

Abstract
CD4+ T cells -- often referred to as T-helper cells -- play a central role in immune defense and pathogenesis. Virus infections and vaccines stimulate and expand populations of antigen-specific CD4+ T cells in mice and in man. These virus-specific CD4+ T cells are extremely important in antiviral protection: deficiencies in CD4+ T cells are associated with virus reactivation, generalized susceptibility to opportunistic infections, and poor vaccine efficacy. As described below, CD4+ T cells influence effector and memory CD8+ T cell responses, humoral immunity, and the antimicrobial activity of macrophages and are involved in recruiting cells to sites of infection. This review summarizes a few key points about the dynamics of the CD4+ T cell response to virus infection, the positive role of pro-inflammatory cytokines in the differentiation of virus-specific CD4+ T cells, and new areas of investigation to improve vaccines against virus infection.
Keywords: Virus infection, CD4+ T cell memory, T cell activation, inflammatory cytokines, interferon, B cells
Introduction
Viruses and the immune system are linked: viruses infect and replicate in an environment where cells are devoted to destroying the infection, so viruses have evolved mechanisms to elude the immune response; conversely, viruses have impacted the survival of their hosts, so the genomes of survivors are imprinted by past battles. Viruses and the immune system change in real time during an infection with detectable heritable changes in the viral genome and the selective expansion and differentiation of T cells and B cells with epigenetic changes that carry on for the life of the animal. The study of how the immune system copes with virus infection is key to understanding virus replication strategies and their overall structure, why certain populations of cells are targeted for infection, and the selection of virus mutants with growth advantage. Likewise, one can decipher how the immune system functions by studying how viruses evolve to survive and propagate in hosts.
CD4+ T cells impact antiviral immunity at multiple stages of the immune response. CD4+ T cells influence antiviral cellular and humoral immunity and play a direct role in suppressing virus infection (Figure 1). As discussed below, CD4+ T cells enhance early expansion of virus-specific primary CTL, their subsequent differentiation into memory cells, guide their localization to sites of infection, and drive secondary expansion of memory CD8+ T cells during re-infection. CD4+ T cells are critical for the processes that lead to long-term humoral immunity, which is the basis of protection for many infections and vaccines. People with deficiencies in CD4+ T cell responses or who have mutations in the molecules involved in CD4-B cell interactions show severe defects in cellular and humoral immunity and suffer from recurrent opportunistic infections. Antiviral CD4+ T cells may interact directly with eosinophils and impact their recruitment and function in lung tissues, potentially contributing to the pathogenesis seen after some lung infections. Thus, virus-specific CD4+ T cells form the nexus between immunologic protection and immune-mediated pathogenesis following infection, so a deep understanding of their induction, regulation, and function is needed to develop safe and effective vaccines against ongoing viral threats. A major point in this review is that CD4+ T cell differentiation and activity is controlled by where they reside.
Figure 1. The central role of virus-specific CD4 T cells in immune defense.

Antiviral CD4+ T cells affect multiple wings of the immune system to protect against infection. CD4+ T cells interact directly with antigen presenting cells to enhance their ability to present viral antigen to T cells. They engage B cells and direct their differentiation into memory B cells and plasma cells and affect the kind of antibody that is made. CD4+ T cells can directly suppress virus infection in MHCII+ target cells. CD4+ T cells are essential for the induction of antiviral CD8+ T cell responses to many infections and enhance the protective recall response of memory CD8+ T cells.
