

Abstract
Prostate cancer is a complex disease in which metastasis to the bone is the main cause of death. Initial stages of metastasis are generally similar to those for most solid tumors; however, the mechanisms that underlie the homing of prostate tumor cells to the bone remain incompletely understood. Prostate cancer bone metastasis is also a microenvironment-driven disease, involving bi-directional interactions between the tumor and the bone microenvironment. In this review, we discuss the current understanding of the biologic processes and regulatory factors involved in the metastasis of prostate cancer cells, and their specific properties that promote growth in bone. Although many of these processes still need to be fully elucidated, a better understanding of the complex tumor/microenvironment interplay is slowly leading to more effective therapies for patients with prostate cancer bone metastases.
Keywords: prostate cancer, bone, metastasis, invasion, migration, osteoblast, osteoclast
Prostate cancer is the second most commonly diagnosed form of cancer and the sixth leading cause of cancer-related deaths among men worldwide.1 In patients with localized prostate cancer, the 5-year survival approximates 100%; however, in patients in whom distant metastases have occurred, the 5-year survival drops to 31%.2 Like most other solid malignancies, prostate cancer can metastasize to distant organs such as the liver, lungs and brain, but it has an unusually high propensity for metastasizing to the bone. In one autopsy study, approximately 80% of the men who had died from prostate cancer possessed bone metastases.3 Most current treatments for individuals with bone metastases have only palliative effects, with little effect on long-term survival.4, 5 Thus, gaining a better understanding of the mechanisms by which prostate cancer metastasizes to the bone is critical to the development and use of therapies to improve patients’ survival. In this review, we discuss the steps of prostate cancer metastasis and its growth within the bone.
Events in Prostate Cancer Progression-development of castrate-resistant disease
Androgen receptor (AR)
The dependence of prostate cancer cells on androgen stimulation was first described in a seminal paper by Huggins and Hodges.6 Androgen binds to the AR and translocates to the nucleus, where the binding of this complex to Androgen Responsive Elements affects the transcription of androgen-regulated genes (e.g. prostate-specific antigen, PSA) and ultimately stimulates proliferation and inhibits apoptosis of prostate cancer cells. Therefore, androgen-deprivation therapy by chemical and surgical castration has been the mainstay of the treatment for early metastatic prostate cancer. However, all patients invariably will progress at some point during the course of their disease since their tumor adapts to the androgen-deprived environment and becomes “castrate-resistant”. Several molecular mechanisms for the development of castration-resistant prostate cancer, an important step in progression of the disease to the bone, have been elucidated to date: Prostate cancer cells become “hypersensitive” to androgen stimulation by upregulation of AR expression. Also, autocrine and paracrine production of androgens is upregulated in castrate-resistant prostate cancer. The AR may be activated in this setting by steroids other than androgens such as estrogens, and ligand-independent activation of the AR by receptor tyrosine kinases has also been described. Finally, in castrate-resistant prostate cancer, bypass pathways identified that contribute to AR independent growth of prostate cancer cells, such as interleukin-6 signaling (described in more detail below). Several reviews have described the role of the AR in castration-resistant prostate cancer.7-10
Interleukin-6 (IL-6)
IL-6 is a glycoprotein implicated in progression to castrate-resistant prostate cancer.7 IL-6 is frequently expressed in prostate cancer cell lines, as early as benign hyperplasia,11-13 as well as in sera of prostate cancer patients, in which expression increases in patients with metastatic disease.14, 15 The expression of IL-6 and its receptor has been consistently demonstrated in human prostate cancer cell lines.11 IL-6 activates AR-mediated gene expression by activation of the AR through a STAT3 pathway in androgen-dependent LNCaP cells.16-18 Overexpression of IL-6 increases PSA mRNA in LNCaP cells (cell lines and variants discussed in this review are described in Table 1), partially ablating the requirement for androgen in growth of these cells.19, 20 Other signaling pathways, such as those mediated by Src and insulin-like growth factor (IGF) may also function to allow non-genomic signaling through AR after depletion of androgen,7 and AR amplification is another common mechanism to permit AR signaling in a castrate resistant environment.21, 22 Thus, many of the pathways discussed below not only contribute to classic steps in metastasis, but also promote this process by constitutively activating AR. Details are described in several reviews.22, 23
Table 1.
Cell lines used in experiments described in this review.
| Cell line | Origin (species) |
Androgen receptor status |
Bone lesion | Method of studied | Aspect of metastasis studied |
Refs |
|---|---|---|---|---|---|---|
| ARCaP | Human | + | Orthotopic xenograft | EMT | 32 | |
| Intracardiac xenograft | ||||||
|
| ||||||
| DU145 | Human | − | Invasion assay | Invasion | 85 | |
|
| ||||||
| LNCaP | Human | + | Mixed | Migration assay | Migration | 64 |
|
| ||||||
| LNCaPcol | Human | + | Mixed | Intratibial xenograft | Adhesion | 107 |
|
| ||||||
| C4-2 | Human | + | Osteolytic | Adhesion assay | Adhesion | 99 |
| Intratibial xenograft | Adhesion | 106 | ||||
| Intratibial xenograft | Bone remodeling | 179 | ||||
|
| ||||||
| C4-2B4 | Human | + | Mixed | Coculture, Migration assay | Migration | 111 |
| Intratibial xenograft | Bone remodeling | 160 | ||||
|
| ||||||
| PC3 | Human | − | Osteolytic | Coculture, Bone xenograft | Invasion | 80 |
| Invasion assay | Invasion | 85 | ||||
| Bone xenograft | Invasion | 86 | ||||
| Binding assay | Adhesion | 98 | ||||
| Intracardiac xenograft | Adhesion | 109 | ||||
| Intratibial, Intracardiac xenograft | Bone homing | 118 | ||||
| Adhesion, Migration assay | Extravasation | 119 | ||||
| Intratibial xenograft | Bone remodeling | 179 | ||||
|
| ||||||
| PC3MM2 | Human | − | Osteolytic | Migration assay | Migration | 68 |
| Orthotopic xenograft | Migration | 68 | ||||
|
| ||||||
| PC3M-Pro4 | Human | − | Osteolytic | Intratibial, Intracardiac xenograft | Bone remodeling | 207 |
|
| ||||||
| P69 | Human | + | Adhesion assay | Adhesion | 99 | |
|
| ||||||
| MDA PCa 2b |
Human | + | Osteoblastic | Coculture, Intratibial xenograft | Bone remodeling | 129 |
|
| ||||||
| LuCap 35 | Human | + | Osteoblastic | Bone xenograft | Bone remodeling | 181 |
|
| ||||||
| LuCaP 23.1 | Human | + | Osteoblastic | Bone xenograft | Bone remodeling | 168 |
|
| ||||||
| ACE-1 | Dog | − | Osteoblastic | Intratibial, Intracardiac xenograft | Bone remodeling | 161 |
|
| ||||||
| LAPC-9 | Human | + | Osteoblastic | Intratibial xenograft | Bone remodeling | 169 |
Events in Prostate Cancer Metastatic Process
The classic model of metastasis of solid tumors, including prostate cancer, is guided by the “seed and soil” hypothesis first proposed by Stephen Paget in 1889.24 In Paget’s model, the “seeds” (i.e., tumor cells) metastasize only to “soil” (i.e., specific organs) well suited (“fertile”) for the tumor’s growth. Although this concept remains an excellent guiding principle, it does not entirely explain the molecular bases for organ-specific metastases. Metastasis of prostate cancer, like that of other solid tumors, involves multiple steps, including angiogenesis, local migration, invasion, intravasation, circulation, and extravasation of tumor cells and then angiogenesis and colonization in the new site. We will describe only the hallmarks of these events, which have been reviewed in extensive detail elsewhere.25-28 We will then discuss our emerging understanding of properties of metastatic prostate tumor cells that facilitate their growth in the bone.
Decreased cell adhesion and the epithelial to mesenchymal transition
The initial stages of metastasis involve the detachment and migration of malignant cells from the primary tumor and their entry into the nearby blood or lymphatic vessels. In the normal prostate gland, epithelial cells have restricted migratory capability, in part because the basal cells inside the lumen attach to the basement membrane, forming a cell layer.28 Normal epithelial cells above that basal cell layer adhere to each other as well as to the extracellular matrix (ECM). Cell-to-cell adhesion in normal epithelium is maintained by many different junctions, such as adherens and tight junctions, which are comprised of protein complexes of cell adhesion molecules (CAMs), such as selectin and cadherin. Binding adjacent epithelial cells via cell junctions is critical for providing anchorage and communication between neighboring cells.
During the process of malignant transformation, however, the adhesiveness of epithelial cells decreases. Specifically, early metastatic prostate cancer cells exhibit alterations in the expression of different molecules leading to decreased cell adhesion, such as E-cadherin.28 This process is a major feature of epithelial to mesenchymal transition (EMT), which is now considered by most investigators to be critical in the development of a more migratory and invasive phenotype of epithelial tumor cells.25, 29-31 Experiments using prostate cancer cell lines in immunodeficient mouse models have confirmed that EMT is important in prostate cancer metastasis. For example, the epithelial-like cell line ARCaPE, which is derived from parental androgen-refractory ARCaP prostate cancer cells, was used for successive orthotopic inoculations into nude mice.32 After only one inoculation, the ARCaPE cells from the resulting tumors exhibited EMT-like phenotypic changes. Cells from the tumors harboring those changes were then injected into a second group of mice, and those cells exhibited increased metastasis to several different organs, including the bone, demonstrating—at least in this model—a role for EMT in increasing metastatic potential.
E-cadherin and β-catenin
Specific molecular changes, many of them hallmarks of EMT, have been shown to play roles in adhesion, migration and invasion. E-cadherin and β-catenin are central in these processes. E-cadherin is a member of the cadherin family of CAMs in epithelial cells.33 E-cadherin functions through the interaction of the calcium-dependent extracellular binding domain between adjacent cells, whereas its intracellular domain anchors the actin cytoskeleton through β-catenin, an adherence junction protein.33, 34 Thus, E-cadherin and β-catenin complexes have important functions in maintaining cell-cell adhesions that connect the lateral side of epithelial cells. E-cadherin/β-catenin complexes are also affected by activation of the tyrosine kinase Src,35 as shown in Figure 1. Other functions of Src in promoting invasion and metastasis are described below.
Figure 1. Src stimulation of migration and invasion through the E-cadherin/β-catenin complex.
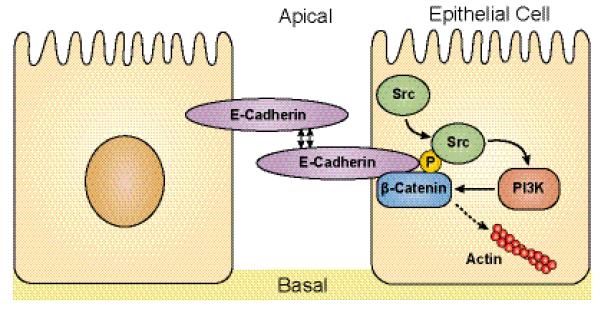
Following engagement, activated Src phosphorylates E-cadherin/β-catenin complexes, with several results including recruitment of PI3K to this complex, activating downstream signaling pathways that lead to unbranching of actin filaments, eventual dissociation of β-catenin from the complex and functional loss of E-cadherin, all processes that promote tumor cell invasion. Src also promotes migration/invasion through focal adhesion kinase, as described in the text.
One of the major features of cells undergoing EMT is cadherin switching, whereby, for example, E-cadherin (characteristically expressed in normal epithelial cells) is downregulated, and N-cadherin (characteristically expressed in mesenchymal cells) is up-regulated. This cadherin switch occurs during prostate tumor progression. Tumor specimens from patients with higher-grade prostate cancer (i.e., a Gleason score ≥ 8) had lower E-cadherin and higher N-cadherin expression than did those from patients with lower-grade disease.36-38 Decreased β-catenin expression was also associated with decreased expression of E-cadherin and also correlated with higher-grade prostate cancer.39
These clinical data are supported by studies from in vitro model systems that demonstrated that E-cadherin expression is reduced in more-invasive human prostate cancer cell lines40 and in invasive rat prostate tumors.41
Although these changes appear to be required for increasing metastatic potential, they may need to be at least partially reversed for outgrowth of tumors at the metastatic site. A recent study performed with clinical specimens showed reduced expression of E-cadherin and β-catenin in the primary prostate tumor but higher expression of both of those factors in metastatic prostate cancer cells in the bone.42 These results suggest that mesenchymal to epithelial transition (MET)is required for the growth of metastatic prostate cancer cells once they reach the bone. An alternative possibility, as suggested by Saha, is that a subpopulation of migrating metastatic prostate cancer cells that overexpresses E-cadherin and β-catenin is responsible for cell adherence and subsequent growth in the bone.42 Further work will be required to distinguish between these (and potentially other) possibilities.
