.4 m/s) above right atrial (RA) pressure. She was transferred to our centre for evaluation of her pulmonary hypertension.
Review of systems revealed 2 years of easy fatigability and cold intolerance. No weight loss, haemoptysis, anaemia, recurrent infections or stigmata of rheumatologic disease were reported. Her past medical history was negative aside from a hospitalisation at 2 years of age for pneumonia associated with mild haemoptysis. Maternal history was negative and paternal history was unknown. The child had no known allergies, recent travel or sick contacts. She had no exposures to secondhand smoke, birds or grain silos.
On exam, the patient was comfortable and afebrile with a heart rate of 108 beats/min, blood pressure of 113/71 mm Hg and respiratory rate of 20 breaths/min. Oxygen saturation was 84% on room air, increasing to 95% on 1 l/min nasal canula oxygen. There was no jugular venous distension. Prominent veins were noted on the upper chest. Her lungs were clear to auscultation. Cardiac exam revealed a prominent S2, a grade II/VI systolic murmur and a soft diastolic murmur at the left upper sternal border. A firm, smooth liver edge extended 4 cm below the right costal margin. She had moderate clubbing.
Investigations
Known causes of pulmonary hypertension were recently categorised according to the Dana Point classification system at the Fourth World Symposium on Pulmonary Arterial Hypertension.4 At this point, an extensive evaluation is needed because secondary causes of pulmonary hypertension are common.5 Laboratory and diagnostic testing should be performed as outlined in table 1.
Table 1.
Diagnostic testing recommended for the evaluation of paediatric pulmonary hypertension
| General lab work | Thromboembolic disease |
| Hypercoagulability panels | |
| Thyroid disease | |
| TSH, free thyroxine | |
| Collagen vascular disease | |
| ANA screen with follow-up testing if positive | |
| CBC | Rheumatoid factor |
| Electrolytes, BUN, creatinine | |
| BNP and NT-proBNP | C3, C4 |
| β-HCG | |
| Uric acid | ESR, CRP |
| Cardiac studies | |
| ECG | Portal hypertension |
| Echocardiogram | |
| Cardiac catheterisation | Hepatic function panel |
| Lung disease | |
| Chest x-Ray | Hepatitis screen |
| Chest CT | |
| Pulmonary function tests | Abdominal/liver ultrasound |
| Ventilation/perfusion scan | |
| Obstructive sleep apnea | Familial disease |
| Polysomnography | |
| HIV infection, drugs, toxins | Genetic analysis for bone morphogenic protein receptor 2 mutations |
| HIV AB 1+2 |
In cases with profound hypoxemia, we strongly recommend high-resolution CT (HRCT) to evaluate parenchymal causes of pulmonary hypertension. A left-sided cardiac catheterisation should be considered to evaluate diastolic dysfunction if the patient does not have an intra-atrial communication, and contrast ECG should be considered to evaluate for intrapulmonary shunt. We recommend pathologic examination of lung tissue if the above studies are suggestive of parenchymal lung disease.
In our case, a detailed laboratory examination was within normal limits (table 2), except for an elevated brain natriuretic peptide (BNP) at 594 pg/ml (normal value <64 pg/ml), a high factor VIII level (218%), and low thyroid stimulating hormone (0.45 μIU/ml).
Table 2.