The T cell response following virus infection consists of several stages (Figure 2). Innate inflammatory processes begin immediately after infection and are largely driven by various pattern recognition receptors, including Toll-Like Receptors (TLR) that recognize viral material at the cell surface or within the endocytic compartment, and RIG-I-like receptors (RLR) and nucleotide-binding domain-leucine-rich repeat-containing molecules (NLR) that recognize viral material in the cytoplasm. These signals induce type 1 interferon production and other pro-inflammatory cytokines; a cascade of signaling events occurs in cells anywhere near the source of IFN. Interferon stimulated cells are induced to express new proteins that have antiviral activities, including ISG15, ISG20, RNAse-L, protein kinase R (PKR), 2′,5′oligoadenylate synthetases, Mx1, TRIM5α, and others (Borrow, Martinez-Sobrido, and de La Torre, 2010; Wilkins and Gale, 2010). The effect of IFN can vary by cell type. Besides direct antiviral effects, IFN transiently retains cells in lymphoid tissue by stimulating CD69 expression, which blocks the activity of sphingosine 1-phosphate receptor-1 (Shiow et al., 2006). IFN acts to enhance MHC presentation of antigens to T cells and modifies the proteolytic machinery involved in producing the peptides that will be recognized by T cells. IFNs stimulate APCs to express costimulatory molecules, potentiating the adaptive T cell response. During this early time, NK cells become activated and release interferon-gamma. The rapid detection of virus material and production of IFNs serves to diminish virus replication and alert the immune system to infection. Within a few hours after infection, viral antigen is presented to T cells and a few days later, there is an explosive expansion of antiviral T cells (Whitmire, Benning, and Whitton, 2006; Whitmire, Eam, and Whitton, 2008). It starts from a minute population of cells that begin dividing at incredibly fast rates with the population doubling at a rate on the order of once per 4– 5 hours, and the responding cells increase in number exponentially during this time (Whitmire, Benning, and Whitton, 2006). Impressively, as the cells accumulate, they also differentiate into different lineages and transition into memory cells, carryout their direct antiviral functions, travel throughout the body, and assist other wings of the immune system (Whitmire, Benning, and Whitton, 2006). These events unfold in an inflammatory environment that guides T cell differentiation and is in turn impacted by their presence. The outcome of this complex network of interactions is initially an expanded population of short-lived virus-reactive T cells, some of which further differentiate and transition into quiescent memory cells, which confer expedited immunity over extended periods of time. A key point is that innate and adaptive immunity are linked: early inflammatory pathways have an immediate impact in limiting infection and a long-term impact on T cell memory, influencing the kind and number of memory cell that survives and the kind of recall response that develops upon re-infection.
Figure 2. The T cell response following acute LCMV infection in mice.
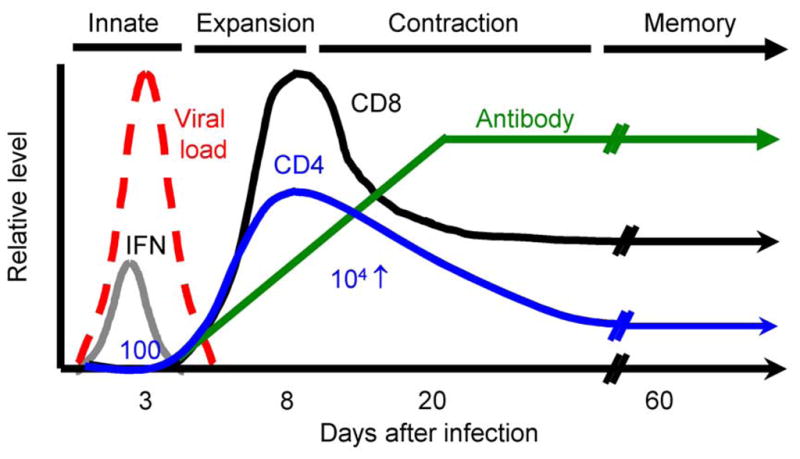
The T cell response following acute LCMV infection provides a well-defined model to study how CD4+ T cells, CD8+ T cells, and B cells responses develop. The graph shows the dramatic increase in number of virus-specific T cells after infection and the establishment of elevated numbers of memory T cells. The innate immune response begins immediately after infection and affects the subsequent T cell response. Responding T cells produce interferon-gamma and express other molecules that impact their expansion in number and differentiation into memory cells. The essential patterns are shown for a robust acute infection, but other infections show smaller or delayed responses that are related to differences in the magnitude or duration of infection and variations in the inflammatory environment.