Integrins
The interaction between normal prostate epithelial cells and the ECM is critical for cell proliferation, survival, and migration.43 Integrins play multiple roles in cells including regulating focal adhesions which are large macromolecular complexes that connect the cell’s cytoskeleton to the ECM. Integrin-mediated signaling through focal adhesions promotes such diverse processes such as cell survival, motility and migration as well as crosstalk with growth factor receptors. Alterations in the expression of the integrins and molecules regulating the downstream signaling events they mediate are associated with prostate cancer metastasis.44
The integrins consist of 24 glycoprotein heterodimers composed of combinations of 18 α and 8 β subunits that span the cell membrane and bind to specific subsets of ECM ligands.45 In normal prostate epithelial cells, the predominant integrins are α5β1, α6β1, and α6β4, but αvβ3 and α3β1 integrins are also expressed.28 Reduced or absent expression of α6 integrins in biopsy samples of prostate tumors correlates with the invasiveness of those cells.46 In normal prostate epithelial cells, the α6β4 integrin forms hemidesmosomes that link the cytoskeleton to laminin-5 in the ECM.47 In prostate cancer tissues, however, the expression of α6β4 integrin is frequently reduced, leading to the loss of hemidesmosomes weakening cell-cell adhesion.48 This change in integrin expression results in decreased adhesion of cells to the ECM, thereby promoting aberrant migration.49
Signaling through Focal Adhesion Kinase (FAK)
Enzymes central to regulation of focal adhesions are the non-receptor tyrosine kinases focal adhesion kinase and Src family kinases. FAK is a key regulator of focal adhesion turnover, and is critical to increasing cell migration.50 During cell migration, focal adhesions are formed at the leading edge of the cell and used as an anchor on which a cell can “pull” itself over the ECM. Then, focal adhesions are disassembled at the rear edge of the cell when it moves forward and withdraws its rear edge. Blockage of focal adhesion turnover is known to increase the number and size of focal adhesions and inhibit cell migration. Alterations in integrin composition and function can thus affect migration through modulation of FAK function. In addition, overexpression of FAK itself is observed in high-grade and metastatic prostate cancer tissues compared to normal prostate tissues.51 Increased levels of FAK in prostate tumor PC3 cells also correlates with increased metastatic potential.52
Src family of kinases (SFKs)
Other key signaling molecules in focal adhesions (but not found exclusively in focal adhesions) include the Src family of non-receptor protein tyrosine kinases. Phosphorylation of FAK following association with integrins leads to Src binding, followed by Src phosphorylation of FAK, recruiting signaling and non-signaling molecules into focal adhesions.53 Many recent studies have shown that aberrant activation of SFKs by multiple mechanisms, including association with FAK, play important roles in prostate cancer progression.54 SFKs may be activated by numerous mechanisms, including constitutive association with FAK as well as with growth factor receptors such as c-Met and insulin-like growth factor I receptor (IGF-IR) that are frequently overexpressed in prostate cancer. Src activation leads to rearrangement of the actin cytoskeleton, and increased focal adhesion turnover and migration.55, 56
Constitutive Src activation (whether through association with FAK or other Src binding partners such as aberrantly expressed growth factor receptors) is sufficient to activate numerous signaling pathways affecting proliferation, apoptosis, angiogenesis, cell cycle regulation and migration. Src/FAK-mediated activation of the phosphatidylinositol 3-kinase (PI3K), Rho, and Ras are important for tumor cell migration. Activation of the PI3K pathway by Src is known to increase cell survival. Activated Rho pathway is responsible for increasing cell motility,57 Ras signaling is involved in increasing cell proliferation, angiogenesis, and migration.58 Specific mechanisms of Src activation have been detailed in many reviews.55, 59, 60 In prostate cancer, Src is highly expressed in patient samples61 and is associated with decreased patient survival and metastases.62 Src activity is increased in castration resistant prostate cancer,62 and is higher in bone metastases than in primary tumors (Parikh and Gallick, unpublished). Aside from affecting focal adhesion turnover, Src activation promotes migration through many other molecules including interleukin 8 (IL-8), which is transcriptionally de-repressed by activated by Src phosphorylation of STAT3, leading to STAT3 binding to IL-8 promoter.63 Inhibition of Src blocked IL-8 induced migration in prostate LNCaP cells.64
Recent work has demonstrated that Src inhibition directly affects metastasis in nude mouse models. SFK kinase inhibitors decreased proliferation, invasion, and migration in prostate cancer cells.65-67 We showed that siRNA knockdown of Src in metastatic prostate PC3MM2 cells inhibited migration in vitro,68 and was sufficient to inhibit metastases to the lymph nodes in orthotopic nude mouse models. In contrast, knockdown of Lyn, another SFK, inhibited proliferation, consistent with previous reports,69 suggesting that in prostate cancer, distinct SFKs play distinct roles in tumor progression and metastasis. Therefore, in mouse models, SFK activation directly contributes to prostate cancer metastasis. As discussed below, Src also affects osteoblast and osteoclast functions that contribute to the growth of prostate cancer in the bone. Clinical trials using Src inhibitors for prostate cancer patients with bone metastases are discussed below.
Prostate cancer invasion: roles of proteases
Prostate cancer invasion requires partial degradation of the ECM. The ECM is comprised of basement membrane and connective tissue. Partial degradation of the ECM is an obligatory step in metastasis. The families of proteinases most associated with ECM degradation in prostate cancer are matrix metalloproteinases and serine proteinases such as urokinase-type plasminogen activator (uPA).
Matrix metalloproteinases (MMPs)
MMPs are a family of zinc-binding pro-enzymes, consisting of 24 different members. The pro-enzymes are inactive until proteolytic cleavage. ProMMP-2 is activated by membrane-type1 MMP.70 Once activated, MMP-2 (as well as MMP-13) activates MMP-9.70 In primary prostate tumor tissues, both the level of MMP-9, and the ratios of MMP-2/-9 to tissue inhibitor of metalloproteinases-1 (TIMP-1) are increased relative to normal prostate epithelium. These levels and ratios are further associated with high Gleason score and poorer patient survival.71-73 Brehmer et al.74 showed that loss of TIMP-1 was correlated with upregulation of MMPs in malignant human prostate cancer tissues. Further, in patients with metastatic disease, high concentrations of MMP-2 and MMP-9 have been observed in plasma.70 Thus, levels of MMP-2 and MMP-9 may be useful prognostic markers in prostate cancer.75 However, a recent prostate tissue microarray study compared the level of MMPs and showed that only MMP-9 expression was prognostic.76 This study did not look at secreted proteins, which may explain the differences in the conclusions with respect to MMPs as prognostic markers.
In vitro studies have demonstrated that deregulation of a number of signaling pathways increase the expression of MMPs.77 MMPs are negatively regulated by TIMPs.77 Culturing minced explants of prostate tumor tissues in medium for 8-10 weeks, Lokeshwar and colleagues78 demonstrated elevated levels of active MMP-2 and reduced levels of TIMP-1 in medium from prostate cancer tissues compared to that from benign prostate hyperplasia, suggesting that there is an imbalance of MMPs and TIMPs, i.e., up-regulation of the former and down-regulation of the latter, in prostate cancer. Other MMPs have different functions that contribute to prostate cancer metastasis. A recent study used a transgenic mouse model for prostate tumor formation and metastasis in mice in which the SV40 large T antigen is overexpressed only in prostatic neuroendocrine cells (Table 2). This mouse strain was then crossed with mice of the same genetic background in which MMP-2 was functionally deleted.79 Regardless of MMP-2 expression (or lack thereof), prostate tumors developed. However, in the mice expressing the SV40 antigen in which MMP-2 was “knocked out”, reduced lung metastases and increased survival were observed. In contrast, using the same SV40 Large T antigen-expressing mice, functional deletion of MMP-7 or MMP-9 did not affect metastasis development, although blood vessel size was reduced.79 Perhaps the most striking report related to roles of MMPs in prostate cancer bone metastasis comes from the study of MMP-12 in co-culture systems. Nabha et al.80 co-cultured PC3 cells with bone marrow stromal (BMS) cells, and demonstrated increased production of MMP-12 in PC3 cells. These results demonstrate that the interaction of BMS cells and PC3 increases MMP-12 expression in the tumor cells. This result was further confirmed by an in vivo study that demonstrated MMP-12 was primarily expressed in PC3 cells injected in human fetal bone xenograft as opposed to subcutaneous PC3 tumors. The authors further demonstrated that decreasing MMP-12 expression with an RNAi strategy reduced the invasiveness of PC3 cells by reducing degradation of type I collagen in the bone, providing convincing evidence that MMP-12 participates in bone-tropic metastasis.80
Table 2.
Transgenic mouse models described in this review.
Urokinase-type plasminogen activator (uPA)
uPA is one of the major serine proteinases involved in facilitating ECM degradation, as well as activating other latent proteinases such as MMPs involved in this process.26 One of the major functions of uPA-uPA receptor (uPAR) binding is to convert plasminogen to plasmin, a broad-spectrum serine protease involved in ECM degradation.81-83 Studies on clinical prostate cancer specimens have demonstrated that high expression of uPA and uPAR are both associated with higher tumor grades (Gleason score ≥ 8), increased invasion and the presence of lymph node metastases.84 Festuccia et al.85 demonstrated that the expression of uPA in prostate PC3 and DU145 cells activates plasmin and leads to activation of MMP-9 and MMP-2, which can be inhibited by neutralizing antibodies to uPA. Additional evidence of the importance of uPA in prostate cancer was derived from experiments of Dong et al.,86 who injected PC3 in which uPA expression was reduced by siRNA cells into fetal bone, followed by implantation of these bones into immunodeficient mice. The authors observed reduced tumor burden and bone degradation relative to PC3 cells in which uPA expression was not decreased. These results provide strong evidence that expression of tumor-derived uPA plays an important role in prostate tumor growth in the bone.
Prostate-Specific Antigen (PSA)
PSA is one of the androgen-regulated genes.87 Its product is a 240 amino acid glycoprotein and belongs to the family of kallikrein-like serine proteases.88 In its catalytic domain, PSA has the His-Asp-Ser sequence, which is characteristic of serine proteases and thus implicates independent protease activity (i.e. not via plasmin) in protein degradation.89 PSA is exclusively secreted by prostate epithelial cells and therefore the most widely used serum marker to diagnose early prostate cancer and also to monitor the course of the disease during and after treatment. PSA is most abundantly found in the seminal plasma, where it mediates the liquefaction of coagulated seminal plasma after ejaculation.90 The degradation of fibronectin by PSA in the seminal plasma indicated a possible role for PSA as a protease in prostate cancer invasion since fibronection is also one of the main components of the ECM. Indeed, in in vitro assays, PSA was shown to degrade fibronectin, and its inhibition with specific antibodies resulted in a dose-dependent decrease of the invasion of PSA-producing LNCaP cells.91 More work is needed to better define the role of PSA in prostate cancer progression.
Homing of prostate cancer cells to the bone
Endothelium attachment
Once cancer cells intravasate into circulation, they must survive in the circulation, attach to the vascular endothelium, and then extravasate into the bone. These later steps in metastasis are the most poorly understood, but likely require interaction of tumor cells with other cells for metastasis to develop. For example, prostate tumor cells bind to human bone marrow endothelial (HBME) cells with higher affinity than to other endothelial cells.92, 93 Although the detailed mechanism involving this preferential endothelial binding is still unknown, a “dock and lock” mechanism has been proposed.94 One aspect of this model is that the endothelial cells in the bone constitutively express adhesion molecules such as P-selectin.95 The sialyl-LewisX carbohydrate on prostate cancer cell surface molecules then associates with P-selectin through a low-affinity docking process.96 The subsequent locking process of prostate cancer cells to endothelial cells is mediated by integrins, including αvβ3, α5β1, and α3β1.97 In a co-culture model, the binding of PC3 cells to HBME cells was inhibited by integrin β1 antibody but not antibodies to other integrins, suggesting that attachment of PC3 cells to bone marrow endothelium is primarily mediated by integrin β1 in this model.98 Other studies showed that human prostate epithelial P69 cells, immortalized with SV40 large T antigen (low tumorigenic potential), showed an increased number of cells bound to the HBME cell layer relative to C4-2 cells. However, as the levels of integrin expressions in C4-2 and LNCaP cells were similar, other factors besides integrins must be involved in attachment of prostate tumor cells to cells in the microenvironment (Fig. 2).99 HBME cells also stimulate proliferation of PC3 cells in co-culture models.100 Therefore, bidirectional paracrine interactions between prostate cancer cells and HBME cells are likely important in metastatic spreading of prostate cancer to the bone.101
Figure 2. Homing and growth of prostate tumor cells in the bone.
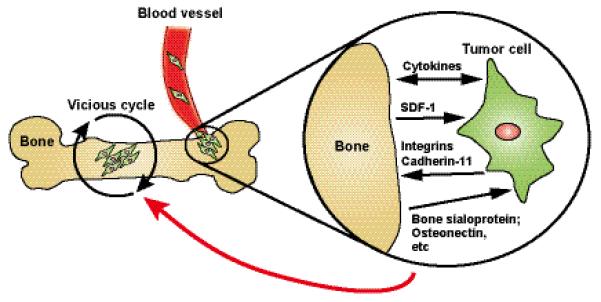
As described in the text, the specific mechanisms by which tumor cells home to the bone remain unknown, but are thought to occur through factors produced or expressed in both bone and tumor. Integrin β1 has been implicated in attachment of prostate cancer cells to the bone epithelium. Cadherin-11 (osteoblast-cadherin) is highly expressed in bone metastases and its knockdown decreases bone homing. Chemotactic factors such as SDF-1 released from the bone are implicated in bone homing of tumor cells. Once Tumor cells reach the bone, the classic vicious cycle occurs, whereby factors from the tumor lead to bone destruction and bone formation, and factors from the bone released in this process promote tumor growth.
Colonization in the bone
While some integrins discussed above are important in the early steps in metastasis, other integrins expressed in prostate cancer cells may be important in colonization of prostate tumor cells in the bone. Prostate cancer cells expressing integrins αvβ3 and α2β1, which are also expressed in osteoclasts,102 facilitate tumor spreading in the bone. Integrin αvβ3 is expressed in metastatic PC3 cells and mediates migration on tissue culture plates coated with vitronectin and osteopontin, components of the bone.103-105 In a mouse xenograft model, intratibial injection of C4-2 cells engineered to ectopically re-express integrin αvβ3 promotes tumor growth in the bone, while other inactive αvβ3 mutants did not.106 Another integrin, α2β1, facilitates cell adhesion on collagen-I in the bone. The binding of metastatic prostate cancer cells to collagen can be inhibited by integrin α2β1 neutralizing antibodies. Intratibial injection of prostate cancer cells overexpressing integrin α2β1 into mice also promoted tumor growth in the bone.107, 108 Thus, expression of the osteoclast integrins αvβ3 and α2β1 by prostate tumor cells may help these cells attach and migrate to the bone matrix, facilitating outgrowth of metastatic tumor cells in the bone.
Alterations in cadherin-11 may also be responsible, in part, for the bone-tropic nature of prostate cancer metastasis. Cadherin-11, also known as osteoblast-cadherin, is highly expressed in human prostatic bone metastases109 as well as in prostate cancer cell lines derived from bone metastases,110 but is not detectable in prostate cancer metastases to other organs.109 Knockdown of cadherin-11 in metastatic prostate PC3 cells using an shRNA strategy led to reduced incidence of metastasis to the bone following their intracardiac injection into immunodeficient mice.109 Importantly, re-expression of cadherin-11 in C4-2B4 cells, a metastatic bone-tropic variant of LNCaP that lacks intrinsic cadherin-11 expression, increases cell migration and invasiveness as well as spreading and intercalation into osteoblast layer.111 These results suggest that functions of cadherin-11 are bone specific and may facilitate the metastatic colonization of prostate cancer cells in the bone.
