

Non-technical summary
Exposure to chronic intermittent hypoxia (CIH) leads to chronically elevated sympathetic nerve activity (SNA) and mean arterial pressure (MAP) with exaggerated rises in SNA and MAP to activation of peripheral chemoreceptors. We show that CIH leads to augmented rises in SNA by stimulation of reflexes from other sensory nerves, suggesting a global change in how the brain processes sensory information. Furthermore, the ability of glutamate in the rostral ventrolateral medulla to increase SNA and MAP is enhanced after CIH, providing a potential brain mechanism for CIH-induced exaggeration of sympathetic reflexes. Paradoxically, exaggerated sympathetic reflexes were accompanied by normal rises in MAP. In agreement, activation of adrenergic vascular receptors yields blunted rises in MAP in rats after CIH. These data suggest that exposure to CIH facilitates rises in SNA, potentially by changes in the brainstem, which are buffered by a reduction in the ability of sympathetic nerves to raise MAP.
Abstract
Abstract
Exposure to chronic intermittent hypoxia (CIH) yields persistent elevations in sympathetic nerve activity (SNA) and mean arterial pressure (MAP) with exaggerated sympathetic chemoreflexes. We examined the impact of CIH upon other sympathoexcitatory reflexes and a potential central mechanism underlying the altered regulation of SNA. Male Sprague–Dawley rats were exposed to CIH for 2 weeks (40 s at 6% O2 every 9 min, 8 h day−1). After exposure to CIH, urethane-anaesthetized, vagotomized, ventilated, paralysed rats had significantly elevated MAP, splanchnic SNA, and rate of phrenic nerve discharge (PND; P < 0.05). Elimination of SNA by ganglionic blockade produced a larger fall in MAP in rats exposed to CIH (P < 0.05). Like acute hypoxia, stimulation of the sciatic nerve or the nasal mucosa evoked greater increases in SNA after exposure to CIH (P < 0.05). In addition, acute hypoxia promoted exaggerated increases in PND amplitude after CIH (P < 0.05). In contrast, the nasopharyngeal reflex evoked exaggerated increases in SNA during apnoea. These sympathoexcitatory reflexes are mediated by glutamatergic activation of the rostral ventrolateral medulla (RVLM), and accordingly, microinjections of glutamate into RVLM evoked larger increases in SNA after CIH (P < 0.05). Paradoxically, none of these exaggerated acute rises in SNA was accompanied by enhanced pressor responses. Reduced adrenergic vascular reactivity may contribute to the blunted sympathetically mediated pressor responses, because bolus doses of phenylephrine evoked attenuated pressor responses after CIH (P < 0.01). These data suggest exposure to CIH facilitates activation of SNA, potentially by changes within the RVLM. However, the exaggerated rises in SNA are not dependent upon stimulation of inspiratory drive. Although elevated SNA may contribute to CIH-induced hypertension, reduced adrenergic vascular reactivity buffers the cardiovascular impact of exaggerated acute rises in SNA.
Introduction
Obstructive sleep apnoea (OSA) in humans promotes chronically elevated sympathetic nerve activity (SNA) and mean arterial pressure (MAP) with exaggerated sympathetic responses to acute hypoxia (Carlson et al. 1993; Imadojemu et al. 2007). Episodes of intermittent hypoxia during brief cessations of breathing with OSA appear to be a critical factor that promotes these deleterious cardiovascular changes (Somers et al. 1995; Imadojemu et al. 2007). Animal models of chronic intermittent hypoxia (CIH) have been developed to mimic the intermittent hypoxaemia produced with OSA and determine mechanisms underlying the disrupted short- and long-term regulation of SNA and MAP. As observed with OSA in humans, exposure to CIH in rats leads to elevated sympathetic vasomotor tone, MAP, and haematocrit with exaggerated sympathetic chemoreflexes (Fletcher et al. 1992; Greenberg et al. 1999a; McGuire & Bradford, 1999; Zoccal et al. 2007). The development of hypertension in patients with OSA and in rodents with CIH appears to be initiated by overstimulation of the carotid sinus nerves, which generate chemoreflex-mediated increases in SNA, because the rise in MAP is prevented by removal of the carotid bodies (Fletcher et al. 1992; Somers & Abboud, 1993). In addition, an intact sympathetic nervous system is also essential for CIH-induced hypertension (Bao et al. 1997; Lesske et al. 1997).
Changes within the central nervous system that contribute to chronically elevated SNA and exaggerated acute sympathetic chemoreflexes with CIH are not well understood. Furthermore, whether these two deficits occur by common or distinct mechanisms is also not known. The carotid sinus afferent nerves themselves are more activated by acute hypoxia after exposure to CIH, suggesting the exaggerated sympathetic response is due to a change in the sensation of the stimulus in the periphery (Peng & Prabhakar, 2004). However, the sympathetic response to hypercapnia is also accentuated after exposure to CIH (Greenberg et al. 1999a), coincident with normal carotid body afferent nerve responses (Peng & Prabhakar, 2004). Because the hypercapnic sympathetic chemoreflex is initiated by chemoreceptors within the brain (Guyenet et al. 2010), these data suggest that altered sympathetic regulation with CIH includes changes in central mechanisms. The common link for sympathoactivation by these two types of chemoreflexes is excitation of presympathetic neurones in the rostral ventrolateral medulla (RVLM; Guyenet, 2000; Guyenet et al. 2010). Moreover, the RVLM appears to be more activated under resting conditions after exposure to CIH as indicated by increased expression of c-fos (Greenberg et al. 1999b). Together, these data suggest the RVLM plays an important role in CIH-induced alterations in the short- and long-term regulation of SNA.
Presympathetic neurones in the RVLM and SNA are prominently modulated by the central respiratory cycle (Haselton & Guyenet, 1989), and exposure to CIH increases ventilatory drive and respiratory-related modulation of SNA (Reeves et al. 2003; Zoccal et al. 2008). Nevertheless, contributions of altered central respiratory regulatory mechanisms to CIH-induced elevation in SNA have not been elucidated. After exposure to CIH, acute hypoxia evokes enhanced increases in ventilatory tidal volume in conscious rats (Reeves et al. 2003) and central respiratory drive, as estimated by amplitude of phrenic nerve discharge (PND), in anaesthetized, ventilated, vagotomized rats (Ling et al. 2001; Peng & Prabhakar, 2003). However, it is not known whether stimulated central respiratory drive is necessary to evoke acute exaggerated sympathetic responses after exposure to CIH.
One aim of this study was to determine whether sympathetic reflexes initiated by peripheral nerves other than the carotid sinus nerve also produce exaggerated sympathetic responses in rats exposed to CIH. Specifically, we hypothesized that the nasopharyngeal reflex and the somatosympathetic reflex, which both increase SNA by glutamatergic activation of the RVLM (Kiely & Gordon, 1994; McCulloch et al. 1999), would be exaggerated after exposure to CIH. In addition, we sought to examine whether exposure to CIH enhances the sympathoactivation evoked by glutamatergic stimulation of the RVLM. Lastly, we examined whether an exaggerated increase in central respiratory drive was required to evoke enhanced sympathetic responses after exposure to CIH. Together these experiments begin to unravel changes in central regulatory mechanisms for SNA that occur with exposure to CIH.
Methods
Ethical approval
All experiments were performed using male Sprague–Dawley rats purchased at 8 weeks of age from Harlan Laboratories Inc. (Indianapolis, IN, USA; http://www.harlan.com) in accordance with the National Institutes of Health's Guide for Care and Use of Laboratory Animals. The animal protocols were reviewed and approved by the Medical College of Georgia Institutional Animal Care and Use Committee. Experiments complied with the policies and regulations set forth by The Journal of Physiology and UK regulations (Drummond, 2009).