Initial laboratory evaluation
| Complete blood count | Hypercoagulability studies | ||
|---|---|---|---|
| White cell count | 9300/mm3 | Antithrombin III | 104% |
| Hematocrit | 39.90% | Factor Vleiden | Normal |
| Platelets | 156 000/mm3 | Factor VIII | 218% |
| Serum chemistries | Protein C | 82% | |
| Sodium | 136 mmol/l | Protein S | 61% |
| Potassium | 3.7 mmol/l | Russell viper venom | Negative |
| Chloride | 106 mmol/l | Homocysteine, S | Negative |
| Bicarbonate | 22 mmol/l | Plasminogen | 97% |
| BUN | 17 mg/dl | Collagen vascular studies | |
| Creatinine | 0.48 mg/dl | ANA Screen | Negative |
| Glucose | 96 mg/dl | P-ANCA | Negative |
| Calcium | 8.8 mg/dl | C-ANCA | Negative |
| Protein | 5.6 g/dl | Cardiolipin antibody | <4 g/l |
| Albumin | 3.4 g/dl | Rheumatoid Factor | <15 IU/ml |
| Total bilirubin | 0.8 mg/dl | Erythrocyte sedimentation rate | 9 mm/h |
| Alkaline phosphatase | 174 U/l | C reactive protein | 0.1 mg/dl |
| Aspartate aminotransferase | 44 U/l | Immunology Studies | |
| Alanine aminotransferase | 27 U/l | Immunoglobulin G | 639 ml/dl |
| B-type natriuretic peptide | 594 pg/ml | Immunoglobulin A | 77 mg/dl |
| Thyroid panel | Immunoglobulin M | 63 mg/dl | |
| Thyroid stimulating hormone | 0.045 µIU/ml | ||
| Free thyroxine | 1.28 ng/dl | ||
Chest radiograph showed mild cardiomegaly with prominent pulmonary arteries, septal lines and interstitial markings (figure 1A). HRCT showed septal thickening and ill-defined nodular opacities in all lung fields (figure 1B). Spirometry was within normal limits. Her liver ultrasound was normal. ECG showed right ventricular hypertrophy and RA enlargement. Echocardiogram showed moderate septal flattening and an estimated RVSP of 77 mm Hg above right atrial pressure (TRJV 4.4 m/s). Cardiac catheterisation revealed no atrial-level shunt, pulmonary venous stenosis or diastolic dysfunction. Systolic pulmonary artery pressure (PAP) was suprasystemic on room air at 95 mm Hg, decreasing to 58 mm Hg with supplemental oxygen and inhaled nitric oxide (iNO; 40 ppm) (table 3).
Figure 1.
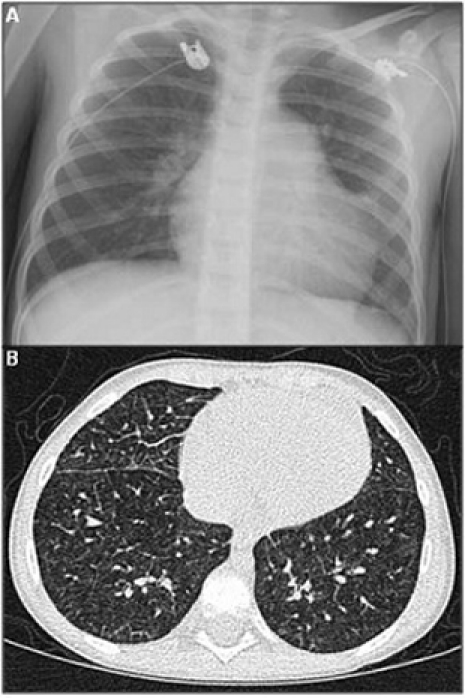
(A) Posteroanterior chest radiograph showing mild cardiomegaly, prominent pulmonary arteries, and interstitial prominence. (B) Chest CT showing septal thickening and multiple ill-defined nodular opacities in both lungs.
Table 3.
Data from cardiac catheterisation with and without supplemental oxygen and nitric oxide at time of diagnosis
| Condition | Arterial partial pressure of oxygen (mm Hg) | Cardiac output (l/min) | Pulmonary artery pressure (mm Hg) | Pulmonary vascular resistance (units×m2) | Systemic vascular resistance (units×m2) | Pulmonary capillary wedge pressure (mm Hg ) |
| Room air | 45 | 1.7 | 95 | 30.0 | 30.5 | 5 |
| 2 l/min supplemental oxygen | 131 | 1.7 | 83 | 27.6 | 32.3 | 6 |
| 100% oxygen + 40 ppm inhaled nitric oxide | 456 | 2.3 | 58 | 12.4 | 24.1 | 6 |
Differential diagnosis
How does the profound hypoxemia limit the differential diagnosis?
The profound hypoxemia seen in this case limits the number of potential diagnoses to those with primary or secondary parenchymal or interstitial lung involvement. Possibilities includes pulmonary hypertension associated with drugs and toxins (generally those with cytotoxic properties), collagen-vascular disease (scleroderma, mixed connective tissue disease, systemic lupus erythematosis (SLE), Sjögren’s syndrome), HIV with lymphocytic interstitial pneumonitis, portal hypertension, diseases with pulmonary venous or capillary involvement, left-sided heart disease, other parenchymal lung diseases causing chronic hypoxemia, thromboembolic disease and rare causes.6
In this case, our initial diagnostic approach failed to identify a cause of pulmonary hypertension. There was no exposure to drugs or toxins and no evidence of collagen vascular disease, HIV, portal hypertension, thrombotic disease, cardiac shunt lesions or left-sided cardiac disease. The most notable finding was the abnormal HRCT suggesting an abnormality within the pulmonary parenchyma.