Chemoattraction to the bone
Evidence from bone xenograft models has demonstrated that prostate cancer cells injected adjacent to the bone migrate toward bone.112 Indeed, extracts from bone can promote chemotaxis and invasion of both PC3 and DU145 prostate tumor cells, while extracts from other organs have no such effect.113 These data suggest that the bone contains chemotactic factors that attract prostate tumor cells. The expression of two such chemotactic factors, CXCR4 and stromal-derived factor-1 (SDF-1), are elevated in metastatic prostate cancer cell lines and in bone metastases.114-116 Experiments support SDF-1 as a homing signal for metastatic prostate cancer cells to bone. Prostate cancer cell lines that localize to the bone express CXCR4 and also migrate across bone marrow endothelial monolayers toward a gradient of SDF-1 in vitro.114, 117 These observations are further supported by an in vivo study demonstrating that injection of neutralizing CXCR4 antibodies reduced bone metastases following intracardiac and intratibial injection of prostate PC3 cells into immunocompromised mice.118 SDF-1 also increases PC3 cell adhesion to human umbilical vein endothelial cell monolayers and enhances trans-endothelial migration.119 In contrast, other studies have provided evidence that CXCR4 expression alone is insufficient to promote bone metastases and requires other factors. When Hart et al.120 used a CXCR4 antagonist peptide at maximal SDF-1 inhibiting concentration, invasion of prostate cancer cells on bone marrow endothelial barrier was not completely inhibited, suggesting that the CXCR4/SDF-1 axis is not the only chemotactic factor involved in bone metastasis. Other factors, such as epidermal growth factor (EGF), IGF, and hepatocyte growth factor have also been shown to increase chemomigration or chemoinvasion of prostate cancer cells and are involved in chemoattractant mechanisms.26 In addition to these molecules, the newly formed bone contains extracellular matrix proteins such as type I collagen, osteonectin, and bone sialoprotein, which also act as chemoattractants for prostate cancer cells migrating to bone.121, 122
Pathophysiology of Bone Metastasis
Early explanations for the preferential bone metastasis of prostate cancer were that prostate tumors have specific phenotypes not found in other tumors that facilitate growth in the bone.123 However, other cancers also metastasize to bone.124 Thus, more recent studies have highlighted the importance of the bone microenvironment and the bidirectional interactions with prostate cancer cells.101 The bone matrix is comprised of 95% of type I collagen and 5% of remaining non-collagen proteins and proteoglycans.125 These non-collagen matrix proteins include osteopontin, bone sialoprotein, and osteonectin, which are the preferred ECM molecules for attachment and growth by prostate cancer cells.126, 127 The cells in the bone marrow comprise not only osteoblasts and osteoclasts but also hematopoietic cells, adipocytes and immune cells. Together, the bone matrix and abundant growth factors secreted by these cells make the bone microenvironment a complex space and fertile “soil” for tumor growth. Current studies suggest that many of the processes that regulate bone cell maturation also promote prostate cancer bone metastasis.128 Here, we discuss the role of cells in the bone microenvironment in facilitating prostate tumor growth in the bone and metastasis, with the understanding that the balance of growth factors and tumor microenvironment/tumor interactions contributes to either osteolytic or osteoblastic lesions.
Osteoblasts and Osteoclasts
The bidirectional interactions of bone cells with prostate cancer cells suggest that not only can tumor-derived growth factors affect bone cells, but cells in the bone microenvironment also stimulate metastatic tumor growth. Numerous recent experiments have supported the importance of this interaction. One such study used primary mouse osteoblasts in co-culture with MDA PCa 2b prostate tumor cells. Under these co-culture conditions, MDA PCa 2b cell proliferation was stimulated, indicating the importance of paracrine interaction of the two cell types in promoting tumor growth in the bone.129 Osteoblasts, derived from mesenchymal stem cells in the bone marrow stroma, are responsible for bone formation.130 Osteoblasts become osteocytes when embedded in the bone or, alternatively, will undergo apoptosis if deposited to new bone matrix.131 The newly formed bone matrix induced by osteoblasts will be hardened by mineralization with deposition of hydroxyapatite crystals to increase the resistance to compression.125 The differentiation and growth of osteoblasts are primary regulated by complex signaling pathways including bone morphogenetic proteins (BMPs), IGFs, transforming growth factor-β (TGF-β), and Wnt.132 Differentiated osteoblasts also secrete many of these growth factors, some of which are embedded in the bone matrix and can later be released by osteoclasts during bone resorption.133 When metastatic cancer cells grow in the bone, they also produce many of these growth factors, resulting in stimulation of proliferation and maturation of osteoblasts and osteoclasts that, in turn, produce or release growth factors that further stimulate metastatic growth. The cross-talk between prostate cancer cells and osteoblasts/osteoclasts and other cells in the microenvironment of the bone is commonly termed the “vicious cycle” in which tumor growth affects bone modeling and bone modeling affects tumor growth (Fig. 2). Many signaling pathways contribute to the cycle. One example that has received considerable recent attention is signaling mediated through Src as discussed above. SFK/Abl inhibitors such as dasatinib inhibit tumor growth but also induce osteoblast differentiation and production of osteonectin, which promotes tumor cell migration and invasion.134 Further, studies have demonstrated that Src activity is critical to osteoclast function.135 Src−/− mice suffer from osteopetrosis due to lack of functional osteoclasts.136 Phase 1 trials with the Src family kinase inhibitor saracatinib (or AZD0530) led to thickening of bones, and decreasing in bone turnover markers.137 Thus Src inhibitors are in clinical trial for prostate cancer bone metastases both for their ability to affect tumor growth and the ability to interrupt the vicious cycle by affecting functions of numerous cells in the microenvironment.138 Encouraging preliminary results from clinical trials using Src inhibitors in combination with chemotherapeutics agents emphasize the importance of targeting the microenvironment in which tumor cells reside as well as markers of osteoblast or osteoclast function as targets. Dasatinib is being used currently in a randomized phase 3 trial in combination with docetaxel for men with metastatic castrate-resistant prostate cancer (ClinicalTrials.gov Identifier: NCT00744497). Results from a different phase 2 trial with dasatinib alone showed decrease in osteolysis,139 demonstrating that targeting of SFKs affects multiple pathways important to development of metastasis, and growth of metastatic tumor cells in the bone. However, the numerous and redundant mechanisms by which the microenvironment contributes to tumor growth in the bone emphasizes the need for a more complete understanding of the tumor-bone microenvironment relationship.
As described above, functions of osteoclasts are critical for releasing growth factors from bone matrix. Osteoclasts are derived from monocytes in the bone marrow.125 The differentiation and maturation of osteoclasts is regulated primarily through the cytokines released by osteoblasts. Macrophage colony stimulating factor-1 and receptor activator of nuclear factor-κB ligand (RANKL) promote fusion of monocytes to form multinucleated mature osteoclasts.140 Mature osteoclasts bind to the surface of bone by αvβ3, αvβ5, and α2β1 integrins, followed by secretion of acid and lysosomal enzymes to degrade bone matrix.125 Hence, bone resorption mediated by osteoclasts then releases several mitogenic growth factors from bone matrix, which is a key step for prostate cancer growth in the bone.141
Bisphosphonates, a class of drugs with structural similarities to pyrophosphate, possess two phosphonate groups, and bind with high affinity to calcium which is abundantly found in the bone. Once ingested by osteoclasts, bisphosphonates induce apoptosis in these cells and thus prevent further bone loss. In patients with castration resistant prostate cancer, a group at high risk for skeletal complications, a large randomized trial showed that treatment with the nitrogen-containing bisphosphonate zoledronic acid resulted in a significant decrease in the number of skeletal-related events which included bone fractures (vertebral or nonvertebral), spinal cord compression, surgery to bone, radiation therapy to bone (including the use of radioisotopes), or a change of antineoplastic therapy to treat bone pain.142 Given the broad and somewhat subjective definition of skeletal-related events in this study, the use of zoledronic acid in this group of patients is not fully endorsed by all physicians.
Bone remodeling
Cancer metastasis in the bone almost invariably leads to an imbalance of bone formation and bone resorption, resulting in osteolytic or osteoblastic lesions.132 Although the underlying mechanism for the imbalance is still not clear, different types of cancer cells have the propensity to secrete more osteoblastic or osteolytic factors. In prostate cancer, bone metastasis is usually osteoblastic with elevated bone formation, resulting in increased bone mineral density.143 Osteoblastic metastasis of prostate cancer may be, in part, due to prostate cancer cells promoting osteoblast proliferation. The number of osteoblasts adjacent to prostate cancer cells is increased in bone metastases, while in osteoclastic tumors an increase in osteoclasts is observed.144 Although bone formation is increased in prostate cancer bone metastases, the tumor-generated bone is abnormal, lacking typical lamellar structure of the normal bone and is thus termed “woven bone.” This type of bone formation easily leads to bone fractures that are frequently seen in prostate cancer patients with bone metastases. However, the increased bone volume may help to confine tumor growth by limiting the space for cancer cells, which delays further progression of prostate cancer metastasis.132 In contrast, bone resorption will increase in response to tumor-derived osteolytic factors, which is an important early step in initiating bone remodeling by creating more space for the tumor. Therefore both osteoblasts and osteoclasts are highly activated, but dysregulated in the process of bone remodeling induced by growth of prostate tumor cells in the bone.
Osteoblast-associated factors
One mechanism by which prostate tumor cells induce osteoblastic lesions is by secreting osteoblastic factors, including endothelin-1, BMPs and IGF.145-147 Here we discuss several osteoblastic factors and their roles in stimulating osteoblast growth and contributing to metastatic prostate tumor growth in the bone. The molecular mechanisms contributing to osteoblastic lesions in other cancers have been reviewed elsewhere.128, 148
Endothelin-1 (ET-1)
ET-1 is a small vasoconstricting peptide produced by the vascular endothelium, which has a key role in vascular homeostasis. ET-1 promotes bone formation by binding to ET receptor subtype A (ETA), which is coupled to heterotrimer G proteins and activates secondary messenger systems to mobilize calcium and stimulate protein kinase C. This process stimulates phosphate transport, and is important for the initiation of bone matrix calcification.149 ET-1 also increases osteoblast proliferation and inhibits osteoclast formation and motility.150-152 The bone formation induced by ET-1 can be inhibited by the ETA atrasentan (pyrrolidine-3-carboxylic acid), in prostate cancer.153, 154 ET-1 may also increase osteoblast proliferation and bone formation by cross-talk with Wnt signaling, leading to the suppression of the inhibitor of Wnt signaling, Dickkopf-1 (DKK1).155
Nelson and Carducci153 first proposed that ET-1 is associated with osteoblastic metastases in prostate cancer, and found that exogenous ET-1 increases proliferation of prostate cancer cells. The level of plasma ET-1 is elevated in patients with osteoblastic prostate cancer metastases.145 Atrasentan is a highly selective and potent ET receptor antagonist that significantly inhibits the development of osteoblastic response to cancer in bone in a variety of model systems. Several phase 2 and 3 trials have evaluated its role in castrate-resistant prostate cancer.156 To date, the clinical experience has shown that atresentan alone or in combination with docetaxel, a chemotherapeutic agent standardly used to treat castrate-resistant prostate cancer, has modest activity in metastatic prostate cancer, and an ongoing phase 3 trial is currently evaluating whether its addition to docetaxel in an earlier stage of disease might be of benefit to patients with prostate cancer (NCT00134056).
Wnt signaling
Wnt is a soluble protein that binds to cell-surface receptors of the Frizzled family, which in turn activate members of the Dishevelled family proteins and subsequently stabilizes β-catenin, which then translocates to the nucleus and promotes multiple effects such as bone formation. Canonical Wnt signaling has been shown to stimulate osteoblast differentiation through β-catenin-induced gene transcription.157 In prostate cancer bone metastasis, Wnt produced by prostate cancer cells stimulates osteoblast differentiation and, in addition, has autocrine effects on tumor proliferation.158 Wnt signaling is also regulated by its antagonist, DKK1, which is primarily expressed in the early stages of prostate cancer, and is decreased in expression in bone metastases.159 Inhibition of DKK1 in osteolytic PC3 cells stimulates their osteoblastic activity, while overexpression of DKK1 in prostate C4-2B cells changes a mixed osteolytic-osteoblastic phenotype to an osteolytic phenotype.160 These results suggest that DKK1 may be one of the molecular switches governing osteolytic metastases becoming osteoblastic in later stages of metastatic growth. In a recent study, overexpression of DKK-1 in prostate cancer Ace-1 cells, derived from a dog prostate carcinoma, increased subcutaneous tumor growth and the incidence of bone metastasis, but significantly decreased the osteoblastic phenotype of bone metastases in an intratibial mouse model. These results suggest DKK-1 has an inhibitory role in bone formation in prostate cancer-induced osteoblastic metastases via the Wnt canonical pathway.161 Other studies suggest that noncanonical Wnt signaling also stimulates osteoblast differentiation, through BMP-dependent and -independent signaling pathways.162
Bone morphogenic proteins (BMPs)
BMPs are members of the TGF-β superfamily163 and are known to be involved in stimulating cancer cell migration. Osteoblast-derived BMP-2 can activate the Akt and ERK pathways, which in turn induce IKKα/β phosphorylation and NF-κB activation, resulting in the activation of β1 and β3 integrins and contributing to the migration of prostate cancer cells.164 BMP signaling also activates the intracellular receptor type I kinase, followed by phosphorylation of SMAD, which translocates to the nucleus and induces the expression of genes important for bone formation.140 In clinical prostate cancer tissues, the expression level of BMP-7 was higher in osteoblastic bone lesions than in normal bone.165 BMPs have been shown to be important factors for initiating osteoblast differentiation in vitro and in vivo.166, 167 Injection of an anti-BMP-6 antibody reduced osteoblast numbers and tumor growth of LuCaP 23.1 prostate cancer cells in implanted human bone in immunodeficient mice.168 The osteoblastic effect of BMPs is further confirmed by expression of Noggin (an antagonist of BMPs) in LAPC-9 osteoblastic prostate cancer cells, which induced osteolytic bone metastases in an intratibial xenograft mouse model.169 Further, ectopic expression of Noggin via a retroviral expression vector together with injection of RANK-Fc delayed the development of lesions induced by osteolytic PC3 cells.170 These results suggest that BMPs play an important role in contributing to osteoblastic phenotype of bone metastasis in prostate cancer. A recent study suggests that BMP-4 signaling to induce apoptosis and Smad-mediated gene expression can be repressed by IGF-I through activating mTOR signaling in prostate epithelial cells (NRP-152), suggesting a crosstalk between BMP and IGF signaling.171
Insulin-like growth factors (IGFs)
Another important growth factor family affecting prostate cancer bone metastasis is the IGF family. IGF-I and II are abundant in the bone matrix and are released during bone resorption.133 IGFs act through binding to the IGF receptors 1 and 2 (classic receptor protein tyrosine kinases) and promote cell proliferation, survival and angiogenesis. When aberrantly expressed, signaling through IGF-IR also promotes malignant transformation of fibroblasts NIH 3T3 cells.172 IGFs promote osteoblasts to increase bone matrix apposition and decrease collagen degradation.173 IGF-I is up-regulated in prostate cancer metastases in the bone, and contributes to cancer cell proliferation and chemotaxis.174, 175 In clinical studies, levels of IGF also correlate with cancer progression, as high levels of IGF-I are associated with a Gleason score ≥ 7.176 The protein level of IGFs and IGF-binding proteins (IGFBPs), which serve as carrier proteins for IGFs, could be mediated by proteolysis of IGFBPs. Indeed, hydrolyzing IGFBPs by uPA increases IGF levels and stimulates osteoblast proliferation.177 The cleavage of IGFBP-3 by PSA also increases IGF-I expression.178 However, other studies suggest that IGF-I is not necessary for inducing the osteoblastic response in prostate cancer.179 When exogenous IGF-I was added to MC3T3 cells (which can be differentiated into osteoblasts), no effect on osteoblast activity was observed, and an antibody to IGF-I did not block the differentiation of MC3T3 cells by prostate tumor cell-conditioned media. Furthermore, overexpression of IGF-I in prostate C4-2 or PC3 cells had no significant effect on osteoblastic growth of tumor in intratibial xenograft model.179 Further studies are required to elucidate the complex roles of IGF in prostate cancer metastasis; however, small molecule IGF-IR inhibitors show promise for inhibiting prostate cancer growth using in vitro models (Dayyani and Gallick, unpublished).