The T cell response following virus infection typically consists of 2 major populations of cells: virus-specific CD8+ T cells and CD4+ T cells
Antiviral CD8+ T cells and CD4+ T cell responses begin from a small number of naïve precursor cells that expand in a non-linear way with roughly similar kinetics of induction and cell-division rate (Whitmire, Benning, and Whitton, 2006; Whitmire, Eam, and Whitton, 2008). There are a number of differences between CD8+ T cells and CD4+ T cells, which should be considered in order to understand why and how viruses avoid them.
Virus-specific CD8+ T cells and CD4+ T cells recognize different aspects of infection and carryout distinct functions. CD4+ T cells recognize viral antigen that is presented by MHCII molecules on a limited number of cells: dendritic cells (DC), B cells, and monocytes/macrophages. CD4+ T cells “see” fragments of extracellular material that professional antigen presenting cells (APC) have taken up, degraded, and presented in the context of MHCII. DC efficiently take-up antigen in the periphery, then travel to the draining lymph nodes, where they process and present antigen to T cells. The separation between scavenging for foreign material and the presentation of that material at different sites is seen for inert antigens, but may or may not hold following live virus infections where there is active recruitment of activated APCs to the site of infection. As discussed below, CD4+ T cells can engage these cells out in peripheral sites and impact their behavior or localization. The extracellular material can include viral particles, viral proteins, and viral debris from necrotic cells. In contrast to CD4+ T cells, CD8+ T cells recognize MHC1 molecules that are loaded with peptides derived from intracellular proteins; these proteins are ubiquitinated and degraded by the proteasome or immunoproteasome into peptides that are shuttled into the secretory pathway, where they are loaded onto MHC1 molecules. The epitopes that are presented on MHC1 molecules are largely derived from viral protein that has entered the cytosol as a result of infection (fusion) or from proteins arising from the de novo synthesis of viral protein from viral nucleic acid sequences. CD8+ T cells can be stimulated by extracellular protein through a process termed “cross-presentation”, but the protective value of this presentation, and the extent to which cross-presentation happens during the course of a natural infection is not clear and could be somewhat less efficient compared with endogenous “classical” pathways of antigen presentation; a recent study of vaccinia virus infection found that direct presentation, and not cross-presentation, plays the major role in the induction of antiviral CD8+ T cells (Xu et al., 2010). There are several recent reviews of cross-presentation (Bevan, 2006; Blanchard and Shastri, 2010; Rock, Farfan-Arribas, and Shen, 2010; Yewdell, 2010; Yewdell and Haeryfar, 2005).
CD4+ T cells can be induced by cells that are not actually infected and recognize antigen from non-replicating virus material, so they can be far more sensitive to foreign antigen than CD8+ T cells. For example, non-replicating or inactivated vaccines composed of viral proteins or bacterial toxins induce CD4+ T cell responses but negligible CD8+ T cell responses. Many viruses effectively inhibit antigen-presentation to CD8+ T cells, but such blockade does not affect CD4+ T cells, since non-infected cells still efficiently take up and present antigen. An example of this effect is seen following Coxsackie virus infection of mice; certain viral proteins (2B, 2BC, 3A) prevent the surface expression of MHC1 (Cornell et al., 2006; Cornell et al., 2007). Minimal to no detectable CD8 response is induced in these mice, even when using extremely sensitive techniques; in contrast, CVB3-specific CD4+ T cells are stimulated and readily measured (Kemball et al., 2009; Kemball, Harkins, and Whitton, 2008). Other viruses, such as myxoma virus (Zuniga et al., 1999), adenovirus (Blair and Blair-Zajdel, 2004; Windheim, Hilgendorf, and Burgert, 2004), HSV (Ahn et al., 1996; Barcy and Corey, 2001; Neumann, Eis-Hubinger, and Koch, 2003; Orr et al., 2005; Sievers et al., 2002; Temme et al., 2010), varicella-zoster virus (Eisfeld et al., 2007), MCMV (del Val et al., 1992; Heise, Connick, and Virgin, 1998; Lemmermann et al.), and HCMV (Jackson, Mason, and Wills, 2010), also have elaborate mechanisms to prevent MHC antigen presentation, thus reducing their visibility to the immune system. It is intrinsically difficult for viruses to dodge CD4+ T cell responses, but several major mechanisms have been identified, including the production of viral homologs of IL-10 (EBV, MCMV) (Kanai et al., 2007; Knappe et al., 2000; Kotenko et al., 2000) to generally suppress MHC expression, targeting DC for depletion by CD8+ T cells (Zuniga et al., 2008), and down-regulating MHCII expression on infected cells (Hegde et al., 2002; Lewandowski, Lo, and Bloom, 1993).