Interpreting the response to pulmonary vasodilators
After catheterisation, our patient was initially treated with nasal nitric oxide (40 ppm) and sildenafil (1 mg/kg three times daily). Over the next 24 h, she developed pulmonary oedema but recovered quickly upon discontinuation of iNO, decreasing the dose of sildenafil (0.5 mg/kg/dose) and the addition of diuretic therapy.
Diagnostically, the response to vasodilators was important and suggested that the aetiology of the pulmonary hypertension was downstream of the pulmonary arterioles. This differential includes left ventricular diastolic dysfunction, large and small vessel pulmonary venous disease and pulmonary capillary disease. In this case, considering the normal left-sided heart function on echo, the lack of evidence for diastolic dysfunction or large pulmonary venous stenosis on catheterisation, and septal thickening and nodular opacities on HRCT, the most likely diagnoses are pulmonary veno-occlusive disease (PVOD) and PCH.
Outcome and follow-up
Based on her physical exam, radiographic findings and response to acute pulmonary vasodilator therapy, a diagnostic lung biopsy was performed. The surface of the lung appeared nodular (figure 2A). Histology showed haemorrhagic regions within the alveolar parenchyma associated with congested vessels (figure 2B). Small pulmonary arteries had striking medial hypertrophy without intimal disease or plexiform lesions (figure 2C). Foci of increased capillary density were present in the distal lung (figure 2D), including cells that were positive for endothelial markers CD31 (figure 2E), CD34 (figure 2F) and Factor VIII (figure 2G). The lack of MIB1 staining suggested low rates of cell proliferation, and CD117 staining showed a uniform distribution of expression. The pulmonary veins were not arterialised, and there was no evidence of veno-occlusive disease. The results were consistent with PCH.
Figure 2.
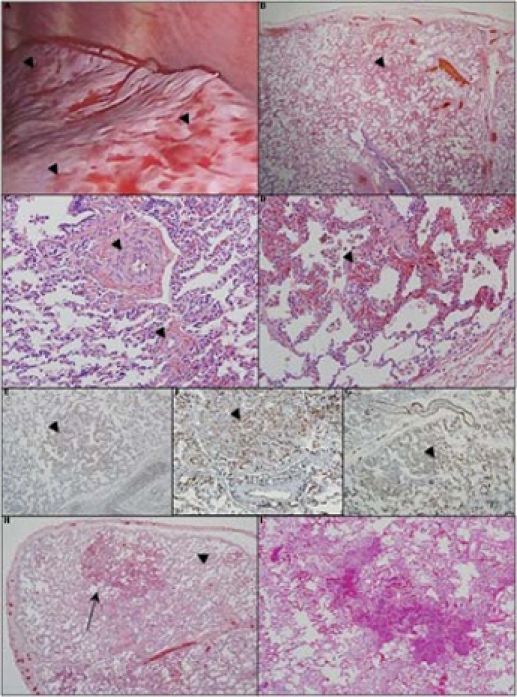
Intraoperative view and photomicrographs of a lung wedge biopsy section. (A) Intraoperative view shows a grossly nodular appearance (arrowheads). (B) Microscopically there is a patchy distribution of dense cellular interstitial infiltrates, haemorrhagic areas and vascular congestion (arrowhead). (C) The pulmonary vasculature shows muscularisation of small and medium arteries with medial hypertrophy (arrowheads). (D) High power views show vascular congestion with foci of increased capillary density (arrowhead) consistent with capillary haemangiomas. The capillary haemangioma appear to be comprised of endothelial cells without nuclear atypia, which on immunohistochemistry, stain positive (arrowheads) for (E) CD-31, (F) CD-34 and (G) Factor VIII staining. The section did not stain positively for MIB-1 (not shown). Comparison of lung biopsy to autopsy histology shows progression of capillary growth over time. (H) Histology at diagnosis showed a heterogeneous distribution of affected areas (arrow) with sparing of parts of the distal lung parenchyma (arrowhead). (I) Despite interferon α 2a therapy, at the time of death, there was progression in the size of the lesions and a decrease in the unaffected areas of lung. Some of the alveolar infiltrate is due to resuscitative efforts near the time of death.