Osteoclast-associated factors
Metastatic cancer cells also produce cytokines and growth factors that activate osteoclast differentiation and bone resorption. In prostate cancer, RANKL, parathyroid hormone related protein (PTHrP), and TGF-β are among the most important osteolytic factors that contribute to bone resorption and osteolytic bone metastasis.
RANK/RANKL/OPG axis
In addition to other factors that activate osteoclasts, prostate cancer cells can secrete soluble RANKL that directly activates osteoclasts.180, 181 RANKL is produced by osteoblasts, and belongs to the TNF superfamily of cytokines. The binding of RANKL to receptor activator of the nuclear factor-κB (RANK) on the surface of osteoclasts activates mitogen-activated protein kinase-related TGF-β-inducible kinase TAK1, along with the TNF receptor-associated factors (TRAF)-binding adapter protein TAB2. TAK1 then induces activation of NF-κB and AP-147, which results in transcription of genes necessary for osteoclast maturation.182 This process is inhibited by osteoprotegerin (OPG) produced by osteoblasts,182 a soluble decoy receptor containing only the extracellular RANK domain for RANKL binding. Hence, interruption of RANK/RANKL/OPG axis has been attempted as a strategy to inhibit metastatic tumor growth in the bone. Experiments using OPG to block RANK/RANKL/OPG axis led to reduced osteoclastogenesis and increased osteoclast apoptosis.183, 184 However, the inhibitory effects of tumor growth by OPG may be compromised, because OPG also protects prostate cancer cells from TRAIL-induced apoptosis.185, 186 Another approach that might potentially avoid this problem is to block the RANK/RANKL/OPG signaling axis using RANK-Fc, a recombinant RANKL antagonist.187 In an in vivo study, after implanted human bones in immunocompromised mice were injected with LuCaP 35 human prostate tumor cells, injection of RANK-Fc reduced osteolytic bone lesions and prostate tumor growth.181 However, blocking RANK/RANKL/OPG axis has only indirect and limited effects on tumor growth, because blocking osteolytic activity may simply reduce the release of growth factors from bone matrix, failing to target tumor proliferation,128 another example of the importance of targeting both the tumor and the microenvironment for therapeutic success.
Denosumab, a fully human monoclonal antibody against RANKL, the primary effector of osteoclast activity, has a high affinity for human RANKL and does not enrich in the bone, as opposed to bisphosphonates. Its role in inhibition of osteoclast activity has been examined in several malignancies, including breast cancer, multiple myeloma, and prostate cancer.188-191 In a wide range of malignancies with bone metastases, denosumab has been shown in a meta-analysis of two trials to delay skeletal-related events (e.g. fracture, spinal cord compression, and radiation or surgery to bone) when compared to the bisphosphonate zoledronic acid. But these trials did not include patients with prostate cancer.192 Since androgen-deprivation therapy is associated with bone loss in men with prostate cancer,193 denosumab has been tested in a large randomized phase 3 trial in men with nonmetastatic prostate cancer who underwent androgen-deprivation therapy. The drug significantly decreased the risk of bone fractures in treated men.194 Trials investigating its role in the treatment and prevention of bone metastasis in prostate cancer are currently under way (NCT00286091, NCT00321620).
Parathyroid hormone related protein (PTHrP)
PTHrP is a homolog of parathyroid hormone, and both share the same parathyroid hormone receptor. PTHrP contributes to osteolytic metastasis by up-regulating RANKL production in osteoblasts and decreasing expression of OPG, a soluble decoy receptor for RANKL binding, which leads to activation of osteoclasts.195, 196 In prostate cancer, tumor-derived PTHrP can enhance osteoclastogenesis in vitro and in vivo.195 In clinical specimens from prostate cancer, PTHrP levels are elevated in osteoblastic bone metastases.197 PTHrP also induces osteoblast differentiation195 and might protect prostate cancer cells and osteoblasts from apoptosis.198 These results indicate that PTHrP produced by prostate cancer cells can enhance both osteoclastogenesis and osteoblastogenesis.195 In addition, PTHrP can be cleaved by PSA, leading to reduced bone resorption and increase in osteoblastic response, which suggests that the degradation of PTHrP may cause the shifting of osteoclastic to osteoblastic processes in prostate tumor bone metastasis.199, 200
Transforming growth factor β (TGF-β)
TGF-β plays an important role as a tumor suppressor in normal epithelial cells. However, prostate epithelial cells (and many other types of tumor cells) become insensitive to growth inhibition of TGF-β during tumor progression.201, 202 TGF-β signaling is initiated by the binding of TGF-β ligands to type II TGF-β receptor which recruits and phosphorylates type I TGF-β receptor (TGFBR1), resulting in phosphorylation of SMADs, which translocate into the nucleus and induce transcription of TGF-β-responsive genes. Activated TGFBR1 also induces activation of SMAD-independent pathways, such as PI3K, AKT, and/or mitogen-activated protein kinase pathways. TGF-β is activated by cleavage of its precursor by osteoclast-derived proteases or by cancer-secreted PSA and uPA.203, 204 TGF-β regulates bone development and bone resorption by promoting expression of PTHrP and IL-11 in cancer cells.148, 205, 206 The most striking evidence for TGF-β promoting prostate cancer metastasis is derived from overexpression of TGF-β signaling inhibitors. The antagonist of TGF-β signaling, BMP-7, inhibits osteolytic metastases in a xenograft model of intratibially injected prostate PC3M-Pro4 cells, a variant of PC3 cells.207 However, BMP-7 fails to inhibit the growth of PC3M-Pro4 cells when inoculated intraprostatically in nude mice, suggesting the osteolytic effect of TGF-β is bone-specific.207
Conclusion and Prospects
Metastasis to the bone remains the major cause of death in advanced prostate cancer. Current treatments for bone metastasis mostly are palliative with little effect on long-term survival. Thus, a better understanding of the biologic mechanisms of cancer metastasis can and is leading to more effective therapies for prostate cancer patients with bone metastases. Many of the factors in the metastatic cascade have been suggested to contribute to the bone-tropic nature of metastasis of prostate cancer. However, it seems likely that several of these functions are redundant, and many of the processes work in concert to increase the metastatic potential of tumor cells and promote metastasis in the bone, complicating the development of successful therapeutic strategies for patients with bone metastases. An increasing amount of evidence supports the essential roles of cells in the microenvironment in the promotion of metastasis, and the plasticity of both tumor cells and microenvironment cells. Thus, a recent hypothesis for selection of increasingly metastatic cells posits that circulating cancer cells can reseed the primary site and gain more metastatic potential during this process.208 Understanding the tumor/microenvironment interactions that lead to increased metastatic potential is critical to devising new therapeutic strategies, and improving on agents that currently show promise in clinical trials.
Acknowledgements
The authors wish to acknowledge the excellent scientific editing of Karen Phillips. This work was supported by NIH T32 CA009666 (FD); NIH/NCI P50 CA140388 (GEG).
Grant sponsor: NIH/NCI; Grant number: T32 CA009666 (FD); P50 CA140388 (GEG)
Abbreviations
- AR
androgen receptor
- BMP
bone morphogenetic protein
- BMS
bone marrow stromal
- CAM
cell adhesion molecule
- DKK1
Dickkopf-1
- ECM
extracellular matrix
- EMT
epithelial to mesenchymal transition
- ET-1
endothelin-1
- ETA
endothelin-1 receptor subtype A
- FAK
focal adhesion kinase
- HBME
human bone marrow endothelium
- IGF
insulin-like growth factor
- IGFBP
IGF-binding protein
- IGF-IR
IGF I receptor
- IL
interleukin
- MMP
matrix metalloproteinase
- OPG
osteoprotegerin
- PI3K
phosphatidylinositol 3-kinase
- PSA
prostate-specific antigen
- PTHrP
parathyroid hormone-related protein
- RANKL
receptor activator of nuclear factor-κB ligand
- SDF-1
stromal-derived factor-1
- SFK
Src family kinases
- TGF-β
transforming growth factor-β
- TGFBR1
type I TGF-β receptor
- TIMP
tissue inhibitor of metalloproteinases
- uPA
urokinase-type plasminogen activator
- uPAR
uPA receptor
References
- 1.Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. GLOBOCAN 2008, cancer incidence and mortality worldwide. International Agency for Research on Cancer. 2010 http://globocan.iarc.fr/factsheets/cancers/prostate.asp.