Exposure to chronic intermittent hypoxia (CIH)
Groups of four to six rats were housed in Plexiglas cages with open wire tops (167.5 sq inches or 425.5 cm2, Super rat 1400) equipped for access to food and water ad libitum. Two animal cages were placed in each chamber (76.2 × 50.8 × 50.8 cm, BioSpherix, Lacona, NY, USA; http://www.biospherix.com) for exposure to CIH or ambient levels of oxygen. All chambers were equipped with gas injectors and sensors for oxygen levels, humidity and temperature. The large chamber size was used to minimize the perception of forced air flows, which were infused near the top of the chambers. The chambers for CIH were infused with 100% nitrogen for 4 min to reduce the concentration of oxygen briefly to 6% (∼40 s; Fig. 1). Then, the chambers were infused with 100% oxygen for 5 min to restore oxygen to ambient levels of 21% until the start of the next cycle. These 9 min cycles were repeated for 8 h each day during the rat sleep period (09.00 h to 17.00 h) for 14 days, as reported by other studies utilizing this CIH model (Braga et al. 2006; Kline et al. 2007). Control chambers were infused with room air and oxygen to maintain 21% oxygen and humidity levels (51–61%) consistent with chambers for CIH. The air flows were regulated by solenoid valves that were controlled by a computerized system programmed to impose the intended oxygen levels and cycles (OxyCycler system, Biospherix). An ActiVent regulator (Model A84CA-A, Biospherix) imposed equivalent timing and force of air flows in control chambers. During the other 16 h of the day, both chambers were flushed with room air to maintain oxygen levels at 21%. The temperature inside the chambers ranged from 21 to 23°C, consistent with room conditions. The concentrations of oxygen in all chambers were continuously recorded by the computer and displayed to ensure accuracy of cycles (Fig. 1).
Figure 1. Representative recordings of oxygen levels for rats exposed to chronic intermittent hypoxia (CIH) or ambient oxygen.
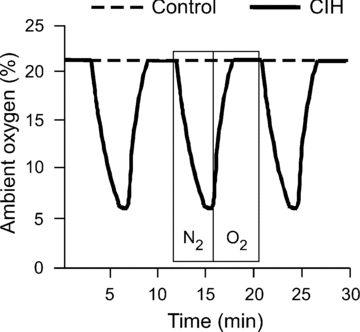
This 30 min period depicts 3 complete cycles of CIH in a chamber containing experimental rats, and a constant level of 21% oxygen in a chamber containing control rats. Boxes represent the periods of infusion of 100% nitrogen or oxygen in the CIH chamber, corresponding to 1 cycle.
Acute surgical procedures
After 14–21 days of exposure to CIH or 21% oxygen, rats were anaesthetized with isoflurane (Webster Veterinary, Sterling, MA, USA; http://www.webstervet.com) in 100% O2. Anaesthesia was induced with 5% isoflurane and maintained for surgery at 1.9–2.1%. Adequate anaesthesic level was confirmed by absence of leg flexion in response to a firm toe pinch. Catheters were placed into the femoral artery and vein to measure MAP and administer drugs, respectively. Catheters were placed rostrally into the trachea to evoke the nasopharyngeal reflex (PE-205), and caudally for artificial ventilation (Model 683, Harvard Apparatus, Holliston, MA, USA; http://www.harvardapparatus.com). Rats were ventilated with 100% O2 (1 ml 100 (g body weight)−1), and respiratory frequency was adjusted to maintain end-tidal CO2 at 3.8–4.2% (CapStar-100, Charles Ward Electronics, Ardmore, PA, USA; http://www.cwe-inc.com). After confirmation of an adequate level of anaesthesia (<10 mmHg change in MAP to firm toe pinch), the neuromuscular blocker, pancuronium, was administered (i.v., 1 mg kg−1, supplemented hourly at 1/3 the dose). The cervical vagus nerves were sectioned bilaterally to allow for the measurement of central respiratory drive independently of artificial ventilation as described previously (Mandel & Schreihofer, 2006, 2009). The animals were placed on a stereotaxic apparatus (David Kopf Instruments, Tujunga, CA, USA; http://www.kopfinstruments.com) with the bite bar at −11 mm. The left splanchnic sympathetic nerve was isolated, placed on two silver wires (Teflon-coated and bared 250 μm at tips, A-M Systems, Carlsborg, WA, USA; http://www.a-msystems.com) and covered with kwik-sil (World Precision Instruments, Sarasota, FL, USA http://www.wpiinc.com). The left phrenic nerve was isolated, distally cut, placed on two silver wires and covered in kwik-sil. For microinjections of drugs into the brain, a partial occipital craniotomy was performed to expose the dorsal surface of the brainstem caudal to the cerebellum. For stimulation of the spinal cord a dorsal midline incision was made to expose the spinal column. The dorsal processes of lower thoracic vertebrae were clamped to slightly elevate the rat. This served to minimize movements due to ventilation and allow for insertion of the stimulating electrode between two vertebrae. After excision of the disc and overlying dura mater, a bipolar stimulating electrode was placed in the dorsolateral funiculus between the first and second thoracic vertebrae. The region was coated in mineral oil to prevent desiccation. After completion of all surgical procedures, the animals were infused with urethane (Sigma, 1.5 mg kg−1, i.v.) whilst isoflurane administration was gradually terminated. Rectal temperature was monitored to maintain core temperature at 37°C (Homeothermic blanket control unit, Harvard apparatus) throughout surgical and experimental procedures.
Activation of sympathoexcitatory reflexes
The peripheral chemoreflex was activated by intravenous injection of potassium cyanide (200 μg kg−1) or by acute hypoxia. To induce acute hypoxia, the O2 flowing through the ventilation tubing was reduced from 100% to 10% for 60 s. The use of these two methods enabled the stimulation of SNA with different changes in MAP. Injections of cyanide evoked sympathetically mediated pressor responses, whereas the pressor response to hypoxia was reduced by direct vasodilatory effects of the stimulus (e.g. Marshall et al. 1993).
As previously described (Stornetta et al. 1989), the somatosympathetic reflex was activated by stimulating a sciatic nerve (33 Hz for 5 s at 1, 2, 4, 6, 8, and 10 mV with 3 min between each stimulus). To evoke the nasopharyngeal or simulated diving response (McCulloch & West, 1992; McCulloch et al. 1999), the nasal mucosa was stimulated by passing water (2 ml) through the nose via the rostral tracheal catheter. Because the rats were vagotomized to investigate changes in central respiratory drive (Mandel & Schreihofer, 2006), the robust vagally mediated bradycardia normally observed with the nasopharyngeal reflex was minimized (see Fig. 5A). Thus, the bradycardia-induced reduction in MAP often observed in the intact state (McCulloch & West, 1992) was reversed to a pressor response due to the reflex-induced rise in SNA (McCulloch et al. 1999). In contrast to hypoxia-induced stimulation of central respiratory drive, nasopharyngeal sympathoexcitatory stimulus occurs coincident with apnoea, allowing for the examination of the sympathetic response in the absence of activated central inspiratory drive.
Figure 5. Physiological responses to stimulation of the nasopharyngeal reflex.
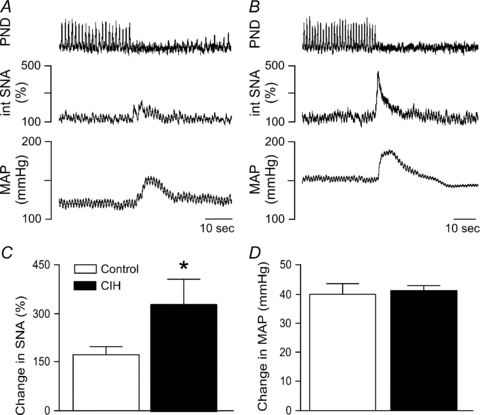
Stimulation of the nasopharyngeal reflex (2 ml of water passed through the nose) evoked rises in SNA and MAP in a control rat (A) and a rat exposed to CIH (B). The rises in SNA were exaggerated in rats exposed to CIH (n = 22) compared to control rats (n = 17, C). *Significantly different from control rats, P < 0.05, unpaired t test.