After diagnosis was confirmed, the patient was started on PEG-interferon α-2a and doxycycline. She remained on sildenafil 20 mg three times daily, aldactazide 12.5 mg daily and 1 l/min nasal canula oxygen. She was referred for lung transplant. At the transplant centre (~500 ft above sea level), she made significant clinical progress with a cardiac catheterisation that demonstrated RV pressures 60% systemic on 1 l/min nasal canula oxygen. After 3 months of therapy, her WHO functional class was I-II and transplantation was deferred. She returned to Colorado, where ECG showed a RVSP of 44 mm Hg above RA pressure (TRJV 3.4 m/s) on 1 l/min nasal canula oxygen. A polysomnogram showed a waking room air saturation of 87% and a sleeping room air saturation of 84% rising to 93% on 1/8 l/min nasal canula oxygen.
Over the next 2 months, she experienced frequent desaturation events and poor weight gain. She developed anaemia, a side effect of interferon therapy. She was admitted 6 months after diagnosis with progressive pulmonary hypertension and pulmonary oedema. At that time, her RVSP was estimated at 103 mm Hg above RA pressure (TRJV 5.06 m/s). She was started on intravenous furosemide in addition to her sildenafil, interferon α-2a and doxycycline. Her condition rapidly deteriorated, and she required mechanical ventilation, inotropic support and emergent atrial septostomy secondary to severe pulmonary hypertension and systemic hypotension. She died several days later within the time frame previously reported for PCH (figure 3).1 An autopsy was performed which was notable for progression of capillary haemangiomas (figure 2I).
Figure 3.
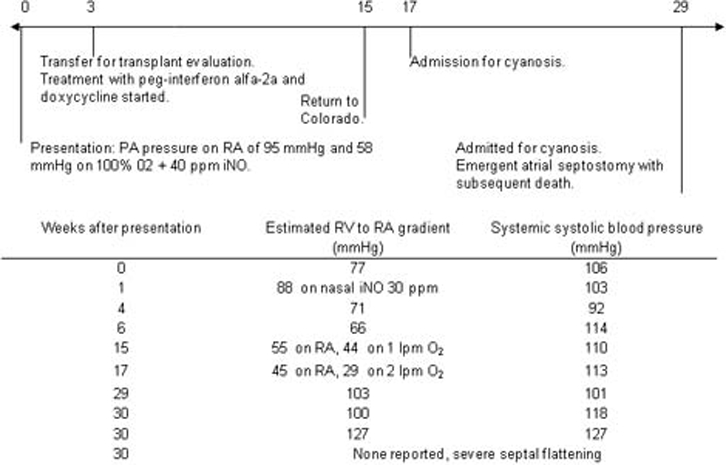
Timeline of disease progression from diagnosis to death with changes over time in right ventricular pressure, estimated by ECG. iNO, inhaled nitric oxide.
Discussion
The diagnosis of PCH is based on characteristic lung histology in a patient with pulmonary hypertension. Most PCH patients are aged 10–40,1 with rare cases reported in neonates7 and the elderly. PCH has been reported in association with hereditary haemorrhagic telangiectasia,8 SLE,9 hypertrophic cardiomyopathy,10 as an incidental finding during surgery11 12 and at autopsy.13 14 The most common presenting symptoms of PCH are dyspnoea, haemoptysis and cough. Physical exam often demonstrates cyanosis, clubbing and stigmata of right ventricular failure.1 Pulmonary function testing most commonly shows restriction with a decreased diffusion capacity in the lung for carbon monoxide.1 Several HRCT findings have been noted, including centrilobular nodular opacities, ground glass attenuation, interstitial infiltrates and septal thickening.1 15 16 Angiography and pulmonary capillary wedge pressure are often normal.1
Pathologically, PCH is characterised by diffuse, patchy distribution of hyperplastic, non-clonal endothelial cells invading the alveolar walls, perivascular spaces and, at times, small airways. Associated arteriolar medial hypertrophy is common.17 A genetic basis for the disease is not yet known, although there is a report of a family with three affected children in an autosomal recessive pattern.18 There is increased expression of vascular endothelial growth factor (VEGF), platelet-derived growth factor-B, platelet-derived growth factor receptor α and β, angiopoetin 1 and CD-117.19–21 Decreased nitric oxide synthase III expression has been reported in PCH patients with severe pulmonary hypertension versus those with normal PAP.22
The mortality is high (80–90%) with death often occurring within 6 months of presentation.1 There are fewer than 60 cases reported in the literature, and the majority of cases were diagnosed at autopsy.1 Given this, we recommend early consideration of lung biopsy.