Another key difference between virus-specific CD8+ T cells and CD4+ T cells is the efficiency with which they kill target cells: this is done expeditiously by antigen-experienced CD8+ T cells, whereas such killing by CD4+ T cells is sluggish. This can be demonstrated using an “in vivo CTL assay”. In this assay, target cells are loaded with specific or irrelevant peptides and then differentially labeled with a dye (CFSE) and intravenously transferred into the same host; after a short period of time, flow cytometry is used to compare the relative loss of the specific peptide-coated cells compared to the control target cells. When target cells are loaded with an LCMV peptide that binds MHCI and is recognized by immunodominant LCMV-specific CD8+ T cells, they are completely eliminated within minutes upon transfer into an LCMV-immune mouse. By comparison, only a small fraction of GP61–80-loaded target cells are killed in these mice over a time span of one day. Thus cytolytic CD4+ T cells emerge after infection (Jellison, Kim, and Welsh, 2005), but their protective role based on direct killing of target cells after infection pales in comparison with virus-specific CD8+ T cells that kill within minutes (Yates et al., 2007). This is underscored by the inability of CD8-deficient mice to resolve LCMV infection despite the presence of large numbers of virus-specific CD4+ T cells. The mechanisms of killing are also very different: CD8+ CTL rely upon preformed perforin and granzyme molecules to poke holes into the target cells; CD4+ T cells utilize FAS-L and TRAIL to induce a caspase-dependent apoptosis. The dichotomy in rapid killing ability makes sense given that cytolytic cells are restricted to targets that are actually infected as opposed to nearby antigen presenting cells that have taken up antigen and are involved in alerting the immune system to infection. Such restricted killing may be beneficial by preserving APC whose depletion would cause generalized immune incompetence. An example of the detrimental effects of APC loss are seen following infection of mice with LCMV variants that selectively infect DC; the resulting CTL response eliminates these cells and the mice become susceptible to other infections (Sevilla et al., 2000; Sevilla et al., 2003; Zuniga et al., 2008). Similar pathogenesis may occur in people exposed to influenza, who then acquire lethal bacterial infections. Virus-specific CD4+ and CD8+ T cells share the ability to directly suppress virus replication by bathing target cells in IFNγ, but CD4+ T cells accomplish this without the collateral damage caused by perforin-dependent cell killing.
CD4+ T cells help CD8+ T cell-dependent antiviral defenses
For many virus infections CD4+ T cells are needed to drive the initial expansion of virus-specific CD8+ T cells (Williams and Bevan, 2007). In the absence of CD4+ T cells there is minimal expansion of CD8+ T cells and deficient cellular immunity. Virus-specific CD4+ T cells engage DCs that are shared with reactive CD8+ T cells; the CD4+ T cells stimulate the APCs to express costimulatory molecules and to increase the presentation of viral antigen so that the APCs better stimulate CD8+ T cells. CD4+ T cells are efficient producers of IL-2, which is a growth factor for CD8+ T cells and encourages improved cell-division and cell survival. It is plausible that virus-specific CD4+ T cell production of IL-2 (and IFNγ, see below) acts on nearby virus-specific CD8+ T cells. Such interactions might occur when both cells are adjacent to a virus infected APC.