Microinjections into the RVLM
Although the three sympathoexcitatory reflexes examined in the present study are initiated by different afferent nerves, the peripheral chemoreflex, the somatosympathetic reflex, and the nasopharyngeal reflex all increase SNA and MAP by glutamatergic activation of the RVLM (Kiely & Gordon, 1994; Sun & Reis, 1995; McCulloch et al. 1999). Therefore, we investigated the ability of glutamate in the RVLM to increase SNA and MAP in control rats and rats exposed to CIH. The RVLM was located by mapping the region for maximal pressor responses to microinjections of glutamate (1 nmol in 100 nl of artificial cerebrospinal fluid) as previously described (Schreihofer et al. 2005b). The optimal coordinates for increasing SNA and MAP were 1.7 mm lateral from the midline, 1.6 mm rostral to the calamus scriptorius and 2.7 mm below the dorsal surface of the brainstem. Microinjections were performed by using glass pipettes pulled to 50 μm tip diameter with the tip positioned 20 deg rostrally. Glutamate was injected bilaterally (1 nmol in 100 nl), and the physiological responses were averaged for each rat. Green latex microspheres (5%, Lumiphore) were mixed into injectate to allow for histological confirmation of injection sites. After the completion of the experimental procedures, the anaesthetized rats were perfused transcardially with phosphate-buffered saline (250 ml, pH 7.4) followed by 4% formaldehyde (500 ml, pH 7.4). The brains were removed and stored in the fixative for 48 h and then sectioned using a Vibratome™ (50 μm coronal sections). The sections were mounted onto glass slides, and coverslips were applied with Krystalon. The microinjection sites were visualized via epifluorescence (Olympus BX40) and were verified to be in the region of the RVLM.
Spinal cord stimulation
To eliminate the possibility that exaggerated sympathetic responses in rats exposed to CIH may be due to spinal or sympathetic ganglionic mechanisms, we measured sympathetic responses to direct stimulation of presympathetic axonal fibres in the upper thoracic spinal cord. The spinal cord was electrically stimulated at 300 μA with 1 ms pulses for 5 s at five intensities (10, 20, 50, 75, and 100 Hz) with 5 min between each stimulus.
Blood volume measurements
A separate set of rats was anaesthetized with isoflurane for implantation of catheters into a femoral artery and vein. As previously described (Schreihofer et al. 2005a), Evans Blue dye (Sigma) was injected intravenously (200 μl of 0.5% solution in saline). After 10 min, 1 ml of blood was withdrawn from the arterial catheter to measure haematocrit (I-Stat Clinical Analyzer System, Heska Corporation, Loveland, CO, USA) and the dilution of dye in plasma. The blood sample was centrifuged (532 g for 5 min), and 200 μl of plasma was diluted into 1.8 ml of saline in duplicate. The absorbance was measured at 605 nm, and the two values were averaged from each rat (Wang, 1959). A standard curve was generated by using 10 ml of donor blood. One of seven amounts of dye (0–200 μg) was added to each of the 1 ml aliquots of donor blood. After centrifugation, 200 μl of plasma was diluted into 1.8 ml of saline for measurement of absorbance at 605 nm. Blood volume was calculated from the plasma volume in relation to the percentage of packed cells in the haematocrit (blood volume = plasma volume (100 − haematocrit)−1) (Wang, 1959).
Whole body pressor reactivity to phenylephrine
After estimation of blood volume, rats were artificially ventilated and the splanchnic nerve was prepared for recording. The autonomic ganglionic antagonist mecamylamine (10 mg kg−1, Sigma), was administered intravenously to eliminate endogenous SNA and baroreceptor-mediated changes in SNA due to evoked changes in MAP (Schreihofer et al. 2005a). Bolus doses of phenylephrine (1–200 μg kg−1 in 50 μl of saline) were administered in a random sequence to evaluate the effects of α1-adrenoreceptor-mediated whole body pressor reactivity as an index of α-adrenergic vascular reactivity (Schreihofer et al. 2005a,b;). The interval between injections was 5 min or until MAP returned to 95% of baseline for 5 min. We examined a subset of rats that did not undergo the previous experiment to estimate blood volume to eliminate the possibility that the previous blood draw would affect the magnitude of the pressor responses. No differences were observed, so the data from these rats were pooled.
Data recording and analyses
The analog signals were converted to a digital output (Micro1401, Cambridge Electronic Design, Cambridge, UK; http://www.ced.co.uk) and all physiological variables were simultaneously viewed and recorded online (Spike2 software, Cambridge Electronic Design). The pulsatile arterial pressure and MAP were sampled at 100 Hz. The HR was triggered from the rising phase of the arterial pressure pulse (spike trigger). The splanchnic SNA was filtered (10 Hz to 3 kHz), amplified (×29,000), and sampled at 4 kHz. Raw SNA was processed for quantification by activating the functions DC Remove (0.01 time constant), full-wave rectification, and smoothing (0.01 time constant) using Spike2 software. The PND was filtered (3 Hz to 3 kHz), amplified (×98,000), and sampled at 4 kHz. The PND was rectified and smoothed (0.1 s bins) using Spike2 software.
Initial baseline values were a 5 min average of the variables before the performance of the first experimental protocol. The responses evoked by the tested reflexes were measured as the difference between baseline (mean of the immediate 30 s prior to these procedures) and the peak responses. For quantification of changes in SNA, the baseline integrated SNA was set at 100%. The minimum SNA (0%) was the noise voltage after administration of the ganglionic antagonist mecamylamine (10 mg kg−1). Data are presented as means ±s.e.m with statistical significance considered at P < 0.05, using Student's unpaired t test for pairwise comparisons or repeated measures ANOVA with the Tukey–Kramer post hoc test for multiple comparisons. Microsoft Excel was used to calculate means and standard errors of the mean, and for t tests. Prism software (GraphPad Software Inc., La Jolla, CA, USA) was used to perform ANOVA with post hoc tests.
Results
Physiological effects of exposure to CIH
Urethane-anaesthetized rats previously exposed to CIH had a significantly higher baseline SNA and MAP compared to control rats (Table 1), in agreement with previous reports in conscious and anaesthetized rats. Administration of an autonomic ganglionic antagonist (mecamylamine, 10 mg kg−1, i.v.) eliminated SNA and reduced MAP to comparable levels in control and experimental rats. The decrease in MAP was significantly greater in CIH rats (Table 1), suggesting an enhanced contribution of SNA to resting MAP (Santajuliana et al. 1996). In addition, the decrease in HR with ganglionic blockade was also greater in CIH rats (Table 1).
Table 1.
Effects of exposure to chronic intermittent hypoxia (CIH) on mean arterial pressure (MAP), heart rate (HR), splanchnic sympathetic nerve activity (SNA) and autonomic contributions to MAP and HR
| Group | n | SNA (μV) | MAP (mmHg) | HR (bpm) | Change in MAP (mmHg) | Change in HR (bpm) |
|---|---|---|---|---|---|---|
| Control | 16 | 2.4 ± 0.1 | 113 ± 4 | 355 ± 7 | −48 ± 4 | −68 ± 6 |
| CIH | 19 | 3.6 ± 0.3* | 122 ± 3* | 357 ± 6 | −60 ± 3* | −110 ± 4* |
All values are expressed as means ±s.e.m. Changes in MAP and HR are decreases with autonomic ganglionic blockade.
Significantly different from control rats, P < 0.05, unpaired t test.
Despite comparable initial body weights for CIH and control rats (213 ± 4 g vs. 215 ± 6 g), after exposure to CIH, rats weighed significantly less than control rats (Table 2), likely to be due to a reduction in food intake (Singh & Selvamurthy, 1993). As shown previously in humans and rats (McGuire & Bradford, 1999; Silverberg et al. 2002), exposure to CIH increased haematocrit (Table 2). Plasma volume was reduced in CIH rats, contributing to a lower total blood volume (Table 2). However, control and CIH rats had comparable blood volumes in relation to body weight (Table 2). Therefore, bolus intravenous drug doses were administered by body weight.
Table 2.