Regarding therapies, a small series of patients improved with interferon α-2a,2 which disrupts basic fibroblast growth factor (bFGF) and VEGF signalling pathways, theoretically interfering with neoangiogenesis.23 24 There is a report of successful doxycycline therapy, which may inhibit neoangiogenesis through inhibition of matrix metalloproteinase 9 activity.3 Mortality in PCH is due to right heart failure from progressive pulmonary hypertension, presumably caused by a compensatory arteriolar response to limit flow to the pulmonary capillary haemangiomas. For this reason, pulmonary vasodilator therapy should be considered. However, fatal pulmonary oedema has been reported in response to pulmonary vasodilators in patients with PCH.25 Despite pharmacologic treatment, disease resolution is extremely rare. Pharmacologic treatment should be utilised as a bridge to lung transplantation, which remains the only definitive therapy for PCH.
This case demonstrates PCH is difficult to treat medically and that current markers of disease progression are not sufficiently sensitive. We initially wondered if the return to altitude complicated treatment as this child showed improved haemodynamics during the first 3 months of therapy and later deteriorated, but it was clear from the autopsy findings that primary disease progression was the main cause of death. In this case, there was extensive spread of disease despite the use of interferon α-2a and doxycycline. Progressive anaemia, likely caused by interferon therapy, led to a high cardiac output state, which contributed to progressive cardiac dysfunction. In retrospect, we misinterpreted her initial haemodynamic improvement as reflecting a treatment effect of the interferon and doxycycline on her PCH, whereas it was likely secondary to the pulmonary vasodilator therapy. We depended on echocardiographic estimations of RVSP to indirectly assess secondary vascular changes (figure 3). Urinary bFGF has been suggested as a potential marker of disease progression, but is not a commonly available assay.3
Our experience has led us to recommend lung biopsy in patients with parenchymal lung disease, pulmonary hypertension and normal cardiac function. In patients who present with severe pulmonary hypertension, many of the arteriolar changes may be irreversible at the time of presentation. Severe, and somewhat unpredictable, decompensation can occur. In patients with the biopsy-proven diagnosis of PCH, interferon α-2a and doxycycline should be considered as a bridge to lung transplant. The combination of poor markers of disease severity, rapid disease progression and a variable response to available therapies underscore the urgency to proceed with lung transplantation.
Learning points.
-
▶
Early lung biopsy should be considered in cases with parenchymal lung disease, pulmonary hypertension and normal cardiac function.
-
▶
Echocardiographic estimations of right ventricular pressure may not be representative of the degree of paediatric lung disease or pulmonary hypertension.
-
▶
PCH is a fatal disease, and lung transplantation is the only definitive treatment at this time.
Footnotes
Competing interests None.
Patient consent Not obtained.
References
- 1.Almagro P, Julià J, Sanjaume M, et al. Pulmonary capillary hemangiomatosis associated with primary pulmonary hypertension: report of 2 new cases and review of 35 cases from the literature. Medicine (Baltimore) 2002;81:417–24 [DOI] [PubMed] [Google Scholar]
- 2.White CW, Sondheimer HM, Crouch EC, et al. Treatment of pulmonary hemangiomatosis with recombinant interferon alfa-2a. N Engl J Med 1989;320:1197–200 [DOI] [PubMed] [Google Scholar]
- 3.Ginns LC, Roberts DH, Mark EJ, et al. Pulmonary capillary hemangiomatosis with atypical endotheliomatosis: successful antiangiogenic therapy with doxycycline. Chest 2003;124:2017–22 [DOI] [PubMed] [Google Scholar]
- 4.Simonneau G, Robbins IM, Beghetti M, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 2009;54(1 Suppl):S43–54 [DOI] [PubMed] [Google Scholar]
- 5.Hoeper MM. Definition, classification, and epidemiology of pulmonary arterial hypertension. Semin Respir Crit Care Med 2009;30:369–75 [DOI] [PubMed] [Google Scholar]