- 2.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 3.Bubendorf L, Schopfer A, Wagner U, Sauter G, Moch H, Willi N, Gasser TC, Mihatsch MJ. Metastatic patterns of prostate cancer: an autopsy study of 1,589 patients. Hum Pathol. 2000;31:578–83. doi: 10.1053/hp.2000.6698. [DOI] [PubMed] [Google Scholar]
- 4.Costa L, Major PP. Effect of bisphosphonates on pain and quality of life in patients with bone metastases. Nat Clin Pract Oncol. 2009;6:163–74. doi: 10.1038/ncponc1323. [DOI] [PubMed] [Google Scholar]
- 5.Lee RJ, Saylor PJ, Smith MR. Treatment and prevention of bone complications from prostate cancer. Bone. 2011;48:88–95. doi: 10.1016/j.bone.2010.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huggins C, Hodges CV. Studies on prostatic cancer. I. The effect of castration, of estrogen and androgen injection on serum phosphatases in metastatic carcinoma of the prostate. CA Cancer J Clin. 1972;22:232–40. doi: 10.3322/canjclin.22.4.232. [DOI] [PubMed] [Google Scholar]
- 7.Dutt SS, Gao AC. Molecular mechanisms of castration-resistant prostate cancer progression. Future Oncol. 2009;5:1403–13. doi: 10.2217/fon.09.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bonkhoff H, Berges R. From pathogenesis to prevention of castration resistant prostate cancer. Prostate. 2010;70:100–12. doi: 10.1002/pros.21042. [DOI] [PubMed] [Google Scholar]
- 9.Knudsen KE, Penning TM. Partners in crime: deregulation of AR activity and androgen synthesis in prostate cancer. Trends Endocrinol Metab. 2010;21:315–24. doi: 10.1016/j.tem.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shen MM, Abate-Shen C. Molecular genetics of prostate cancer: new prospects for old challenges. Genes Dev. 2010;24:1967–2000. doi: 10.1101/gad.1965810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Siegall CB, Schwab G, Nordan RP, FitzGerald DJ, Pastan I. Expression of the interleukin 6 receptor and interleukin 6 in prostate carcinoma cells. Cancer Res. 1990;50:7786–8. [PubMed] [Google Scholar]
- 12.Siegsmund MJ, Yamazaki H, Pastan I. Interleukin 6 receptor mRNA in prostate carcinomas and benign prostate hyperplasia. J Urol. 1994;151:1396–9. doi: 10.1016/s0022-5347(17)35267-9. [DOI] [PubMed] [Google Scholar]
- 13.Hobisch A, Rogatsch H, Hittmair A, Fuchs D, Bartsch G, Jr., Klocker H, Bartsch G, Culig Z. Immunohistochemical localization of interleukin-6 and its receptor in benign, premalignant and malignant prostate tissue. J Pathol. 2000;191:239–44. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH633>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 14.Adler HL, McCurdy MA, Kattan MW, Timme TL, Scardino PT, Thompson TC. Elevated levels of circulating interleukin-6 and transforming growth factor-beta1 in patients with metastatic prostatic carcinoma. J Urol. 1999;161:182–7. [PubMed] [Google Scholar]
- 15.Drachenberg DE, Elgamal AA, Rowbotham R, Peterson M, Murphy GP. Circulating levels of interleukin-6 in patients with hormone refractory prostate cancer. Prostate. 1999;41:127–33. doi: 10.1002/(sici)1097-0045(19991001)41:2<127::aid-pros7>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 16.Hobisch A, Eder IE, Putz T, Horninger W, Bartsch G, Klocker H, Culig Z. Interleukin-6 regulates prostate-specific protein expression in prostate carcinoma cells by activation of the androgen receptor. Cancer Res. 1998;58:4640–5. [PubMed] [Google Scholar]
- 17.Chen T, Wang LH, Farrar WL. Interleukin 6 activates androgen receptor-mediated gene expression through a signal transducer and activator of transcription 3-dependent pathway in LNCaP prostate cancer cells. Cancer Res. 2000;60:2132–5. [PubMed] [Google Scholar]
- 18.Lou W, Ni Z, Dyer K, Tweardy DJ, Gao AC. Interleukin-6 induces prostate cancer cell growth accompanied by activation of stat3 signaling pathway. Prostate. 2000;42:239–42. doi: 10.1002/(sici)1097-0045(20000215)42:3<239::aid-pros10>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 19.Lee SO, Lou W, Hou M, de Miguel F, Gerber L, Gao AC. Interleukin-6 promotes androgen-independent growth in LNCaP human prostate cancer cells. Clin Cancer Res. 2003;9:370–6. [PubMed] [Google Scholar]
- 20.Lee SO, Lou W, Johnson CS, Trump DL, Gao AC. Interleukin-6 protects LNCaP cells from apoptosis induced by androgen deprivation through the Stat3 pathway. Prostate. 2004;60:178–86. doi: 10.1002/pros.20045. [DOI] [PubMed] [Google Scholar]
- 21.Haapala K, Kuukasjarvi T, Hyytinen E, Rantala I, Helin HJ, Koivisto PA. Androgen receptor amplification is associated with increased cell proliferation in prostate cancer. Hum Pathol. 2007;38:474–8. doi: 10.1016/j.humpath.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 22.Attar RM, Takimoto CH, Gottardis MM. Castration-resistant prostate cancer: locking up the molecular escape routes. Clin Cancer Res. 2009;15:3251–5. doi: 10.1158/1078-0432.CCR-08-1171. [DOI] [PubMed] [Google Scholar]
- 23.Mellado B, Codony J, Ribal MJ, Visa L, Gascon P. Molecular biology of androgen-independent prostate cancer: the role of the androgen receptor pathway. Clin Transl Oncol. 2009;11:5–10. doi: 10.1007/s12094-009-0304-3. [DOI] [PubMed] [Google Scholar]
- 24.Paget S. The distribution of secondary growth in cancer of the breast. Lancet. 1889;1:571–3. [PubMed] [Google Scholar]
- 25.Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009;9:265–73. doi: 10.1038/nrc2620. [DOI] [PubMed] [Google Scholar]
- 26.Arya M, Bott SR, Shergill IS, Ahmed HU, Williamson M, Patel HR. The metastatic cascade in prostate cancer. Surg Oncol. 2006;15:117–28. doi: 10.1016/j.suronc.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 27.Clarke NW, Hart CA, Brown MD. Molecular mechanisms of metastasis in prostate cancer. Asian J Androl. 2009;11:57–67. doi: 10.1038/aja.2008.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mol AJ, Geldof AA, Meijer GA, van der Po H, van Moorselaar RJ. New experimental markers for early detection of high-risk prostate cancer: role of cell-cell adhesion and cell migration. J Cancer Res Clin Oncol. 2007;133:687–95. doi: 10.1007/s00432-007-0235-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–54. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 30.Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell. 2008;14:818–29. doi: 10.1016/j.devcel.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 31.Tsuji T, Ibaragi S, Hu GF. Epithelial-mesenchymal transition and cell cooperativity in metastasis. Cancer Res. 2009;69:7135–9. doi: 10.1158/0008-5472.CAN-09-1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.He H, Yang X, Davidson AJ, Wu D, Marshall FF, Chung LW, Zhau HE, Wang R. Progressive epithelial to mesenchymal transitions in ARCaP E prostate cancer cells during xenograft tumor formation and metastasis. Prostate. 2010;70:518–28. doi: 10.1002/pros.21086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Roy F, Berx G. The cell-cell adhesion molecule E-cadherin. Cell Mol Life Sci. 2008;65:3756–88. doi: 10.1007/s00018-008-8281-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Takeichi M. Cadherin cell adhesion receptors as a morphogenetic regulator. Science. 1991;251:1451–5. doi: 10.1126/science.2006419. [DOI] [PubMed] [Google Scholar]
- 35.Guarino M. Src signaling in cancer invasion. J Cell Physiol. 2010;223:14–26. doi: 10.1002/jcp.22011. [DOI] [PubMed] [Google Scholar]
- 36.Gravdal K, Halvorsen OJ, Haukaas SA, Akslen LA. A switch from E-cadherin to N-cadherin expression indicates epithelial to mesenchymal transition and is of strong and independent importance for the progress of prostate cancer. Clin Cancer Res. 2007;13:7003–11. doi: 10.1158/1078-0432.CCR-07-1263. [DOI] [PubMed] [Google Scholar]
- 37.Umbas R, Schalken JA, Aalders TW, Carter BS, Karthaus HF, Schaafsma HE, Debruyne FM, Isaacs WB. Expression of the cellular adhesion molecule E-cadherin is reduced or absent in high-grade prostate cancer. Cancer Res. 1992;52:5104–9. [PubMed] [Google Scholar]
- 38.Umbas R, Isaacs WB, Bringuier PP, Schaafsma HE, Karthaus HF, Oosterhof GO, Debruyne FM, Schalken JA. Decreased E-cadherin expression is associated with poor prognosis in patients with prostate cancer. Cancer Res. 1994;54:3929–33. [PubMed] [Google Scholar]
- 39.Jaggi M, Johansson SL, Baker JJ, Smith LM, Galich A, Balaji KC. Aberrant expression of E-cadherin and beta-catenin in human prostate cancer. Urol Oncol. 2005;23:402–6. doi: 10.1016/j.urolonc.2005.03.024. [DOI] [PubMed] [Google Scholar]
- 40.Tran NL, Nagle RB, Cress AE, Heimark RL. N-Cadherin expression in human prostate carcinoma cell lines. An epithelial-mesenchymal transformation mediating adhesion withStromal cells. Am J Pathol. 1999;155:787–98. doi: 10.1016/S0002-9440(10)65177-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bussemakers MJ, van Moorselaar RJ, Giroldi LA, Ichikawa T, Isaacs JT, Takeichi M, Debruyne FM, Schalken JA. Decreased expression of E-cadherin in the progression of rat prostatic cancer. Cancer Res. 1992;52:2916–22. [PubMed] [Google Scholar]
- 42.Saha B, Arase A, Imam SS, Tsao-Wei D, Naritoku WY, Groshen S, Jones LW, Imam SA. Overexpression of E-cadherin and beta-catenin proteins in metastatic prostate cancer cells in bone. Prostate. 2008;68:78–84. doi: 10.1002/pros.20670. [DOI] [PubMed] [Google Scholar]
- 43.Guo W, Giancotti FG. Integrin signalling during tumour progression. Nat Rev Mol Cell Biol. 2004;5:816–26. doi: 10.1038/nrm1490. [DOI] [PubMed] [Google Scholar]
- 44.Slack-Davis JK, Parsons JT. Emerging views of integrin signaling: implications for prostate cancer. J Cell Biochem. 2004;91:41–6. doi: 10.1002/jcb.10665. [DOI] [PubMed] [Google Scholar]
- 45.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–87. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 46.Schmelz M, Cress AE, Scott KM, Burger F, Cui H, Sallam K, McDaniel KM, Dalkin BL, Nagle RB. Different phenotypes in human prostate cancer: alpha6 or alpha3 integrin in cell-extracellular adhesion sites. Neoplasia. 2002;4:243–54. doi: 10.1038/sj.neo.7900223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cress AE, Rabinovitz I, Zhu W, Nagle RB. The alpha 6 beta 1 and alpha 6 beta 4 integrins in human prostate cancer progression. Cancer Metastasis Rev. 1995;14:219–28. doi: 10.1007/BF00690293. [DOI] [PubMed] [Google Scholar]
- 48.Davis TL, Cress AE, Dalkin BL, Nagle RB. Unique expression pattern of the alpha6beta4 integrin and laminin-5 in human prostate carcinoma. Prostate. 2001;46:240–8. doi: 10.1002/1097-0045(20010215)46:3<240::aid-pros1029>3.0.co;2-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hao J, Yang Y, McDaniel KM, Dalkin BL, Cress AE, Nagle RB. Differential expression of laminin 5 (alpha 3 beta 3 gamma 2) by human malignant and normal prostate. Am J Pathol. 1996;149:1341–9. [PMC free article] [PubMed] [Google Scholar]
- 50.Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol. 2005;6:56–68. doi: 10.1038/nrm1549. [DOI] [PubMed] [Google Scholar]
- 51.Rovin JD, Frierson HF, Jr., Ledinh W, Parsons JT, Adams RB. Expression of focal adhesion kinase in normal and pathologic human prostate tissues. Prostate. 2002;53:124–32. doi: 10.1002/pros.10114. [DOI] [PubMed] [Google Scholar]
- 52.Tremblay L, Hauck W, Aprikian AG, Begin LR, Chapdelaine A, Chevalier S. Focal adhesion kinase (pp125FAK) expression, activation and association with paxillin and p50CSK in human metastatic prostate carcinoma. Int J Cancer. 1996;68:164–71. doi: 10.1002/(sici)1097-0215(19961009)68:2<169::aid-ijc4>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 53.Mitra SK, Schlaepfer DD. Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr Opin Cell Biol. 2006;18:516–23. doi: 10.1016/j.ceb.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 54.Fizazi K. The role of Src in prostate cancer. Ann Oncol. 2007;18:1765–73. doi: 10.1093/annonc/mdm086. [DOI] [PubMed] [Google Scholar]
- 55.Summy JM, Gallick GE. Src family kinases in tumor progression and metastasis. Cancer Metastasis Rev. 2003;22:337–58. doi: 10.1023/a:1023772912750. [DOI] [PubMed] [Google Scholar]
- 56.Kim MP, Park SI, Kopetz S, Gallick GE. Src family kinases as mediators of endothelial permeability: effects on inflammation and metastasis. Cell Tissue Res. 2009;335:249–59. doi: 10.1007/s00441-008-0682-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Parri M, Chiarugi P. Rac and Rho GTPases in cancer cell motility control. Cell Commun Signal. 2010;8:23. doi: 10.1186/1478-811X-8-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Summy JM, Gallick GE. Treatment for advanced tumors: SRC reclaims center stage. Clin Cancer Res. 2006;12:1398–401. doi: 10.1158/1078-0432.CCR-05-2692. [DOI] [PubMed] [Google Scholar]
- 59.Frame MC. Newest findings on the oldest oncogene; how activated src does it. J Cell Sci. 2004;117:989–98. doi: 10.1242/jcs.01111. [DOI] [PubMed] [Google Scholar]
- 60.Yeatman TJ. A renaissance for SRC. Nat Rev Cancer. 2004;4:470–80. doi: 10.1038/nrc1366. [DOI] [PubMed] [Google Scholar]