Determination of haematocrit (Ht), plasma volume (PV) and blood volume (BV) with Evans Blue in chronic intermittent hypoxia and control rats
| Group | n | BW (g) | Ht (%) | PV (ml) | BV (ml) | BV/100 g (ml) |
|---|---|---|---|---|---|---|
| Control | 8 | 335 ± 12 | 38 ± 0.9 | 22.4 ± 1 | 37.3 ± 1.8 | 11.2 ± 0.6 |
| CIH | 12 | 258 ± 8* | 43 ± 0.7* | 17.7 ± 0.5* | 31.3 ± 1* | 12.2 ± 0.4 |
All values are expressed as means ±s.e.m.
P < 0.05, unpaired t test.
Effects of exposure to CIH on peripheral chemoreflexes
Stimulation of peripheral chemoreceptors with injection of potassium cyanide (200 μg kg−1, i.v.) evoked a larger increase in SNA in CIH rats (n = 7) compared to control rats (n = 8, Fig. 2A), as observed previously (Braga et al. 2006). The bradycardia normally observed with cyanide was prevented because the rats were vagotomized. This preparation allowed for the observation of the underlying sympathetically mediated rise in HR with cyanide. Under these conditions, the cyanide-induced increase in HR in CIH rats was significantly larger than in control rats (Fig. 2B). Despite the clearly exaggerated sympathoactivation, the cyanide-induced pressor response was not enhanced by exposure to CIH (Fig. 2C).
Figure 2. Physiological responses to activation of the peripheral chemoreflex with cyanide.

Injection of cyanide (200 μg kg−1, i.v.) increased splanchnic sympathetic nerve activity (SNA, A), heart rate (HR, B), and mean arterial pressure (MAP, C) in all rats. In rats exposed to CIH (n = 7), the rise in SNA and HR was exaggerated compared to control rats (n = 8). *Significantly different from control rats, P < 0.05, unpaired t test.
Stimulation of peripheral chemoreceptors with acute hypoxia (10% oxygen for 60 s) facilitated examination of chemoreflex-induced changes in central respiratory drive in addition to the sympathoactivation (Fig. 3A and B). At the onset of acute hypoxia the end tidal CO2 levels were comparable in control and CIH rats (4.01%versus 4.05%). The basal rate of PND was higher in CIH rats (23 ± 1 cycles (30 s)−1 in control rats versus 29 ± 1 cycles (30 s)−1 in rats after CIH, P < 0.05), as previously reported in conscious rats (Reeves et al. 2003). However, the increase in PND rate with acute hypoxia was not different between groups (Fig. 3C). In contrast, although the basal amplitude of PND was not different between groups (2.2 ± 0.5 μV in control rats and 3.9 ± 1.1 μV in CIH rats), acute hypoxia evoked a significantly larger rise in the amplitude of PND in CIH rats compared to control rats (Fig. 3D). As observed with cyanide, acute hypoxia induced a greater increase in SNA in CIH rats (Fig. 3A, B and E). Whether the enhanced increase in central respiratory drive contributed to the exaggerated sympathetic response in CIH rats was not determined. The changes in MAP were variable due to simultaneous sympathetically mediated vasoconstriction and opposing vasodilatory effects of hypoxia upon the vasculature (Marshall et al. 1993).
Figure 3. Physiological responses to inspiration of hypoxic air.
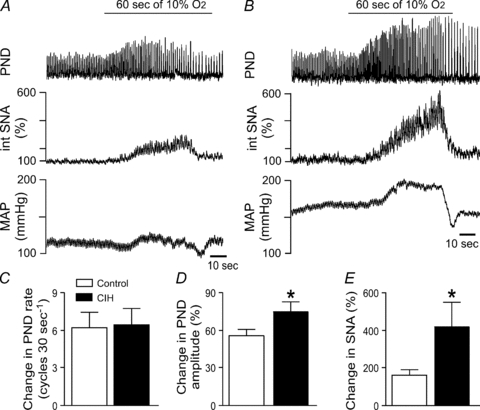
Acute hypoxia (10% oxygen for 60 s) increased phrenic nerve activity (PND) and integrated sympathetic nerve activity (int. SNA), with variable effects on MAP in a control rat (A) and a rat exposed to CIH (B). The increase in PND rate was comparable between groups (C). In contrast, the increases in PND amplitude (D) and in SNA (E) were exaggerated in rats exposed to CIH (n = 11) compared to control rats (n = 9). *Significantly different from control rats, P < 0.05, unpaired t test.
Effects of exposure to CIH on somatosympathetic and nasopharyngeal reflexes
As observed with the peripheral chemoreflex, stimulation of the sciatic nerve induced a larger increase in SNA in CIH rats (n = 11) compared to control rats (n = 10, Fig. 4A–C). However, as observed with the responses to cyanide, the exaggerated sympathoactivation was accompanied by pressor responses that were comparable in control and CIH rats (Fig. 4D). Likewise, stimulation of the nasopharyngeal reflex evoked a greater increase in SNA in CIH rats (n = 22) compared to control rats (n = 17, Fig. 5A–C), but the rise in MAP was similar between the two groups (Fig. 5D). Immediately preceding production of the nasopharyngeal reflex, baseline MAP was somewhat elevated, but not significantly different (135 ± 4 mmHg in controls versus 142 ± 4 mmHg in rats after CIH), suggesting the lack of exaggerated pressor response in the rats after CIH was not due to a ceiling effect. In contrast to acute hypoxia, stimulation of the nasopharyngeal reflex silenced the PND in both groups (Fig. 5A and B), suggesting that exaggerated increases in central respiratory drive are not necessary to evoke enhanced sympathetic responses in CIH rats.
Figure 4. Physiological responses to stimulation of the sciatic nerve.
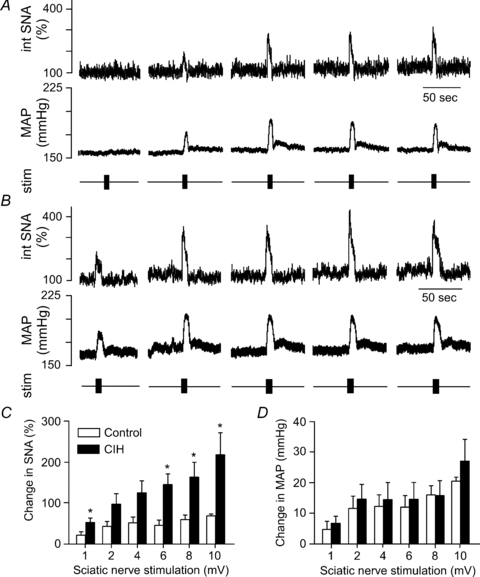
Stimulation of sciatic nerve (5 s at 33 Hz, 1, 2, 4, 6, 8, and 10 mV with 3 min between stimuli) evoked brief rises in SNA and MAP in a control rat (A) and a rat exposed to CIH (B). The rises in SNA were exaggerated in rats exposed to CIH (n = 11) compared to control rats (n = 10, C). *Significantly different from control rats at that stimulus intensity, P < 0.05, 2- way repeated measures ANOVA with Tukey–Kramer post hoc tests.
Effects of microinjections into the RVLM
Microinjections of glutamate into the RVLM evoked increases in SNA and MAP in both groups (Fig. 6). As observed with initiation of peripheral sympathoexcitatory reflexes, the increases in SNA were enhanced in CIH rats (n = 8) compared to control rats (n = 6, Fig. 6A), but the pressor responses were comparable (Fig. 6B).
Figure 6. Physiological responses to activation of the rostral ventrolateral medulla.
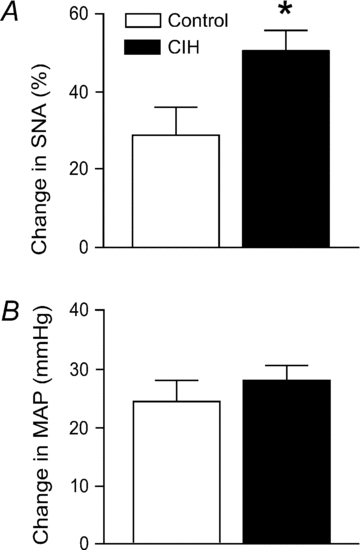
Activation of neurones in the rostral ventrolateral medulla with microinjections of glutamate (1 nmol in 100 nl) evoked exaggerated rises in SNA (A), but not in MAP (B) in rats exposed to CIH (n = 8) compared to control rats (n = 6). *Significantly different from control rats, P < 0.05, unpaired t test.