- 6.Simonneau G, Galiè N, Rubin LJ, et al. Clinical classification of pulmonary hypertension. J Am Coll Cardiol 2004;43(12 Suppl S):5S–12S [DOI] [PubMed] [Google Scholar]
- 7.Oviedo A, Abramson LP, Worthington R, et al. Congenital pulmonary capillary hemangiomatosis: Report of two cases and review of the literature. Pediatr Pulmonol 2003;36:253–6 [DOI] [PubMed] [Google Scholar]
- 8.Varnholt H, Kradin R. Pulmonary capillary hemangiomatosis arising in hereditary hemorrhagic telangiectasia. Hum Pathol 2004;35:266–8 [DOI] [PubMed] [Google Scholar]
- 9.Fernández-Alonso J, Zulueta T, Reyes-Ramirez JR, et al. Pulmonary capillary hemangiomatosis as cause of pulmonary hypertension in a young woman with systemic lupus erythematosus. J Rheumatol 1999;26:231–3 [PubMed] [Google Scholar]
- 10.Jing X, Yokoi T, Nakamura Y, et al. Pulmonary capillary hemangiomatosis: a unique feature of congestive vasculopathy associated with hypertrophic cardiomyopathy. Arch Pathol Lab Med 1998;122:94–6 [PubMed] [Google Scholar]
- 11.Cioffi U, De Simone M, Pavoni G, et al. Pulmonary capillary hemangiomatosis in an asymptomatic elderly patient. Int Surg 1999;84:168–70 [PubMed] [Google Scholar]
- 12.Moritani S, Ichihara S, Seki Y, et al. Pulmonary capillary hemangiomatosis incidentally detected in a lobectomy specimen for a metastatic colon cancer. Pathol Int 2006;56:350–7 [DOI] [PubMed] [Google Scholar]
- 13.Umezu H, Naito M, Yagisawa K, et al. An autopsy case of pulmonary capillary hemangiomatosis without evidence of pulmonary hypertension. Virchows Arch 2001;439:586–92 [DOI] [PubMed] [Google Scholar]
- 14.Havlik DM, Massie LW, Williams WL, et al. Pulmonary capillary hemangiomatosis-like foci. An autopsy study of 8 cases. Am J Clin Pathol 2000;113:655–62 [DOI] [PubMed] [Google Scholar]
- 15.Dufour B, Maître S, Humbert M, et al. High-resolution CT of the chest in four patients with pulmonary capillary hemangiomatosis or pulmonary venoocclusive disease. AJR Am J Roentgenol 1998;171:1321–4 [DOI] [PubMed] [Google Scholar]
- 16.Frazier AA, Franks TJ, Mohammed TL, et al. From the Archives of the AFIP: pulmonary veno-occlusive disease and pulmonary capillary hemangiomatosis. Radiographics 2007;27:867–82 [DOI] [PubMed] [Google Scholar]
- 17.Pietra GG, Capron F, Stewart S, et al. Pathologic assessment of vasculopathies in pulmonary hypertension. J Am Coll Cardiol 2004;43(12 Suppl S):25S–32S [DOI] [PubMed] [Google Scholar]
- 18.Langleben D, Heneghan JM, Batten AP, et al. Familial pulmonary capillary hemangiomatosis resulting in primary pulmonary hypertension. Ann Intern Med 1988;109:106–9 [DOI] [PubMed] [Google Scholar]
- 19.Kawut SM, Assaad AM, Arcasoy SM, et al. Pulmonary capillary hemangiomatosis: results of gene expression analysis. Chest 2005;128(6 Suppl):575S–5766S [DOI] [PubMed] [Google Scholar]
- 20.Assaad AM, Kawut SM, Arcasoy SM, et al. Platelet-derived growth factor is increased in pulmonary capillary hemangiomatosis. Chest 2007;131:850–5 [DOI] [PubMed] [Google Scholar]
- 21.Sullivan A, Chmura K, Cool CD, et al. Pulmonary capillary hemangiomatosis: an immunohistochemical analysis of vascular remodeling in a fatal case. Chest 2005;128(6 Suppl):576S. [DOI] [PubMed] [Google Scholar]
- 22.Kradin R, Matsubara O, Mark EJ. Endothelial nitric oxide synthase expression in pulmonary capillary hemangiomatosis. Exp Mol Pathol 2005;79:194–7 [DOI] [PubMed] [Google Scholar]
- 23.Wu WZ, Sun HC, Shen YF, et al. Interferon alpha 2a down-regulates VEGF expression through PI3 kinase and MAP kinase signaling pathways. J Cancer Res Clin Oncol 2005;131:169–78 [DOI] [PubMed] [Google Scholar]
- 24.Torcia M, Lucibello M, De Chiara G, et al. Interferon-alpha-induced inhibition of B16 melanoma cell proliferation: interference with the bFGF autocrine growth circuit. Biochem Biophys Res Commun 1999;262:838–44 [DOI] [PubMed] [Google Scholar]
- 25.Gugnani MK, Pierson C, Vanderheide R, et al. Pulmonary edema complicating prostacyclin therapy in pulmonary hypertension associated with scleroderma: a case of pulmonary capillary hemangiomatosis. Arthritis Rheum 2000;43:699–703 [DOI] [PubMed] [Google Scholar]