- 61.Paronetto MP, Farini D, Sammarco I, Maturo G, Vespasiani G, Geremia R, Rossi P, Sette C. Expression of a truncated form of the c-Kit tyrosine kinase receptor and activation of Src kinase in human prostatic cancer. Am J Pathol. 2004;164:1243–51. doi: 10.1016/S0002-9440(10)63212-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tatarov O, Mitchell TJ, Seywright M, Leung HY, Brunton VG, Edwards J. SRC family kinase activity is up-regulated in hormone-refractory prostate cancer. Clin Cancer Res. 2009;15:3540–9. doi: 10.1158/1078-0432.CCR-08-1857. [DOI] [PubMed] [Google Scholar]
- 63.Trevino JG, Gray MJ, Nawrocki ST, Summy JM, Lesslie DP, Evans DB, Sawyer TK, Shakespeare WC, Watowich SS, Chiao PJ, McConkey DJ, Gallick GE. Src activation of Stat3 is an independent requirement from NF-kappaB activation for constitutive IL-8 expression in human pancreatic adenocarcinoma cells. Angiogenesis. 2006;9:101–10. doi: 10.1007/s10456-006-9038-9. [DOI] [PubMed] [Google Scholar]
- 64.Lee LF, Louie MC, Desai SJ, Yang J, Chen HW, Evans CP, Kung HJ. Interleukin-8 confers androgen-independent growth and migration of LNCaP: differential effects of tyrosine kinases Src and FAK. Oncogene. 2004;23:2197–205. doi: 10.1038/sj.onc.1207344. [DOI] [PubMed] [Google Scholar]
- 65.Nam S, Kim D, Cheng JQ, Zhang S, Lee JH, Buettner R, Mirosevich J, Lee FY, Jove R. Action of the Src family kinase inhibitor, dasatinib (BMS-354825), on human prostate cancer cells. Cancer Res. 2005;65:9185–9. doi: 10.1158/0008-5472.CAN-05-1731. [DOI] [PubMed] [Google Scholar]
- 66.Recchia I, Rucci N, Festuccia C, Bologna M, MacKay AR, Migliaccio S, Longo M, Susa M, Fabbro D, Teti A. Pyrrolopyrimidine c-Src inhibitors reduce growth, adhesion, motility and invasion of prostate cancer cells in vitro. Eur J Cancer. 2003;39:1927–35. doi: 10.1016/s0959-8049(03)00394-0. [DOI] [PubMed] [Google Scholar]
- 67.Slack JK, Adams RB, Rovin JD, Bissonette EA, Stoker CE, Parsons JT. Alterations in the focal adhesion kinase/Src signal transduction pathway correlate with increased migratory capacity of prostate carcinoma cells. Oncogene. 2001;20:1152–63. doi: 10.1038/sj.onc.1204208. [DOI] [PubMed] [Google Scholar]
- 68.Park SI, Zhang J, Phillips KA, Araujo JC, Najjar AM, Volgin AY, Gelovani JG, Kim SJ, Wang Z, Gallick GE. Targeting SRC family kinases inhibits growth and lymph node metastases of prostate cancer in an orthotopic nude mouse model. Cancer Res. 2008;68:3323–33. doi: 10.1158/0008-5472.CAN-07-2997. [DOI] [PubMed] [Google Scholar]
- 69.Goldenberg-Furmanov M, Stein I, Pikarsky E, Rubin H, Kasem S, Wygoda M, Weinstein I, Reuveni H, Ben-Sasson SA. Lyn is a target gene for prostate cancer: sequence-based inhibition induces regression of human tumor xenografts. Cancer Res. 2004;64:1058–66. doi: 10.1158/0008-5472.can-03-2420. [DOI] [PubMed] [Google Scholar]
- 70.Morgia G, Falsaperla M, Malaponte G, Madonia M, Indelicato M, Travali S, Mazzarino MC. Matrix metalloproteinases as diagnostic (MMP-13) and prognostic (MMP-2, MMP-9) markers of prostate cancer. Urol Res. 2005;33:44–50. doi: 10.1007/s00240-004-0440-8. [DOI] [PubMed] [Google Scholar]
- 71.Wood M, Fudge K, Mohler JL, Frost AR, Garcia F, Wang M, Stearns ME. In situ hybridization studies of metalloproteinases 2 and 9 and TIMP-1 and TIMP-2 expression in human prostate cancer. Clin Exp Metastasis. 1997;15:246–58. doi: 10.1023/a:1018421431388. [DOI] [PubMed] [Google Scholar]
- 72.Lichtinghagen R, Musholt PB, Lein M, Romer A, Rudolph B, Kristiansen G, Hauptmann S, Schnorr D, Loening SA, Jung K. Different mRNA and protein expression of matrix metalloproteinases 2 and 9 and tissue inhibitor of metalloproteinases 1 in benign and malignant prostate tissue. Eur Urol. 2002;42:398–406. doi: 10.1016/s0302-2838(02)00324-x. [DOI] [PubMed] [Google Scholar]
- 73.Trudel D, Fradet Y, Meyer F, Harel F, Tetu B. Significance of MMP-2 expression in prostate cancer: an immunohistochemical study. Cancer Res. 2003;63:8511–5. [PubMed] [Google Scholar]
- 74.Brehmer B, Biesterfeld S, Jakse G. Expression of matrix metalloproteinases (MMP-2 and -9) and their inhibitors (TIMP-1 and -2) in prostate cancer tissue. Prostate Cancer Prostatic Dis. 2003;6:217–22. doi: 10.1038/sj.pcan.4500657. [DOI] [PubMed] [Google Scholar]
- 75.Zhong WD, Han ZD, He HC, Bi XC, Dai QS, Zhu G, Ye YK, Liang YX, Qin WJ, Zhang Z, Zeng GH, Chen ZN. CD147, MMP-1, MMP-2 and MMP-9 protein expression as significant prognostic factors in human prostate cancer. Oncology. 2008;75:230–6. doi: 10.1159/000163852. [DOI] [PubMed] [Google Scholar]
- 76.Boxler S, Djonov V, Kessler TM, Hlushchuk R, Bachmann LM, Held U, Markwalder R, Thalmann GN. Matrix metalloproteinases and angiogenic factors: predictors of survival after radical prostatectomy for clinically organ-confined prostate cancer? Am J Pathol. 2010;177:2216–24. doi: 10.2353/ajpath.2010.091190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Overall CM, Lopez-Otin C. Strategies for MMP inhibition in cancer: innovations for the post-trial era. Nat Rev Cancer. 2002;2:657–72. doi: 10.1038/nrc884. [DOI] [PubMed] [Google Scholar]
- 78.Lokeshwar BL, Selzer MG, Block NL, Gunja-Smith Z. Secretion of matrix metalloproteinases and their inhibitors (tissue inhibitor of metalloproteinases) by human prostate in explant cultures: reduced tissue inhibitor of metalloproteinase secretion by malignant tissues. Cancer Res. 1993;53:4493–8. [PubMed] [Google Scholar]
- 79.Littlepage LE, Sternlicht MD, Rougier N, Phillips J, Gallo E, Yu Y, Williams K, Brenot A, Gordon JI, Werb Z. Matrix metalloproteinases contribute distinct roles in neuroendocrine prostate carcinogenesis, metastasis, and angiogenesis progression. Cancer Res. 2010;70:2224–34. doi: 10.1158/0008-5472.CAN-09-3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nabha SM, dos Santos EB, Yamamoto HA, Belizi A, Dong Z, Meng H, Saliganan A, Sabbota A, Bonfil RD, Cher ML. Bone marrow stromal cells enhance prostate cancer cell invasion through type I collagen in an MMP-12 dependent manner. Int J Cancer. 2008;122:2482–90. doi: 10.1002/ijc.23431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Testa JE, Quigley JP. The role of urokinase-type plasminogen activator in aggressive tumor cell behavior. Cancer Metastasis Rev. 1990;9:353–67. doi: 10.1007/BF00049524. [DOI] [PubMed] [Google Scholar]
- 82.Sheng S. The urokinase-type plasminogen activator system in prostate cancer metastasis. Cancer Metastasis Rev. 2001;20:287–96. doi: 10.1023/a:1015539612576. [DOI] [PubMed] [Google Scholar]
- 83.Li Y, Cozzi PJ. Targeting uPA/uPAR in prostate cancer. Cancer Treat Rev. 2007;33:521–7. doi: 10.1016/j.ctrv.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 84.Kumano M, Miyake H, Muramaki M, Furukawa J, Takenaka A, Fujisawa M. Expression of urokinase-type plasminogen activator system in prostate cancer: correlation with clinicopathological outcomes in patients undergoing radical prostatectomy. Urol Oncol. 2009;27:180–6. doi: 10.1016/j.urolonc.2008.01.012. [DOI] [PubMed] [Google Scholar]
- 85.Festuccia C, Dolo V, Guerra F, Violini S, Muzi P, Pavan A, Bologna M. Plasminogen activator system modulates invasive capacity and proliferation in prostatic tumor cells. Clin Exp Metastasis. 1998;16:513–28. doi: 10.1023/a:1006590217724. [DOI] [PubMed] [Google Scholar]
- 86.Dong Z, Saliganan AD, Meng H, Nabha SM, Sabbota AL, Sheng S, Bonfil RD, Cher ML. Prostate cancer cell-derived urokinase-type plasminogen activator contributes to intraosseous tumor growth and bone turnover. Neoplasia. 2008;10:439–49. doi: 10.1593/neo.08106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Christensson A, Lilja H. Complex formation between protein C inhibitor and prostate-specific antigen in vitro and in human semen. Eur J Biochem. 1994;220:45–53. doi: 10.1111/j.1432-1033.1994.tb18597.x. [DOI] [PubMed] [Google Scholar]
- 88.Watt KW, Lee PJ, M’Timkulu T, Chan WP, Loor R. Human prostate-specific antigen: structural and functional similarity with serine proteases. Proc Natl Acad Sci U S A. 1986;83:3166–70. doi: 10.1073/pnas.83.10.3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Christensson A, Laurell CB, Lilja H. Enzymatic activity of prostate-specific antigen and its reactions with extracellular serine proteinase inhibitors. Eur J Biochem. 1990;194:755–63. doi: 10.1111/j.1432-1033.1990.tb19466.x. [DOI] [PubMed] [Google Scholar]
- 90.Lilja H, Oldbring J, Rannevik G, Laurell CB. Seminal vesicle-secreted proteins and their reactions during gelation and liquefaction of human semen. J Clin Invest. 1987;80:281–5. doi: 10.1172/JCI113070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Webber MM, Waghray A, Bello D. Prostate-specific antigen, a serine protease, facilitates human prostate cancer cell invasion. Clin Cancer Res. 1995;1:1089–94. [PubMed] [Google Scholar]
- 92.Cooper CR, McLean L, Walsh M, Taylor J, Hayasaka S, Bhatia J, Pienta KJ. Preferential adhesion of prostate cancer cells to bone is mediated by binding to bone marrow endothelial cells as compared to extracellular matrix components in vitro. Clin Cancer Res. 2000;6:4839–47. [PubMed] [Google Scholar]
- 93.Lehr JE, Pienta KJ. Preferential adhesion of prostate cancer cells to a human bone marrow endothelial cell line. J Natl Cancer Inst. 1998;90:118–23. doi: 10.1093/jnci/90.2.118. [DOI] [PubMed] [Google Scholar]
- 94.Honn KV, Tang DG. Adhesion molecules and tumor cell interaction with endothelium and subendothelial matrix. Cancer Metastasis Rev. 1992;11:353–75. doi: 10.1007/BF01307187. [DOI] [PubMed] [Google Scholar]
- 95.Mazo IB, von Andrian UH. Adhesion and homing of blood-borne cells in bone marrow microvessels. J Leukoc Biol. 1999;66:25–32. doi: 10.1002/jlb.66.1.25. [DOI] [PubMed] [Google Scholar]
- 96.Martensson S, Bigler SA, Brown M, Lange PH, Brawer MK, Hakomori S. Sialyl-Lewis(x) and related carbohydrate antigens in the prostate. Hum Pathol. 1995;26:735–9. doi: 10.1016/0046-8177(95)90220-1. [DOI] [PubMed] [Google Scholar]
- 97.Romanov VI, Goligorsky MS. RGD-recognizing integrins mediate interactions of human prostate carcinoma cells with endothelial cells in vitro. Prostate. 1999;39:108–18. doi: 10.1002/(sici)1097-0045(19990501)39:2<108::aid-pros5>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 98.Scott LJ, Clarke NW, George NJ, Shanks JH, Testa NG, Lang SH. Interactions of human prostatic epithelial cells with bone marrow endothelium: binding and invasion. Br J Cancer. 2001;84:1417–23. doi: 10.1054/bjoc.2001.1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sikes RA, Nicholson BE, Koeneman KS, Edlund NM, Bissonette EA, Bradley MJ, Thalmann GN, Cecchini MG, Pienta KJ, Chung LW. Cellular interactions in the tropism of prostate cancer to bone. Int J Cancer. 2004;110:497–503. doi: 10.1002/ijc.20153. [DOI] [PubMed] [Google Scholar]
- 100.Barrett JM, Mangold KA, Jilling T, Kaul KL. Bi-directional interactions of prostate cancer cells and bone marrow endothelial cells in three-dimensional culture. Prostate. 2005;64:75–82. doi: 10.1002/pros.20206. [DOI] [PubMed] [Google Scholar]
- 101.Efstathiou E, Logothetis CJ. A new therapy paradigm for prostate cancer founded on clinical observations. Clin Cancer Res. 2010;16:1100–7. doi: 10.1158/1078-0432.CCR-09-1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nesbitt S, Nesbit A, Helfrich M, Horton M. Biochemical characterization of human osteoclast integrins. Osteoclasts express alpha v beta 3, alpha 2 beta 1, and alpha v beta 1 integrins. J Biol Chem. 1993;268:16737–45. [PubMed] [Google Scholar]
- 103.Cooper CR, Chay CH, Pienta KJ. The role of alpha(v)beta(3) in prostate cancer progression. Neoplasia. 2002;4:191–4. doi: 10.1038/sj.neo.7900224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zheng DQ, Woodard AS, Fornaro M, Tallini G, Languino LR. Prostatic carcinoma cell migration via alpha(v)beta3 integrin is modulated by a focal adhesion kinase pathway. Cancer Res. 1999;59:1655–64. [PubMed] [Google Scholar]
- 105.Zheng DQ, Woodard AS, Tallini G, Languino LR. Substrate specificity of alpha(v)beta(3) integrin-mediated cell migration and phosphatidylinositol 3-kinase/AKT pathway activation. J Biol Chem. 2000;275:24565–74. doi: 10.1074/jbc.M002646200. [DOI] [PubMed] [Google Scholar]
- 106.McCabe NP, De S, Vasanji A, Brainard J, Byzova TV. Prostate cancer specific integrin alphavbeta3 modulates bone metastatic growth and tissue remodeling. Oncogene. 2007;26:6238–43. doi: 10.1038/sj.onc.1210429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hall CL, Dai J, van Golen KL, Keller ET, Long MW. Type I collagen receptor (alpha 2 beta 1) signaling promotes the growth of human prostate cancer cells within the bone. Cancer Res. 2006;66:8648–54. doi: 10.1158/0008-5472.CAN-06-1544. [DOI] [PubMed] [Google Scholar]