Many virus infections grow to high titer in vivo and, due to efficient MHCI presentation, readily stimulate virus-specific CD8+ T cells. Virus-specific CD4+ T cells often are not needed for the initial expansion of CD8+ T cells in these models. However, CD4+ T cells affect the subsequent ability of memory CD8+ T cells to undergo vigorous secondary responses upon re-challenge with infection (Hamilton et al., 2006; Janssen et al., 2003; Shedlock and Shen, 2003; Sun and Bevan, 2003; Sun, Williams, and Bevan, 2004). Evidence indicates that early CD4− dependent signals leave a lasting effect on memory cell formation; help-less CD8 T cells expand poorly compared to those that differentiate in the presence of CD4+ T cells. Other evidence implicates memory CD4+ T cell effects during the actual recall response itself (Agnellini et al., 2008). In this case, CD4+ T cells deliver signals – and it is not clear what they are – that further the ability of responding memory CD8+ T cells to accumulate in number (Sun, Williams, and Bevan, 2004). The simultaneous activation of memory CD4+ T cells and memory CD8+ T cells results in a more protective CTL response compared to the activation of CD8+ T cells alone.
Antiviral T cells circulate widely after infection (Marshall et al., 2001; Masopust et al., 2001; Whitmire, Benning, and Whitton, 2006). In several model systems, antiviral CD4+ T cells are needed to recruit CD8+ T cells and other cell types to peripheral sites of infection. For example, CD4+ T cells recruit mouse hepatitis virus-specific CD8+ T cells into the CNS (Stohlman et al., 1998). CD4+ T cells recruit influenza-specific CD8+ T cells into the lung (Teijaro et al., 2010). HSV2-specific CD4+ T cells enhance CD8+ T cell movement to HSV2 infected tissues, and the effect is dependent upon IFNγ production by the CD4+ T cells and the local induction of CXCR3 that allows circulating CD8+ T cells to enter the tissue (Nakanishi et al., 2009). Perhaps the local production of IFNαβ caused by infection results in an influx of cells, including those with antigen presenting functions and CD4+ T cells. Once there, CD4+ T cells direct other responses locally and guide other cell types into the infected tissue (Lane et al., 2000).
Virus-specific CD4+ T cells sustain antiviral CD8+ T cell responses during protracted infections (Khanolkar, Fuller, and Zajac, 2004; Kumaraguru, Banerjee, and Rouse, 2005; Wherry et al., 2007; Zajac et al., 1998a). For example, variants of LCMV will grow to high titer and target new populations of cells and disseminate throughout the body of the mouse. The CD8+ T cell response against these variants is strained and eventually virus-specific CD8+ T cells either undergo apoptosis or become dysfunctional (exhausted), losing their ability to proliferate and produce cytokines when exposed to their cognate antigen. Eventually, wildtype mice reduce the viral load to levels at or below the detection limits of plaque assay in most tissues but not mice that are depleted of CD4+ T cells (Battegay et al., 1994; Leist, Kohler, and Zinkernagel, 1989; Matloubian, Concepcion, and Ahmed, 1994). Transient depletion of CD4+ T cells is sufficient to prevent long-term control of these variants. In wildtype mice, functional cytolytic CD8+ T cells can be recovered once the infection is controlled, but in CD4-deficient mice, functional CD8+ T cells are not recovered. Dysfunctional CD8+ T cells can be rescued by the adoptive transfer of functional CD4+ T cells (Homann et al., 1998; Kumaraguru, Banerjee, and Rouse, 2005). The mechanism by which CD4+ T cells rescue virus-specific CD8+ T cells is elusive; it could be related to the release of copious amounts of IL-2 that acts on CD8+ T cells, the ability of CD4+ T cells to stimulate APC activity or survival that indirectly sustains CD8+ T cells, or possibly modulation of inhibitory receptor-ligand interactions between CD8+ T cells and APCs. Virus-specific CD4+ T cell production of IL-21 sustains antiviral CD8+ T cells during persistent virus infection (Elsaesser, Sauer, and Brooks, 2009; Frohlich et al., 2009; Melief and Schoenberger, 2010; Yi, Du, and Zajac, 2009; Yi, Ingram, and Zajac, 2010). CD4+ T cells may also protect APCs from CTL-mediated killing (Mueller et al., 2006), thus preserving their immune-stimulatory functions.