Effects of spinal cord stimulation
Stimulation of the spinal cord evoked comparable increases in SNA in all rats tested. The rises in SNA were similar between control rats and rats exposed to CIH, suggesting that the exaggerated rises in SNA evoked by reflexes or activation of the RVLM are likely to be mediated by supraspinal mechanisms. Consistent with the observed reflex responses, rises in SNA were accompanied by attenuated increases in MAP in the CIH-exposed rats (n = 5) compared to controls (n = 5, Fig. 7) at some stimulation intensities.
Figure 7. Physiological responses to stimulation of the spinal cord.
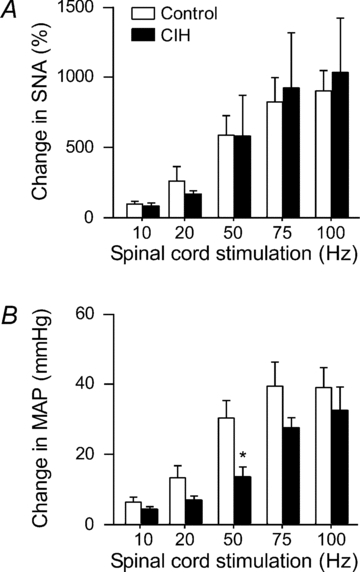
Stimulation of the spinal cord (10, 20, 50, 75 or 100 Hz, at 300 μA, 1 ms pulse for 5 s) evoked comparable rises in SNA (A), accompanied by attenuated increases in MAP (B) in rats exposed to CIH (n = 5) compared to control rats (n = 5). **Significantly different from control rats, P < 0.01, unpaired t test.
Whole body pressor reactivity to phenylephrine
After ganglionic blockade (mecamylamine, 10 mg kg−1, i.v.) to eliminate endogenous SNA and equalize MAP, a series of injections of phenylephrine was performed to evaluate α-adrenergic pressor reactivity in control rats (n = 16) and CIH rats (n = 19). Phenylephrine induced dose-dependent increases in MAP, and these responses were significantly attenuated in the CIH compared to control rats at most of the doses examined, including the maximally effective dose (Fig. 8). These data suggest that exposure to CIH chronically elevates SNA whilst blunting the ability of sympathetic nerves to constrict vessels and raise MAP.
Figure 8. Physiological responses to stimulation of adrenergic receptors.
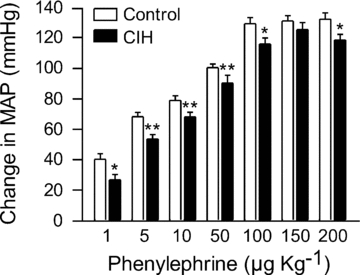
Stimulation of α1-adrenergic receptors with injections of phenylephrine (1, 5, 10, 50, 100, 150, and 200 μg kg−1, i.v.) evoked attenuated rises in MAP in rats exposed to CIH (n = 19) compared to control rats (n = 16). *P < 0.05 and **P < 0.01, significant difference from control rats, 2-way repeated measures ANVOA with Tukey–Kramer post hoc tests.
Discussion
Exposing rats to CIH promotes persistent elevations in SNA, MAP and haematocrit with exaggerated sympathetic chemoreflexes. Previous studies have suggested that enhanced afferent nerve activity from the carotid bodies contributes to the elevated SNA and exaggerated sympathetic response to acute hypoxia after CIH (Fletcher et al. 1992; Peng & Prabhakar, 2004). A major finding of the present study was that sympathoexcitatory reflexes initiated by other peripheral nerves also evoked exaggerated increases in SNA after exposure to CIH. Furthermore, stimulation of the final common pathway for these reflexes, the RVLM, evoked an augmented rise in SNA after CIH. Altered regulation by central respiratory neurones has been suggested as a contributor to the augmented basal SNA and sympathetic reflexes observed after CIH (Zoccal et al. 2009). In agreement, in the present study the rate of PND was elevated under resting conditions and the rise in amplitude of PND with acute hypoxia was exaggerated after CIH. Paradoxically, none of the exaggerated sympathoexcitatory responses were accompanied by enhanced pressor responses. After exposure to CIH rats showed attenuated whole body pressor reactivity to adrenergic stimulation, which may underlie the reduced ability of SNA to raise MAP acutely. Together these data suggest that after exposure to CIH sympathoexcitatory reflexes may be exaggerated due to enhanced excitability of the RVLM, and that acute exaggerated rises in SNA are buffered by reduced adrenergic vascular reactivity.
Numerous protocols for exposure to CIH with slight variations in parameters have been developed to investigate mechanisms underlying altered autonomic regulation of the circulation. Hypertension-inducing animal models of CIH vary in terms of frequency of cycles (30 s to 10 min), the level of hypoxia (5–10%), the duration of the hypoxic period (5 s to 5 min), number of days of exposure (10–40 days), and timing (day or night). The range of these parameters utilized for CIH across studies appear to effectively exaggerate sympathetic chemoreflexes and lead to increased haematocrit, SNA and MAP, suggesting the intermittent nature of the exposure and not the precise parameters of the CIH is the critical independent variable. The CIH model used in the present study mimicked timing and degree of hypoxia used by others (Braga et al. 2006; Kline et al. 2007) and yielded the previously reported modest hypertension with exaggerated sympathetic response to activation of peripheral chemoreceptors. The duration of the cycles, which was longer than those used by some laboratories (Fletcher et al. 1992; Greenberg et al. 1999a,b;), was partly a consequence of slower turnover of gases with housing the rats in large chambers. The rats were housed in cages placed within the large chambers to minimize their potential detection of air flows associated with the gas exchanges and prevent air jet stress-induced changes in cardiovascular function. In addition, the use of larger chambers allowed rats to be housed in groups with no limitations on naturally occurring behaviours during the extended period of exposure to CIH.
In the present study we observed elevated splanchnic SNA under resting, anaesthetized conditions that occurred with significantly elevated MAP. Acute ganglionic blockade with the nicotinic cholinergic antagonist mecamylamine abolished SNA and produced larger decreases in MAP, suggesting an augmented contribution of SNA to resting MAP (Santajuliana et al. 1996). These data are in agreement with a previous report utilizing ganglionic blockade (Zoccal et al. 2007), with a slightly different interpretation. In the previous study SNA was not directly measured, and hexamethonium was used as the ganglionic blocker. The conscious Wistar rats exposed to CIH had elevated resting MAP and plasma noradrenaline, but treatment with hexamethonium produced comparable falls in MAP in control and CIH-exposed rats. To observe a difference with ganglionic blockade prior antagonism of angiotensin II type 1 receptors was required. A plausible explanation for differences between these studies is the selection of the ganglionic antagonist. Although hexamethonium is commonly used as a ganglionic blocker in rats, it is ineffective in eliminating sympathetic vasomotor tone or sympathetically mediated reflexes (Fadhel & Seager, 1969; Abdel-Rahman, 1989; authors’ unpublished observations). In contrast, chlorisondamine and mecamylamine are quite effective for acutely eliminating SNA and blocking sympathetic reflex responses (Abdel-Rahman, 1989; Schreihofer et al. 2007). In studies utilizing ganglionic antagonists when SNA is not directly measured, some verification of the efficacy of the blockade is important for accurate interpretation of results, especially if conditions of elevated SNA may require a higher effective dose.
Central mechanisms generating the chronically elevated SNA with CIH are not well understood. Exposure to CIH in rats promotes increased expression of c-fos, a marker of neuronal activation, in several brainstem regions that are critical for normal regulation of SNA, such as the nucleus of the solitary tract and the ventrolateral medulla (Greenberg et al. 1999b). In addition, forebrain regions that affect SNA such as medial prefrontal, cingulate and insular corticies also express c-fos after CIH (Sica et al. 2000), suggesting the CIH-induced alterations in SNA involves multiple levels of the CNS. The initiation of hypertension with CIH appears to require neural inputs from the carotid bodies (Fletcher et al. 1992; Lesske et al. 1997), but whether removing this input preserves MAP due to normalization of SNA is not known. Carotid body denervation promotes a state of chronic room air alveolar hypoventilation with changes in arterial blood gases that may directly affect vascular function to reduce MAP (Fletcher et al. 1992; Eickhoff et al. 2008). Other studies measuring MAP as the dependent variable suggest that activation of the renin–angiotensin system (Fletcher et al. 1999, 2002) and reactive oxygen species (Troncoso Brindeiro et al. 2007) are important for CIH-induced hypertension. Future studies will be necessary to determine whether the brain contributes as a critical site of action for these factors via the sympathetic nervous system.