- 108.Hall CL, Dubyk CW, Riesenberger TA, Shein D, Keller ET, van Golen KL. Type I collagen receptor (alpha2beta1) signaling promotes prostate cancer invasion through RhoC GTPase. Neoplasia. 2008;10:797–803. doi: 10.1593/neo.08380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chu K, Cheng CJ, Ye X, Lee YC, Zurita AJ, Chen DT, Yu-Lee LY, Zhang S, Yeh ET, Hu MC, Logothetis CJ, Lin SH. Cadherin-11 promotes the metastasis of prostate cancer cells to bone. Mol Cancer Res. 2008;6:1259–67. doi: 10.1158/1541-7786.MCR-08-0077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bussemakers MJ, Van Bokhoven A, Tomita K, Jansen CF, Schalken JA. Complex cadherin expression in human prostate cancer cells. Int J Cancer. 2000;85:446–50. [PubMed] [Google Scholar]
- 111.Huang CF, Lira C, Chu K, Bilen MA, Lee YC, Ye X, Kim SM, Ortiz A, Wu FL, Logothetis CJ, Yu-Lee LY, Lin SH. Cadherin-11 increases migration and invasion of prostate cancer cells and enhances their interaction with osteoblasts. Cancer Res. 2010;70:4580–9. doi: 10.1158/0008-5472.CAN-09-3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Tsingotjidou AS, Zotalis G, Jackson KR, Sawyers C, Puzas JE, Hicks DG, Reiter R, Lieberman JR. Development of an animal model for prostate cancer cell metastasis to adult human bone. Anticancer Res. 2001;21:971–8. [PubMed] [Google Scholar]
- 113.Jacob K, Webber M, Benayahu D, Kleinman HK. Osteonectin promotes prostate cancer cell migration and invasion: a possible mechanism for metastasis to bone. Cancer Res. 1999;59:4453–7. [PubMed] [Google Scholar]
- 114.Taichman RS, Cooper C, Keller ET, Pienta KJ, Taichman NS, McCauley LK. Use of the stromal cell-derived factor-1/CXCR4 pathway in prostate cancer metastasis to bone. Cancer Res. 2002;62:1832–7. [PubMed] [Google Scholar]
- 115.Sun YX, Wang J, Shelburne CE, Lopatin DE, Chinnaiyan AM, Rubin MA, Pienta KJ, Taichman RS. Expression of CXCR4 and CXCL12 (SDF-1) in human prostate cancers (PCa) in vivo. J Cell Biochem. 2003;89:462–73. doi: 10.1002/jcb.10522. [DOI] [PubMed] [Google Scholar]
- 116.Arya M, Patel HR, McGurk C, Tatoud R, Klocker H, Masters J, Williamson M. The importance of the CXCL12-CXCR4 chemokine ligand-receptor interaction in prostate cancer metastasis. J Exp Ther Oncol. 2004;4:291–303. [PubMed] [Google Scholar]
- 117.Hirbe AC, Morgan EA, Weilbaecher KN. The CXCR4/SDF-1 chemokine axis: a potential therapeutic target for bone metastases? Curr Pharm Des. 2010;16:1284–90. doi: 10.2174/138161210791034012. [DOI] [PubMed] [Google Scholar]
- 118.Sun YX, Schneider A, Jung Y, Wang J, Dai J, Wang J, Cook K, Osman NI, Koh-Paige AJ, Shim H, Pienta KJ, Keller ET, et al. Skeletal localization and neutralization of the SDF-1(CXCL12)/CXCR4 axis blocks prostate cancer metastasis and growth in osseous sites in vivo. J Bone Miner Res. 2005;20:318–29. doi: 10.1359/JBMR.041109. [DOI] [PubMed] [Google Scholar]
- 119.Kukreja P, Abdel-Mageed AB, Mondal D, Liu K, Agrawal KC. Up-regulation of CXCR4 expression in PC-3 cells by stromal-derived factor-1alpha (CXCL12) increases endothelial adhesion and transendothelial migration: role of MEK/ERK signaling pathway-dependent NF-kappaB activation. Cancer Res. 2005;65:9891–8. doi: 10.1158/0008-5472.CAN-05-1293. [DOI] [PubMed] [Google Scholar]
- 120.Hart CA, Brown M, Bagley S, Sharrard M, Clarke NW. Invasive characteristics of human prostatic epithelial cells: understanding the metastatic process. Br J Cancer. 2005;92:503–12. doi: 10.1038/sj.bjc.6602325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Jacob K, Webber M, Benayahu D, Kleinman HK. Osteonectin promotes prostate cancer cell migration and invasion: a possible mechanism for metastasis to bone. Cancer Res. 1999;59:4453–7. [PubMed] [Google Scholar]
- 122.Stewart DA, Cooper CR, Sikes RA. Changes in extracellular matrix (ECM) and ECM-associated proteins in the metastatic progression of prostate cancer. Reprod Biol Endocrinol. 2004;2:2. doi: 10.1186/1477-7827-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mundy GR. Mechanisms of bone metastasis. Cancer. 1997;80:1546–56. doi: 10.1002/(sici)1097-0142(19971015)80:8+<1546::aid-cncr4>3.3.co;2-r. [DOI] [PubMed] [Google Scholar]
- 124.Coleman RE. Metastatic bone disease: clinical features, pathophysiology and treatment strategies. Cancer Treat Rev. 2001;27:165–76. doi: 10.1053/ctrv.2000.0210. [DOI] [PubMed] [Google Scholar]
- 125.Bussard KM, Gay CV, Mastro AM. The bone microenvironment in metastasis; what is special about bone? Cancer Metastasis Rev. 2008;27:41–55. doi: 10.1007/s10555-007-9109-4. [DOI] [PubMed] [Google Scholar]
- 126.Romanowski R, Jundt G, Termine JD, von der Ma K, Schulz A. Immunoelectron microscopy of osteonectin and type I collagen in osteoblasts and bone matrix. Calcif Tissue Int. 1990;46:353–60. doi: 10.1007/BF02554964. [DOI] [PubMed] [Google Scholar]
- 127.Pinero GJ, Farach-Carson MC, Devoll RE, Aubin JE, Brunn JC, Butler WT. Bone matrix proteins in osteogenesis and remodelling in the neonatal rat mandible as studied by immunolocalization of osteopontin, bone sialoprotein, alpha 2HS-glycoprotein and alkaline phosphatase. Arch Oral Biol. 1995;40:145–55. doi: 10.1016/0003-9969(94)00144-z. [DOI] [PubMed] [Google Scholar]
- 128.Virk MS, Lieberman JR. Tumor metastasis to bone. Arthritis Res Ther. 2007;9(Suppl 1) doi: 10.1186/ar2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Fizazi K, Yang J, Peleg S, Sikes CR, Kreimann EL, Daliani D, Olive M, Raymond KA, Janus TJ, Logothetis CJ, Karsenty G, Navone NM. Prostate cancer cells-osteoblast interaction shifts expression of growth/survival-related genes in prostate cancer and reduces expression of osteoprotegerin in osteoblasts. Clin Cancer Res. 2003;9:2587–97. [PubMed] [Google Scholar]
- 130.Minguell JJ, Erices A, Conget P. Mesenchymal stem cells. Exp Biol Med (Maywood) 2001;226:507–20. doi: 10.1177/153537020122600603. [DOI] [PubMed] [Google Scholar]
- 131.Kanis JA, McCloskey EV. Bone turnover and biochemical markers in malignancy. Cancer. 1997;80:1538–45. doi: 10.1002/(sici)1097-0142(19971015)80:8+<1538::aid-cncr3>3.3.co;2-v. [DOI] [PubMed] [Google Scholar]
- 132.Ibrahim T, Flamini E, Mercatali L, Sacanna E, Serra P, Amadori D. Pathogenesis of osteoblastic bone metastases from prostate cancer. Cancer. 2010;116:1406–18. doi: 10.1002/cncr.24896. [DOI] [PubMed] [Google Scholar]
- 133.Hauschka PV, Mavrakos AE, Iafrati MD, Doleman SE, Klagsbrun M. Growth factors in bone matrix. Isolation of multiple types by affinity chromatography on heparin-Sepharose. J Biol Chem. 1986;261:12665–74. [PubMed] [Google Scholar]
- 134.Lee YC, Huang CF, Murshed M, Chu K, Araujo JC, Ye X, deCrombrugghe B, Yu-Lee LY, Gallick GE, Lin SH. Src family kinase/abl inhibitor dasatinib suppresses proliferation and enhances differentiation of osteoblasts. Oncogene. 2010;29:3196–207. doi: 10.1038/onc.2010.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Saad F, Lipton A. SRC kinase inhibition: targeting bone metastases and tumor growth in prostate and breast cancer. Cancer Treat Rev. 2010;36:177–84. doi: 10.1016/j.ctrv.2009.11.005. [DOI] [PubMed] [Google Scholar]
- 136.Soriano P, Montgomery C, Geske R, Bradley A. Targeted disruption of the c-src proto-oncogene leads to osteopetrosis in mice. Cell. 1991;64:693–702. doi: 10.1016/0092-8674(91)90499-o. [DOI] [PubMed] [Google Scholar]
- 137.Hannon RA, Clack G, Rimmer M, Swaisland A, Lockton JA, Finkelman RD, Eastell R. Effects of the Src kinase inhibitor saracatinib (AZD0530) on bone turnover in healthy men: a randomized, double-blind, placebo-controlled, multiple-ascending-dose phase I trial. J Bone Miner Res. 2010;25:463–71. doi: 10.1359/jbmr.090830. [DOI] [PubMed] [Google Scholar]
- 138.Araujo J, Logothetis C. Dasatinib: a potent SRC inhibitor in clinical development for the treatment of solid tumors. Cancer Treat Rev. 2010;36:492–500. doi: 10.1016/j.ctrv.2010.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Yu EY, Wilding G, Posadas E, Gross M, Culine S, Massard C, Morris MJ, Hudes G, Calabro F, Cheng S, Trudel GC, Paliwal P, et al. Phase II study of dasatinib in patients with metastatic castration-resistant prostate cancer. Clin Cancer Res. 2009;15:7421–8. doi: 10.1158/1078-0432.CCR-09-1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Rentsch CA, Cecchini MG, Thalmann GN. Loss of inhibition over master pathways of bone mass regulation results in osteosclerotic bone metastases in prostate cancer. Swiss Med Wkly. 2009;139:220–5. doi: 10.4414/smw.2009.12284. [DOI] [PubMed] [Google Scholar]
- 141.Taichman RS, Loberg RD, Mehra R, Pienta KJ. The evolving biology and treatment of prostate cancer. J Clin Invest. 2007;117:2351–61. doi: 10.1172/JCI31791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Saad F, Gleason DM, Murray R, Tchekmedyian S, Venner P, Lacombe L, Chin JL, Vinholes JJ, Goas JA, Chen B, CN - ZAPCSG A randomized, placebo-controlled trial of zoledronic acid in patients with hormone-refractory metastatic prostate carcinoma. J Natl Cancer Inst. 2002;94:1458–68. doi: 10.1093/jnci/94.19.1458. [DOI] [PubMed] [Google Scholar]
- 143.Urwin GH, Percival RC, Harris S, Beneton MN, Williams JL, Kanis JA. Generalised increase in bone resorption in carcinoma of the prostate. Br J Urol. 1985;57:721–3. doi: 10.1111/j.1464-410x.1985.tb07040.x. [DOI] [PubMed] [Google Scholar]
- 144.Logothetis CJ, Lin SH. Osteoblasts in prostate cancer metastasis to bone. Nat Rev Cancer. 2005;5:21–8. doi: 10.1038/nrc1528. [DOI] [PubMed] [Google Scholar]
- 145.Nelson JB, Hedican SP, George DJ, Reddi AH, Piantadosi S, Eisenberger MA, Simons JW. Identification of endothelin-1 in the pathophysiology of metastatic adenocarcinoma of the prostate. Nat Med. 1995;1:944–9. doi: 10.1038/nm0995-944. [DOI] [PubMed] [Google Scholar]
- 146.Autzen P, Robson CN, Bjartell A, Malcolm AJ, Johnson MI, Neal DE, Hamdy FC. Bone morphogenetic protein 6 in skeletal metastases from prostate cancer and other common human malignancies. Br J Cancer. 1998;78:1219–23. doi: 10.1038/bjc.1998.658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Lee Y, Schwarz E, Davies M, Jo M, Gates J, Wu J, Zhang X, Lieberman JR. Differences in the cytokine profiles associated with prostate cancer cell induced osteoblastic and osteolytic lesions in bone. J Orthop Res. 2003;21:62–72. doi: 10.1016/S0736-0266(02)00095-5. [DOI] [PubMed] [Google Scholar]
- 148.Casimiro S, Guise TA, Chirgwin J. The critical role of the bone microenvironment in cancer metastases. Mol Cell Endocrinol. 2009;310:71–81. doi: 10.1016/j.mce.2009.07.004. [DOI] [PubMed] [Google Scholar]
- 149.Guise TA, Yin JJ, Mohammad KS. Role of endothelin-1 in osteoblastic bone metastases. Cancer. 2003;97:779–84. doi: 10.1002/cncr.11129. [DOI] [PubMed] [Google Scholar]
- 150.Chiao JW, Moonga BS, Yang YM, Kancherla R, Mittelman A, Wu-Wong JR, Ahmed T. Endothelin-1 from prostate cancer cells is enhanced by bone contact which blocks osteoclastic bone resorption. Br J Cancer. 2000;83:360–5. doi: 10.1054/bjoc.2000.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Nelson JB, Chan-Tack K, Hedican SP, Magnuson SR, Opgenorth TJ, Bova GS, Simons JW. Endothelin-1 production and decreased endothelin B receptor expression in advanced prostate cancer. Cancer Res. 1996;56:663–8. [PubMed] [Google Scholar]
- 152.Yin JJ, Mohammad KS, Kakonen SM, Harris S, Wu-Wong JR, Wessale JL, Padley RJ, Garrett IR, Chirgwin JM, Guise TA. A causal role for endothelin-1 in the pathogenesis of osteoblastic bone metastases. Proc Natl Acad Sci U S A. 2003;100:10954–9. doi: 10.1073/pnas.1830978100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Nelson JB. Endothelin inhibition: novel therapy for prostate cancer. J Urol. 2003;170:S65–7. doi: 10.1097/01.ju.0000096372.07687.86. discussion S67–8. [DOI] [PubMed] [Google Scholar]
- 154.Kopetz ES, Nelson JB, Carducci MA. Endothelin-1 as a target for therapeutic intervention in prostate cancer. Invest New Drugs. 2002;20:173–82. doi: 10.1023/a:1015630513908. [DOI] [PubMed] [Google Scholar]
- 155.Clines GA, Mohammad KS, Bao Y, Stephens OW, Suva LJ, Shaughnessy JD, Jr., Fox JW, Chirgwin JM, Guise TA. Dickkopf homolog 1 mediates endothelin-1-stimulated new bone formation. Mol Endocrinol. 2007;21:486–98. doi: 10.1210/me.2006-0346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Russo A, Bronte G, Rizzo S, Fanale D, Di Gaudio F, Gebbia N, Bazan V. Anti-endothelin drugs in solid tumors. Expert Opin Emerg Drugs. 2010;15:27–40. doi: 10.1517/14728210903571667. [DOI] [PubMed] [Google Scholar]
- 157.Behrens J, von Kries JP, Kuhl M, Bruhn L, Wedlich D, Grosschedl R, Birchmeier W. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature. 1996;382:638–42. doi: 10.1038/382638a0. [DOI] [PubMed] [Google Scholar]
- 158.Hall CL, Kang S, MacDougald OA, Keller ET. Role of Wnts in prostate cancer bone metastases. J Cell Biochem. 2006;97:661–72. doi: 10.1002/jcb.20735. [DOI] [PubMed] [Google Scholar]
- 159.Hall CL, Daignault SD, Shah RB, Pienta KJ, Keller ET. Dickkopf-1 expression increases early in prostate cancer development and decreases during progression from primary tumor to metastasis. Prostate. 2008;68:1396–404. doi: 10.1002/pros.20805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Hall CL, Bafico A, Dai J, Aaronson SA, Keller ET. Prostate cancer cells promote osteoblastic bone metastases through Wnts. Cancer Res. 2005;65:7554–60. doi: 10.1158/0008-5472.CAN-05-1317. [DOI] [PubMed] [Google Scholar]