Big responses from small numbers
The T cell Receptor consists of several subunits, and the regions involved in binding peptide and MHC complexes are highly variable and lead to a diverse array of pro-T cells that travel from the bone marrow to the thymus; a subset of the cells go on to differentiate into single positive CD4+ or CD8+ T cells that populate the peripheral lymphoid organs. Based on the number of domains, their random juxtaposition and additional sequences that are added at the joining sites, the theoretical variety of unique TCRs is enormous and far greater than the actual number of cells in a mouse. Until recently, the frequency of any given antigen-specific T cell in the naïve pool was unknown, but it was estimated by CDR3-length measurements of different classes of TCR molecules that there are approximately 2×106 distinct T cell precursor pools, each containing an average of 10 cells (Casrouge et al., 2000). This estimate was largely borne-out by direct measurements of antigen-specific cells using either functional methods or tetramer-enrichment techniques. For example, in an uninfected wildtype (B6) mouse, there are approximately 100 CD4+ T cells with the potential to respond to an immuno-dominant I-Ab-restricted epitope of LCMV (GP61–80) (MacLeod et al., 2008; Whitmire, Benning, and Whitton, 2006). A similar frequency for an immuno-dominant LCMV-specific CD8+ T cell population (Blattman et al., 2002) indicates that there is overlap in the frequencies of precursors between immuno-dominant CD4+ and CD8+ T cells. The frequency for other epitope-specific T cell precursors ranges from ~10– 1,000 per mouse (Hataye et al., 2006; Moon et al., 2007; Obar, Khanna, and Lefrancois, 2008). There is a trend where the initial naïve precursor frequency correlates with the subsequent immunodominance hierarchy that is seen following infection, but this pattern does not always hold.
The induction phase of the response commences immediately upon infection and involves viral antigen along with inflammatory signals and costimulatory signals. Within a few hours after infection, viral antigen is presented to T cells at sufficient levels to stimulate antigen-experienced cells to make cytokine (Whitmire, Eam, and Whitton, 2008). This can be shown experimentally by re-challenging immune mice, which contain large numbers of memory T cells, and then directly injecting BFA soon afterwards. The BFA acts to prevent T cell secretion of IFNγ, so reactive cells accumulate this cytokine following antigen exposure in vivo. The cells are isolated and immediately stained for intracellular IFNγ without further ex-vivo manipulation. A large fraction of virus-specific CD4+ T cells will express IFNγ 6– 12 hours after infection, indicating that those cells recognized their cognate antigen in vivo. The actual speed with which antigen is presented may be much faster and on a timescale of minutes. From the standpoint of virus infection, it is important to have an adaptive response that matches the rate at which viruses replicate and spread. These events can happen quickly (such as LCMV replication and spread in mice) and the T cell response occurs quickly; other infections are slow and the resulting T cell response dithers. For example, the T cell responses against mycobacteria peak one month after infection; the T cell responses against the hepatotropic viruses HAV and HCV only emerge 4– 6 weeks after infection. It is unclear why the kinetics vary so dramatically – different viruses and other pathogens influence the kind of milieu that forms during these critical stages of the T cell response, and this may explain the variation in kinetics, quality and magnitude of subsequent memory T cells. It is also possible that some viruses have evolved mechanisms to delay the kinetics to facilitate virus survival and spread. For example, the liver environment suppresses immune responses, which may explain why viruses targeting that location induce small and delayed immune responses.