A potentiated sympathetic chemoreflex is a hallmark of exposure to CIH and has been attributed to enhanced responsiveness of carotid body afferents to the brain (Peng & Prabhakar, 2004). Although this mechanism is likely to contribute, it cannot explain the exaggerated sympathoexcitatory response to hypercapnia after CIH, which occurs with normal carotid body afferent nerve activity (Peng & Prabhakar, 2004). Furthermore, the present study showed that exposure to CIH promotes augmented sympathoexcitatory responses from reflexes initiated by other peripheral nerves, namely the nasopharyngeal and somatosympathetic reflexes. These data suggest CIH induces lasting changes within the central nervous system that utilize pathways common to these reflexes. Glutamatergic activation of the RVLM plays an essential role in evoking the acute rise in SNA with the peripheral chemoreflex, the nasopharyngeal reflex and the somatosympathetic reflex (Kiely & Gordon, 1994; Sun & Reis, 1995; McCulloch et al. 1999). In agreement, direct activation of the RVLM by microinjection of glutamate also evoked exaggerated increases in SNA in rats exposed to CIH. Whether activation of the carotid body afferents by CIH is essential to promote this persistent generalized enhancement of sympathoexcitatory reflexes is not known. Further study will be necessary to delineate the basis for and cellular mechanisms of changes to RVLM neurones induced by exposure to CIH.
Central respiratory regulatory neurones are potential contributors for altered regulation of SNA after CIH. The sympathetic nerves that target the cardiovascular system are prominently modulated by the central respiratory cycle in all species examined. These respiratory-related patterns of activity have also been reported in the cardiovascular-related neurones of the caudal and rostral ventrolateral medulla of the rat (Haselton & Guyenet, 1989; Mandel & Schreihofer, 2006). Acute hypoxia alters the activity of these medullary neurones to increase SNA in a pattern related to the central respiratory cycle, specifically with a prominent rise during the expiratory period (Dick et al. 2004; Mandel & Schreihofer, 2009). Interestingly, after exposure to CIH, SNA is elevated particularly during the expiratory period (Zoccal et al. 2008), suggesting a potential common underlying mechanism for the acute and chronic state. In addition to changes in patterning of SNA with hypoxia, exposure to CIH also promotes enduring changes in central respiratory drive. In the present study we observed an increase in the rate of PND in rats after CIH, as reported by others (Reeves et al. 2003), which may also contribute to the elevated basal SNA after CIH.
Enhanced changes in central respiratory drive evoked by acute hypoxia may also contribute to the heightened sympathetic response after CIH. In agreement, the present study reports hypoxia-induced exaggerated increases in the amplitude of PND after CIH, as seen by others (Ling et al. 2001; Reeves et al. 2003). Whether altered cardio-respiratory coupling contributes to augmented sympathoexcitatory responses after CIH is not known. Our observation that the augmented nasopharyngeal reflex after CIH occurs during apnoea suggests stimulated inspiratory drive is not essential for the enhanced sympathetic response. Nevertheless, changes in central respiratory neurones that are not reflected in the final motor output observed in the PND may play important roles in the cardio-respiratory coupling after CIH. Future studies are essential to elucidate how central respiratory neurones impact those that regulate SNA and whether this interaction is altered by exposure to CIH.
Although the sympathoexcitatory responses examined in the present study were consistently augmented by exposure to CIH, the acute pressor responses were comparable to control rats. We observed reduced whole body adrenergic pressor reactivity in rats after CIH that is likely to contribute to the attenuated ability of sympathetic nerves to constrict vessels and raise MAP. In agreement, in isolated gracilis arteries constriction to noradrenaline was blunted after rats were exposed to CIH (Phillips et al. 2006). However, reduced adrenergic vascular reactivity may not be generalized to all beds, because constriction to noradrenaline in cremaster arterioles is normal in hypertensive CIH-exposed rats (Tahawi et al. 2001). The chronically elevated SNA with CIH may lead to adrenergic desensitization at cardiovascular targets, but this theory remains to be tested. Exposure to CIH also impairs vasodilatory mechanisms and promotes structural changes in vessels (Tahawi et al. 2001; Phillips et al. 2004, 2010) that are also likely to contribute to cardiovascular deficits.
The resemblance of autonomic and cardiovascular consequences of CIH exposure in rats to the physiological changes observed in patients with OSA is striking. Although most CIH rodent models do not mimic the cessation of breathing or hypercapnia seen with OSA, CIH effectively imitates the intermittent hypoxaemia produced by OSA. As observed with rodent models of CIH, patients with OSA routinely present with elevations in haematocrit, plasma noradrenaline levels, SNA and MAP (Fletcher et al. 1992; Greenberg et al. 1999a; McGuire & Bradford, 1999; Silverberg et al. 2002; Zoccal et al. 2007). In agreement with the range of effective hypertensive rodent models of CIH, the elevated muscle SNA in humans is linked to the presence of OSA, but is not correlated with the severity of the disorder, namely the frequency of the hypoxic episodes or the minimum arterial oxygen saturation (Carlson et al. 1993), suggesting a threshold effect of hypoxic exposure. As observed with our rodent model of CIH, Imadojemu et al. (2007) saw exaggerated hypoxia-induced increases in muscle SNA coincident with normal pressor responses in OSA patients. Consistent with the observation of attenuated sympathetically mediated pressor responses, reduced adrenergic vascular reactivity has been reported in OSA patients with elevated SNA (Grote et al. 2000). Furthermore, like rodent models of CIH, patients with OSA present with exaggerated ventilatory responses to acute hypoxia (Narkiewicz et al. 1999; Imadojemu et al. 2007), but exaggerated hypoxia-induced increases in SNA can be observed during apnoea (Narkiewicz et al. 1999). The numerous common attributes of rodents exposed to CIH and patients with OSA suggests the intermittent hypoxaemia with OSA is a significant contributor to the autonomic, ventilatory, and cardiovascular consequences of the disorder.
Conclusions
In conclusion, our findings show that exposing rats to CIH leads to persistent elevations in SNA and widespread augmentation of acute sympathoexcitatory reflexes. Although CIH-induced changes in carotid body afferent nerves may initiate this cascade of altered autonomic regulation, the established state includes changes within central autonomic regulatory centres that promote increased SNA. Stimulation of the primary driver for SNA, the RVLM, evokes exaggerated increases in SNA after CIH, which is likely to contribute to the observed augmentation of sympathoexcitatory reflexes. Although sympathetic vasomotor tone contributes to elevated basal MAP, acute reflex-mediated rises in SNA are met with muted pressor responses. Whole body adrenergic pressor reactivity may be an adaptive desensitization to chronically elevated SNA that buffers against acute exaggerated rises in SNA. Because CIH produces distinct changes in adrenergic vascular reactivity among beds, the sites critical for the chronically elevated sympathetic contribution to MAP versus the blunted acute sympathetically mediated changes in MAP may differ. Alternatively, the basal SNA may be elevated sufficiently after CIH to raise vascular resistance in the face of reduced adrenergic vascular reactivity, and the development of hypertension may emerge as the disease state progresses. Although the rodent model of CIH does not precisely match all of the events of OSA in humans, there are remarkable similarities between these two conditions that suggest the intermittent episodes of hypoxia in OSA are a primary factor for the observed deleterious cardiovascular consequences. Moreover, the rodent models of CIH are a powerful tool for the dissection of cellular changes within the brain and periphery that are likely to provide important insights into the mechanisms contributing to cardiovascular consequences of OSA in humans.