- 161.Thudi NK, Martin CK, Murahari S, Shu ST, Lanigan LG, Werbeck JL, Keller ET, McCauley LK, Pinzone JJ, Rosol TJ. Dickkopf-1 (DKK-1) stimulated prostate cancer growth and metastasis and inhibited bone formation in osteoblastic bone metastases. Prostate. 2010 doi: 10.1002/pros.21277. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Dai J, Hall CL, Escara-Wilke J, Mizokami A, Keller JM, Keller ET. Prostate cancer induces bone metastasis through Wnt-induced bone morphogenetic protein-dependent and independent mechanisms. Cancer Res. 2008;68:5785–94. doi: 10.1158/0008-5472.CAN-07-6541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Ide H, Yoshida T, Matsumoto N, Aoki K, Osada Y, Sugimura T, Terada M. Growth regulation of human prostate cancer cells by bone morphogenetic protein-2. Cancer Res. 1997;57:5022–7. [PubMed] [Google Scholar]
- 164.Lai TH, Fong YC, Fu WM, Yang RS, Tang CH. Osteoblasts-derived BMP-2 enhances the motility of prostate cancer cells via activation of integrins. Prostate. 2008;68:1341–53. doi: 10.1002/pros.20799. [DOI] [PubMed] [Google Scholar]
- 165.Masuda H, Fukabori Y, Nakano K, Takezawa Y, CSuzuki T, Yamanaka H. Increased expression of bone morphogenetic protein-7 in bone metastatic prostate cancer. Prostate. 2003;54:268–74. doi: 10.1002/pros.10193. [DOI] [PubMed] [Google Scholar]
- 166.Abe E, Yamamoto M, Taguchi Y, Lecka-Czernik B, O’Brien CA, Economides AN, Stahl N, Jilka RL, Manolagas SC. Essential requirement of BMPs-2/4 for both osteoblast and osteoclast formation in murine bone marrow cultures from adult mice: antagonism by noggin. J Bone Miner Res. 2000;15:663–73. doi: 10.1359/jbmr.2000.15.4.663. [DOI] [PubMed] [Google Scholar]
- 167.Gori F, Thomas T, Hicok KC, Spelsberg TC, Riggs BL. Differentiation of human marrow stromal precursor cells: bone morphogenetic protein-2 increases OSF2/CBFA1, enhances osteoblast commitment, and inhibits late adipocyte maturation. J Bone Miner Res. 1999;14:1522–35. doi: 10.1359/jbmr.1999.14.9.1522. [DOI] [PubMed] [Google Scholar]
- 168.Dai J, Keller J, Zhang J, Lu Y, Yao Z, Keller ET. Bone morphogenetic protein-6 promotes osteoblastic prostate cancer bone metastases through a dual mechanism. Cancer Res. 2005;65:8274–85. doi: 10.1158/0008-5472.CAN-05-1891. [DOI] [PubMed] [Google Scholar]
- 169.Feeley BT, Gamradt SC, Hsu WK, Liu N, Krenek L, Robbins P, Huard J, Lieberman JR. Influence of BMPs on the formation of osteoblastic lesions in metastatic prostate cancer. J Bone Miner Res. 2005;20:2189–99. doi: 10.1359/JBMR.050802. [DOI] [PubMed] [Google Scholar]
- 170.Virk MS, Petrigliano FA, Liu NQ, Chatziioannou AF, Stout D, Kang CO, Dougall WC, Lieberman JR. Influence of simultaneous targeting of the bone morphogenetic protein pathway and RANK/RANKL axis in osteolytic prostate cancer lesion in bone. Bone. 2009;44:160–7. doi: 10.1016/j.bone.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Wahdan-Alaswad RS, Song K, Krebs TL, Shola DT, Gomez JA, Matsuyama S, Danielpour D. Insulin-Like Growth Factor I Suppresses Bone Morphogenetic Protein Signaling in Prostate Cancer Cells by Activating mTOR Signaling. Cancer Res. 2010;70:9106–17. doi: 10.1158/0008-5472.CAN-10-1119. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 172.Kaleko M, Rutter WJ, Miller AD. Overexpression of the human insulinlike growth factor I receptor promotes ligand-dependent neoplastic transformation. Mol Cell Biol. 1990;10:464–73. doi: 10.1128/mcb.10.2.464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Koch H, Jadlowiec JA, Campbell PG. Insulin-like growth factor-I induces early osteoblast gene expression in human mesenchymal stem cells. Stem Cells Dev. 2005;14:621–31. doi: 10.1089/scd.2005.14.621. [DOI] [PubMed] [Google Scholar]
- 174.Rubin J, Chung LW, Fan X, Zhu L, Murphy TC, Nanes MS, Rosen CJ. Prostate carcinoma cells that have resided in bone have an upregulated IGF-I axis. Prostate. 2004;58:41–9. doi: 10.1002/pros.10299. [DOI] [PubMed] [Google Scholar]
- 175.Ritchie CK, Andrews LR, Thomas KG, Tindall DJ, Fitzpatrick LA. The effects of growth factors associated with osteoblasts on prostate carcinoma proliferation and chemotaxis: implications for the development of metastatic disease. Endocrinology. 1997;138:1145–50. doi: 10.1210/endo.138.3.4974. [DOI] [PubMed] [Google Scholar]
- 176.Ryan CJ, Haqq CM, Simko J, Nonaka DF, Chan JM, Weinberg V, Small EJ, Goldfine ID. Expression of insulin-like growth factor-1 receptor in local and metastatic prostate cancer. Urol Oncol. 2007;25:134–40. doi: 10.1016/j.urolonc.2006.07.019. [DOI] [PubMed] [Google Scholar]
- 177.Sommerfeldt DW, Rubin CT. Biology of bone and how it orchestrates the form and function of the skeleton. Eur Spine J. 2001;10(Suppl 2):S86–95. doi: 10.1007/s005860100283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Cohen P, Peehl DM, Graves HC, Rosenfeld RG. Biological effects of prostate specific antigen as an insulin-like growth factor binding protein-3 protease. J Endocrinol. 1994;142:407–15. doi: 10.1677/joe.0.1420407. [DOI] [PubMed] [Google Scholar]
- 179.Rubin J, Fan X, Rahnert J, Sen B, Hsieh CL, Murphy TC, Nanes MS, Horton LG, Beamer WG, Rosen CJ. IGF-I secretion by prostate carcinoma cells does not alter tumor-bone cell interactions in vitro or in vivo. Prostate. 2006;66:789–800. doi: 10.1002/pros.20379. [DOI] [PubMed] [Google Scholar]
- 180.Huang L, Cheng YY, Chow LT, Zheng MH, Kumta SM. Tumour cells produce receptor activator of NF-kappaB ligand (RANKL) in skeletal metastases. J Clin Pathol. 2002;55:877–8. doi: 10.1136/jcp.55.11.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Zhang J, Dai J, Yao Z, Lu Y, Dougall W, Keller ET. Soluble receptor activator of nuclear factor kappaB Fc diminishes prostate cancer progression in bone. Cancer Res. 2003;63:7883–90. [PubMed] [Google Scholar]
- 182.Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423:337–42. doi: 10.1038/nature01658. [DOI] [PubMed] [Google Scholar]
- 183.Zhang J, Dai J, Qi Y, Lin DL, Smith P, Strayhorn C, Mizokami A, Fu Z, Westman J, Keller ET. Osteoprotegerin inhibits prostate cancer-induced osteoclastogenesis and prevents prostate tumor growth in the bone. J Clin Invest. 2001;107:1235–44. doi: 10.1172/JCI11685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Miller RE, Roudier M, Jones J, Armstrong A, Canon J, Dougall WC. RANK ligand inhibition plus docetaxel improves survival and reduces tumor burden in a murine model of prostate cancer bone metastasis. Mol Cancer Ther. 2008;7:2160–9. doi: 10.1158/1535-7163.MCT-08-0046. [DOI] [PubMed] [Google Scholar]
- 185.Holen I, Croucher PI, Hamdy FC, Eaton CL. Osteoprotegerin (OPG) is a survival factor for human prostate cancer cells. Cancer Res. 2002;62:1619–23. [PubMed] [Google Scholar]
- 186.Nyambo R, Cross N, Lippitt J, Holen I, Bryden G, Hamdy FC, Eaton CL. Human bone marrow stromal cells protect prostate cancer cells from TRAIL-induced apoptosis. J Bone Miner Res. 2004;19:1712–21. doi: 10.1359/JBMR.040703. [DOI] [PubMed] [Google Scholar]
- 187.Whang PG, Schwarz EM, Gamradt SC, Dougall WC, Lieberman JR. The effects of RANK blockade and osteoclast depletion in a model of pure osteoblastic prostate cancer metastasis in bone. J Orthop Res. 2005;23:1475–83. doi: 10.1016/j.orthres.2005.05.004.1100230634. [DOI] [PubMed] [Google Scholar]
- 188.Bartsch R, Steger GG. Role of denosumab in breast cancer. Expert Opin Biol Ther. 2009;9:1225–33. doi: 10.1517/14712590903146877. [DOI] [PubMed] [Google Scholar]
- 189.Fizazi K, Lipton A, Mariette X, Body JJ, Rahim Y, Gralow JR, Gao G, Wu L, Sohn W, Jun S. Randomized phase II trial of denosumab in patients with bone metastases from prostate cancer, breast cancer, or other neoplasms after intravenous bisphosphonates. J Clin Oncol. 2009;27:1564–71. doi: 10.1200/JCO.2008.19.2146. [DOI] [PubMed] [Google Scholar]
- 190.Vij R, Horvath N, Spencer A, Taylor K, Vadhan-Raj S, Vescio R, Smith J, Qian Y, Yeh H, Jun S. An open-label, phase 2 trial of denosumab in the treatment of relapsed or plateau-phase multiple myeloma. Am J Hematol. 2009;84:650–6. doi: 10.1002/ajh.21509. [DOI] [PubMed] [Google Scholar]
- 191.Stopeck AT, Lipton A, Body JJ, Steger GG, Tonkin K, de Boer RH, Lichinitser M, Fujiwara Y, Yardley DA, Viniegra M, Fan M, Jiang Q, et al. Denosumab compared with zoledronic acid for the treatment of bone metastases in patients with advanced breast cancer: a randomized, double-blind study. J Clin Oncol. 2010;28:5132–9. doi: 10.1200/JCO.2010.29.7101. [DOI] [PubMed] [Google Scholar]
- 192.Lipton A, Stopeck A, von Moos R, Henry DH, Richardson GE, Rodriguez GI, Bourgeois HP, Ke C, Jun S, Dansey RD. A meta-analysis of results from two randomized, double-blind studies of denosumab versus zoledronic acid (ZA) for treatment of bone metastases. ASCO Meeting Abstracts. 2010;28:9015. [Google Scholar]
- 193.Lattouf JB, Saad F. Bone complications of androgen deprivation therapy: screening, prevention, and treatment. Curr Opin Urol. 2010;20:247–52. doi: 10.1097/MOU.0b013e32833835be. [DOI] [PubMed] [Google Scholar]
- 194.Smith MR, Egerdie B, Toriz N Hernandez, Feldman R, Tammela TL, Saad F, Heracek J, Szwedowski M, Ke C, Kupic A, Leder BZ, Goessl C, et al. Denosumab in men receiving androgen-deprivation therapy for prostate cancer. N Engl J Med. 2009;361:745–55. doi: 10.1056/NEJMoa0809003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Liao J, Li X, Koh AJ, Berry JE, Thudi N, Rosol TJ, Pienta KJ, McCauley LK. Tumor expressed PTHrP facilitates prostate cancer-induced osteoblastic lesions. Int J Cancer. 2008;123:2267–78. doi: 10.1002/ijc.23602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Thomas RJ, Guise TA, Yin JJ, Elliott J, Horwood NJ, Martin TJ, Gillespie MT. Breast cancer cells interact with osteoblasts to support osteoclast formation. Endocrinology. 1999;140:4451–8. doi: 10.1210/endo.140.10.7037. [DOI] [PubMed] [Google Scholar]
- 197.Liao J, McCauley LK. Skeletal metastasis: Established and emerging roles of parathyroid hormone related protein (PTHrP) Cancer Metastasis Rev. 2006;25:559–71. doi: 10.1007/s10555-006-9033-z. [DOI] [PubMed] [Google Scholar]
- 198.Vela I, Gregory L, Gardiner EM, Clements JA, Nicol DL. Bone and prostate cancer cell interactions in metastatic prostate cancer. BJU Int. 2007;99:735–42. doi: 10.1111/j.1464-410X.2006.06670.x. [DOI] [PubMed] [Google Scholar]
- 199.Cramer SD, Chen Z, Peehl DM. Prostate specific antigen cleaves parathyroid hormone-related protein in the PTH-like domain: inactivation of PTHrP-stimulated cAMP accumulation in mouse osteoblasts. J Urol. 1996;156:526–31. doi: 10.1097/00005392-199608000-00076. [DOI] [PubMed] [Google Scholar]
- 200.Iwamura M, Hellman J, Cockett AT, Lilja H, Gershagen S. Alteration of the hormonal bioactivity of parathyroid hormone-related protein (PTHrP) as a result of limited proteolysis by prostate-specific antigen. Urology. 1996;48:317–25. doi: 10.1016/S0090-4295(96)00182-3. [DOI] [PubMed] [Google Scholar]
- 201.Danielpour D. Functions and regulation of transforming growth factor-beta (TGF-beta) in the prostate. Eur J Cancer. 2005;41:846–57. doi: 10.1016/j.ejca.2004.12.027. [DOI] [PubMed] [Google Scholar]
- 202.Bello-DeOcampo D, Tindall DJ. TGF-betal/Smad signaling in prostate cancer. Curr Drug Targets. 2003;4:197–207. doi: 10.2174/1389450033491118. [DOI] [PubMed] [Google Scholar]
- 203.Dallas SL, Zhao S, Cramer SD, Chen Z, Peehl DM, Bonewald LF. Preferential production of latent transforming growth factor beta-2 by primary prostatic epithelial cells and its activation by prostate-specific antigen. J Cell Physiol. 2005;202:361–70. doi: 10.1002/jcp.20147. [DOI] [PubMed] [Google Scholar]
- 204.Josson S, Matsuoka Y, Chung LW, Zhau HE, Wang R. Tumor-stroma co-evolution in prostate cancer progression and metastasis. Semin Cell Dev Biol. 2010;21:26–32. doi: 10.1016/j.semcdb.2009.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Yin JJ, Selander K, Chirgwin JM, Dallas M, Grubbs BG, Wieser R, Massague J, Mundy GR, Guise TA. TGF-beta signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J Clin Invest. 1999;103:197–206. doi: 10.1172/JCI3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordon-Cardo C, Guise TA, Massague J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell. 2003;3:537–49. doi: 10.1016/s1535-6108(03)00132-6. [DOI] [PubMed] [Google Scholar]
- 207.Buijs JT, Rentsch CA, van der Ho G, van Overveld PG, Wetterwald A, Schwaninger R, Henriquez NV, Ten Dijke P, Borovecki F, Markwalder R, Thalmann GN, Papapoulos SE, et al. BMP7, a putative regulator of epithelial homeostasis in the human prostate, is a potent inhibitor of prostate cancer bone metastasis in vivo. Am J Pathol. 2007;171:1047–57. doi: 10.2353/ajpath.2007.070168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Kim MY, Oskarsson T, Acharyya S, Nguyen DX, Zhang XH, Norton L, Massague J. Tumor self-seeding by circulating cancer cells. Cell. 2009;139:1315–26. doi: 10.1016/j.cell.2009.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]