Despite the initial presence of LCMV antigen, virus-specific naïve and memory T cells undergo cell division after a delay of up to 3 days, followed by an explosive transition into extremely rapid cell division and accumulation (Whitmire, Eam, and Whitton, 2008). The initiation of the response is governed, at least in part, by the inflammatory environment as opposed to intrinsic limits governing the ability of T cells to transform from quiescent cells to ones that are fully capable of dividing. T cells that are placed within an inflamed environment (i.e. a pre-infected mouse) are able to begin dividing without much delay (2 days as opposed to 3– 4 days), which implies that certain signals beyond antigen, are involved in initiating the rapid response. These signals could include the induction of positive regulators or the elimination of negative regulators or Treg cells.
As the response develops, populations of cells are differentially impacted by various selective forces – including antigen abundance, TCR-peptide/MHC affinity, and cytokines – that determine the cells that preferentially grow out in number and contribute to T cell differentiation and survival into the memory phase. The magnitude of the CD4 response is largely governed by the amount and duration of antigen (Obst et al., 2005; Ravkov and Williams, 2009). The CD4 response truncates when infection is artificially terminated, such as by treating recombinant-Listeria monocytogenes-infected mice with antibiotics. Similar findings were seen following recall responses. These analyses were done in the context of LM-driven T cell responses; whether similar correlations hold for virus infection remains to be shown. Analogous experiments using ribavirin in virus infected mice should be done to evaluate whether antiviral CD4+ T cell responses are also highly dependent on antigen-dose and duration.
Remarkably, much of the virus-specific CD4+ T cell accumulation occurs within a few days, implying cell-division rates on the order of one population doubling every 4–5 hours, but possibly faster if some of the daughter cells undergo apoptosis during the response. Given the speed with which viruses replicate, it is important to have an adaptive immune system that can keep up; on the other hand, a robust autoreactive response or one that is otherwise misdirected could lead to catastrophic immune-mediated pathology. Therefore, the response is heavily governed, but the details of how this occurs are obscure.
Virus infections stimulate distinct lineages of CD4+ T cells that serve unique functions
As antigen-specific CD4+ T cells respond to infection, they differentiate into lineages and produce characteristic sets of cytokines that impact how they influence other components of the immune response. They also express certain chemokine receptors that govern whether they travel to peripheral sites, localize within lymphoid organs in T cell zones, or localize near B cell zones. The overall kinetics of the CD4+ T cell response and their survival/maintenance into to the memory phase has been studied in models that induce predominantly a Th1-type response. Antiviral Th1 cells are commonly found after live virus infections, because target cells typically produce IFNαβ, and DC and NK cells produce IL-12, IL-18 and IFNγ that stimulate cells to differentiate into the Th1 path. Th1 cells are located in lymphoid organs but also travel to peripheral sites. They express IFNγ, which acts on target cells to directly suppress virus replication and augments antigen presentation by DC, B cells, and macrophages. Th1 cells also make IL-2 to support the expansion of other responding T cells. Th1 cells in the periphery produce IFN that modulates chemokine gradients and causes macrophages and CD8+ T cells to be recruited to sites of infection.
Virus-specific TFH cells emerge after infection (Johnston et al., 2009; Yusuf et al., 2010). TFH cells preferentially localize to the B cell boundaries within lymph nodes rather than circulate to peripheral sites (McHeyzer-Williams and McHeyzer-Williams, 2004). Given their proximal location to B cell areas, it is thought that TFH cells are directly involved in B cell differentiation, including the germinal center response and memory B cell formation (McHeyzer-Williams et al., 2009). These CD4+ T cells express CD40L and SAP that are vital for stimulating virus-specific B cells to further differentiate into MBC or plasma cells (Crotty et al., 2003; McHeyzer-Williams and McHeyzer-Williams, 2004). Virus-specific TFH cells express IL-4 (a B cell growth factor and differentiation factor), IFNγ (also a B cell differentiation factor), and CD40L. The ability of TFH cells to simultaneously make IFNγ and IL-4 may explain why IgG1 and IgG2a antibodies are often found together after infection. Many interesting questions remain about this population of cells: are they short-lived or long-lived; do they form memory; how are they related to other populations of virus-specific CD4+ T cells.