Acknowledgments
This work was funded by a grant to A.M.S. from the Heart, Lung, and Blood Institute at the National Institutes of Health (HL075174) and a postdoctoral fellowship from the American Heart Association to A.Q.S. (10POST4720006).
Glossary
Abbreviations
- CIH
chronic intermittent hypoxia
- HR
heart rate
- MAP
mean arterial pressure
- PND
phrenic nerve discharge
- RVLM
rostral ventrolateral medulla
- SNA
sympathetic nerve activity
Author contributions
A.Q.S. and A.M.S. contributed to the conception and the design of the study. The experiments were performed by A.Q.S. with the guidance of A.M.S. The manuscript was prepared and edited by both authors, and both approved the submitted version. The experiments were conducted at the Medical College of Georgia in Augusta.
References
- Abdel-Rahman A-R. Inadequate blockade by hexamethonium of the baroreceptor heart rate response in anesthetized and conscious rats. Arch Int Pharmacodyn Ther. 1989;297:68–85. [PubMed] [Google Scholar]
- Bao G, Metreveli N, Li R, Taylor A, Fletcher EC. Blood pressure response to chronic episodic hypoxia: role of the sympathetic nervous system. J Appl Physiol. 1997;83:95–101. doi: 10.1152/jappl.1997.83.1.95. [DOI] [PubMed] [Google Scholar]
- Braga VA, Soriano RN, Machado BH. Sympathoexcitatory response to peripheral chemoreflex activation is enhanced in juvenile rats exposed to chronic intermittent hypoxia. Exp Physiol. 2006;91:1025–1031. doi: 10.1113/expphysiol.2006.034868. [DOI] [PubMed] [Google Scholar]
- Carlson JT, Hedner J, Elam M, Ejnell H, Sellgren J, Wallin BG. Augmented resting sympathetic activity in awake patients with obstructive sleep apnea. Chest. 1993;103:1763–1768. doi: 10.1378/chest.103.6.1763. [DOI] [PubMed] [Google Scholar]
- Dick TE, Hsieh YH, Morrison S, Coles SK, Prabhakar N. Entrainment pattern between sympathetic and phrenic nerve activities in the Sprague-Dawley rat: hypoxia-evoked sympathetic activity during expiration. Am J Physiol Regul Integr Comp Physiol. 2004;286:R1121–R1128. doi: 10.1152/ajpregu.00485.2003. [DOI] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eickhoff P, Valipour A, Kiss D, Schreder M, Cekici L, Geyer K, Kohansal R, Burghuber OC. Determinants of systemic vascular function in patients with stable chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2008;178:1211–1218. doi: 10.1164/rccm.200709-1412OC. [DOI] [PubMed] [Google Scholar]
- Fadhel N, Seager LD. The influence of hexamethonium intracisternally and of hexamethonium and trimethaphan camphorsulfonate intravenously on the pressor responses to intracisternal veratrine. Proc Soc Exp Biol Med. 1969;131:1263–1266. doi: 10.3181/00379727-131-34083. [DOI] [PubMed] [Google Scholar]
- Fletcher EC, Bao G, Li R. Renin activity and blood pressure in response to chronic episodic hypoxia. Hypertension. 1999;34:309–314. doi: 10.1161/01.hyp.34.2.309. [DOI] [PubMed] [Google Scholar]
- Fletcher EC, Lesske J, Behm R, Miller CC, 3rd, Stauss H, Unger T. Carotid chemoreceptors, systemic blood pressure, and chronic episodic hypoxia mimicking sleep apnea. J Appl Physiol. 1992;75:1978–1984. doi: 10.1152/jappl.1992.72.5.1978. [DOI] [PubMed] [Google Scholar]
- Fletcher EC, Orolinova N, Bader M. Blood pressure response to chronic episodic hypoxia: the renin-angiotensin system. J Appl Physiol. 2002;92:627–633. doi: 10.1152/japplphysiol.00152.2001. [DOI] [PubMed] [Google Scholar]
- Greenberg HE, Sica A, Batson D, Scharf SM. Chronic intermittent hypoxia increases sympathetic responsiveness to hypoxia and hypercapnia. J Appl Physiol. 1999a;86:298–305. doi: 10.1152/jappl.1999.86.1.298. [DOI] [PubMed] [Google Scholar]
- Greenberg HE, Sica AL, Scharf SM, Ruggiero DA. Expression of c-fos in the rat brainstem after chronic intermittent hypoxia. Brain Res. 1999b;816:638–645. doi: 10.1016/s0006-8993(98)01222-0. [DOI] [PubMed] [Google Scholar]
- Grote L, Kraiczi H, Hedner J. Reduced alpha- and beta(2)-adrenergic vascular response in patients with obstructive sleep apnea. Am J Respir Crit Care Med. 2000;162:1480–1487. doi: 10.1164/ajrccm.162.4.9912028. [DOI] [PubMed] [Google Scholar]
- Guyenet PG. Neural structures that mediate sympathoexcitation during hypoxia. Resp Physiol. 2000;121:147–162. doi: 10.1016/s0034-5687(00)00125-0. [DOI] [PubMed] [Google Scholar]
- Guyenet PG, Stornetta RL, Abbott SB, Depuy SD, Fortuna MG, Kanbar R. Central CO2-chemoreception and integrated neural mechanisms of cardiovascular and respiratory control. J Appl Physiol. 2010;108:995–1002. doi: 10.1152/japplphysiol.00712.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haselton JR, Guyenet PG. Central respiratory modulation of medullary sympathoexcitatory neurons in rat. Am J Physiol Regul Integr Comp Physiol. 1989;256:R739–R750. doi: 10.1152/ajpregu.1989.256.3.R739. [DOI] [PubMed] [Google Scholar]
- Imadojemu VA, Mawji Z, Kunselamn A, Gray KS, Hogeman CS, Leuenberger UA. Sympathetic chemoreflex responses in obstructive sleep apnea and effects of continuous positive airway pressure therapy. Chest. 2007;131:1406–1413. doi: 10.1378/chest.06-2580. [DOI] [PubMed] [Google Scholar]
- Kiely JM, Gordon FJ. Role of rostral ventrolateral medulla in centrally mediated pressor responses. Am J Physiol Heart Circ Physiol. 1994;267:H1549–H1556. doi: 10.1152/ajpheart.1994.267.4.H1549. [DOI] [PubMed] [Google Scholar]
- Kline DD, Ramirez-Navarro A, Kunze DL. Adaptive depression in synaptic transmission in the nucleus of the solitary tract after in vivo chronic intermittent hypoxia: evidence for homeostatic plasticity. J Neurosci. 2007;27:4663–4673. doi: 10.1523/JNEUROSCI.4946-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesske J, Fletcher EC, Bao G, Unger T. Hypertension caused by chronic intermittent hypoxia: influence of chemoreceptors and sympathetic nervous system. J Hypertens. 1997;15:1593–1603. doi: 10.1097/00004872-199715120-00060. [DOI] [PubMed] [Google Scholar]
- Ling L, Fuller DD, Bach KB, Kinkead R, Olson EB, Jr, Mitchell GS. Chronic intermittent hypoxia elicits serotonin-dependent plasticity in the central neural control of breathing. J Neurosci. 2001;21:5381–5388. doi: 10.1523/JNEUROSCI.21-14-05381.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandel DA, Schreihofer AM. Central respiratory modulation of barosensitive neurones in rat caudal ventrolateral medulla. J Physiol. 2006;572:881–896. doi: 10.1113/jphysiol.2005.103622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandel DA, Schreihofer AM. Modulation of the sympathetic response to acute hypoxia by the caudal ventrolateral medulla in rats. J Physiol. 2009;587:461–475. doi: 10.1113/jphysiol.2008.161760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall JM, Thomas T, Turner L. A link between adenosine, ATP-sensitive K+ channels, potassium and muscle vasodilatation in the rat in systemic hypoxia. J Physiol. 1993;472:1–9. doi: 10.1113/jphysiol.1993.sp019931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCulloch PF, Panneton WM, Guyenet PG. The rostral ventrolateral medulla mediates the sympathoactivation produced by chemical stimulation of the rat nasal mucosa. J Physiol. 1999;516:471–484. doi: 10.1111/j.1469-7793.1999.0471v.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCulloch PF, West NH. Cardiovascular responses to nasal water flow in rats are unaffected by chemoreceptor drive. Am J Physiol Regul Integr Comp Physiol. 1992;263:R1049–R1056. doi: 10.1152/ajpregu.1992.263.5.R1049. [DOI] [PubMed] [Google Scholar]
- McGuire M, Bradford A. Chronic intermittent hypoxia increases haematocrit and causes right ventricular hypertrophy in the rat. Resp Physiol. 1999;117:53–58. doi: 10.1016/s0034-5687(99)00047-x. [DOI] [PubMed] [Google Scholar]
- Narkiewicz K, van de Borne PJ, Pesek CA, Dyken ME, Montano N, Somers VK. Selective potentiation of peripheral chemoreflex sensitivity in obstructive sleep apnea. Circulation. 1999;99:1183–1189. doi: 10.1161/01.cir.99.9.1183. [DOI] [PubMed] [Google Scholar]
- Peng YJ, Prabhakar NR. Reactive oxygen species in the plasticity of respiratory behavior elicited by chronic intermittent hypoxia. J Appl Physiol. 2003;94:2342–2349. doi: 10.1152/japplphysiol.00613.2002. [DOI] [PubMed] [Google Scholar]
- Peng Y-J, Prabhakar NR. Effect of two paradigms of chronic intermittent hypoxia on carotid body sensory activity. J Appl Physiol. 2004;96:1236–1242. doi: 10.1152/japplphysiol.00820.2003. [DOI] [PubMed] [Google Scholar]
- Philippi NR, Bird CE, Marcus NJ, Olson EB, Chesler NC, Morgan BJ. Time course of intermittent hypoxia-induced impairments in resistance artery structure and function. Respir Physiol Neurobiol. 2010;170:157–163. doi: 10.1016/j.resp.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips SA, Olson EB, Morgan BJ, Lombard JH. Chronic intermittent hypoxia impairs endothelium-dependent dilation in rat cerebral and skeletal muscle resistance arteries. Am J Physiol Heart Circ Physiol. 2004;286:H388–H393. doi: 10.1152/ajpheart.00683.2003. [DOI] [PubMed] [Google Scholar]
- Phillips SA, Olson EB, Lombard JH, Morgan BJ. Chronic intermittent hypoxia alters NE reactivity and mechanics of skeletal muscle resistance arteries. J Appl Physiol. 2006;100:1117–1123. doi: 10.1152/japplphysiol.00994.2005. [DOI] [PubMed] [Google Scholar]
- Reeves SR, Gozal E, Guo SZ, Sachleben LR, Brittian KR, Lipton AJ, Gozal D. Effect of long-term intermittent and sustained hypoxia on hypoxic ventilatory and metabolic responses in the adult rat. J Appl Physiol. 2003;95:1767–1774. doi: 10.1152/japplphysiol.00759.2002. [DOI] [PubMed] [Google Scholar]
- Santajuliana D, Hornfeldt BJ, Osborn JW. Use of ganglionic blockers to assess neurogenic pressor activity in conscious rats. J Pharmacol Toxicol Methods. 1996;35:45–54. doi: 10.1016/1056-8719(95)00132-8. [DOI] [PubMed] [Google Scholar]
- Schreihofer AM, Hair CD, Stepp DW. Reduced plasma volume and mesenteric vascular reactivity in obese Zucker rats. Am J Physiol Regul Integr Comp Physiol. 2005a;288:R253–R261. doi: 10.1152/ajpregu.00498.2004. [DOI] [PubMed] [Google Scholar]
- Schreihofer AM, Mandel DA, Mobley SC, Stepp DW. Impairment of sympathetic baroreceptor reflexes in obese Zucker rats. Am J Physiol Heart Circ Physiol. 2007;293:H2543–H2549. doi: 10.1152/ajpheart.01201.2006. [DOI] [PubMed] [Google Scholar]
- Schreihofer AM, Ito S, Sved AF. Brain stem control of arterial pressure in chronic arterial baroreceptor-denervated rats. Am J Physiol Regul Comp Physiol. 2005b;289:R1746–R1755. doi: 10.1152/ajpregu.00307.2005. [DOI] [PubMed] [Google Scholar]
- Sica AL, Greenberg HE, Scharf SM, Ruggiero DA. Immediate-early gene expression in cerebral cortex following exposure to chronic-intermittent hypoxia. Brain Res. 2000;870:204–210. doi: 10.1016/s0006-8993(00)02170-3. [DOI] [PubMed] [Google Scholar]
- Silverberg DS, Iaina A, Oksenberg A. Treating obstructive sleep apnea improves essential hypertension and quality of life. Am Fam Physician. 2002;65:229–236. [PubMed] [Google Scholar]
- Singh SB, Selvamurthy W. Effect of intermittent chronic exposure to hypoxia on feeding behaviour of rats. Int J Biometeorol. 1993;37:200–202. doi: 10.1007/BF01387523. [DOI] [PubMed] [Google Scholar]
- Somers VK, Abboud FM. Chemoreflexes: responses, interactions and implications for sleep apnea. Sleep. 1993;16:S30–S34. [PubMed] [Google Scholar]
- Somers VK, Dyken ME, Clary MP, Abboud FM. Sympathetic neural mechanisms in obstructive sleep apnea. J Clin Invest. 1995;96:1897–1904. doi: 10.1172/JCI118235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stornetta RL, Morrison SF, Reis DJ. Neurons of rostral ventrolateral medulla mediate somatic pressor reflex. Am J Physiol Regul Integr Comp Physiol. 1989;256:R448–R462. doi: 10.1152/ajpregu.1989.256.2.R448. [DOI] [PubMed] [Google Scholar]
- Sun MK, Reis DJ. NMDA receptor-mediated sympathetic chemoreflex excitation of RVL-spinal vasomotor neurons in rats. J Physiol. 1995;482:53–68. doi: 10.1113/jphysiol.1995.sp020499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahawi Z, Orolinova N, Joshua IG, Bader M, Fletcher EC. Altered vascular reactivity in arterioles of chronic intermittent hypoxic rats. J Appl Physiol. 2001;90:2007–2013. doi: 10.1152/jappl.2001.90.5.2007. [DOI] [PubMed] [Google Scholar]
- Troncoso Brindeiro CM, da Silva AQ, Allahdadi KJ, Youngblood V, Kanagy NL. Reactive oxygen species contribute to sleep apnea-induced hypertension in rats. Am J Physiol Heart Circ Physiol. 2007;293:H2971–H2976. doi: 10.1152/ajpheart.00219.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L. Plasma volume, cell volume, total blood volume and F cells factor in the normal and splenectomized Sherman rat. Am J Physiol. 1959;196:188–192. doi: 10.1152/ajplegacy.1958.196.1.188. [DOI] [PubMed] [Google Scholar]
- Zoccal DB, Bonagamba LG, Oliveira FR, Antunes-Rodrigues J, Machado BH. Increased sympathetic activity in rats submitted to chronic intermittent hypoxia. Exp Physiol. 2007;92:79–85. doi: 10.1113/expphysiol.2006.035501. [DOI] [PubMed] [Google Scholar]
- Zoccal DB, Paton JF, Machado BH. Do changes in the coupling between respiratory and sympathetic activities contribute to neurogenic hypertension? Clin Exp Pharmacol Physiol. 2009;36:1188–1196. doi: 10.1111/j.1440-1681.2009.05202.x. [DOI] [PubMed] [Google Scholar]
- Zoccal DB, Simms AE, Bonagamba LG, Braga VA, Pickering AE, Paton JF, Machado BH. Increased sympathetic outflow in juvenile rats submitted to chronic intermittent hypoxia correlates with enhanced expiratory activity. J Physiol. 2008;586:3253–3265. doi: 10.1113/jphysiol.2008.154187. [DOI] [PMC free article] [PubMed] [Google Scholar]