The patterns and behaviors of the other lineages may be similar or different to Th1 cells, so comparable analyses in other infection models would be useful, as they would provide insights about adjuvant design and overall regulation of immunity and immune-mediated pathogenesis. For example, virus-specific Th2 cells are found after some infections. RSV in particular induces a Th2 response that is associated with pathogenesis and an excessive influx of eosinophils into the lung. Virus-specific Th17, Th21, Th9, and TReg emerge after virus infections, but much remains to be learned about their regulation, function, and longevity. Virus-specific cytotoxic CD4+ T cells are found during some virus infections (HCMV, MCMV, others) (Appay et al., 2002). Cytotoxic CD4+ T cells express IFNγ and granzyme and FasL could be regarded as a subset of Th1 cells, but it is possible that they are a unique lineage, and their relationship to other cells is not clear. These cells hold potential for therapeutic applications: when primed and expanded artificially, they can be adoptively transferred into people with certain tumors to kill transformed cells.
It is important to consider that live virus infections create unique milieus that result in complex mixtures of responses, so in an individual there can be a mixture of CD4+ T cells from different lineages (Marzo et al., 2002; Roman et al., 2002). CD4+ T cells tend to fall neatly into distinct lineages, but recent data suggest there is pliability: Th2 cells can be “re-programmed” into Th1 when exposed to the interferons that are expressed during virus infection (Hegazy et al., 2010); other data indicate that IFNγ – the proto-typical Th1-inducing cytokine – is also important for Th2 cell formation (Bocek, Foucras, and Paul, 2004). The environment in which the virus grows changes with time, so it is plausible that CD4 differentiation is flexible and varies according to the local abundances of cytokines and antigen that occur throughout the response.
Memory T cells are a first line of defense against re-infection
CD4 T cell memory is typically seen after infection or vaccination and, where examined, it is long-lived. There are some examples of memory waning over time (Homann, Teyton, and Oldstone, 2001), but other data show fairly stable levels over time (Hammarlund et al., 2003; Harrington et al., 2002; Varga and Welsh, 1998; Whitmire et al., 1998). Evidence from studies utilizing “marked effector cells” indicate that memory T cells are derived from antigen-experienced effector cells in a linear differentiation process from naïve to effector to memory cell (Harrington et al., 2008; Hu et al., 2001; Jacob and Baltimore, 1999; Opferman, Ober, and Ashton-Rickardt, 1999). These studies used transgenic T cells that contain an inactive marker, which recombines when the T cells are stimulated by antigen; those T cells and all of their progeny can be identified by flow cytometry. Several studies using this approach show that memory cells that form after infection are marked, indicating a direct relationship between the effector cells and memory cells.
Memory precursors can be identified early on based on their expression of IL-7R and their ability to bind tetramers (Kaech et al., 2003; Kondrack et al., 2003; Lenz et al., 2004; Li, Huston, and Swain, 2003; Seddon, Tomlinson, and Zamoyska, 2003). During the initial response memory precursors emerge. Virus-specific CD4+ T cells down-regulate IL-7R, but there are cells that re-express IL-7R (Kaech et al., 2003; Kondrack et al., 2003; Lenz et al., 2004; Li, Huston, and Swain, 2003; Seddon, Tomlinson, and Zamoyska, 2003) and down-regulate KLRG1 (Hand, Morre, and Kaech, 2007; Joshi et al., 2007). After the infection is resolved, the swollen number of effector cells is no longer needed and it is energetically expensive to sustain such a vast population of cells. Therefore, most of the cells (~90%) are purged during the contraction phase and the survivors have the characteristics of memory cells (described below). The other effector cells undergo cell death and the IL-7R+ population accumulates (or at least doesn’t die); over time, only IL-7R+ cells remain and they behave like memory cells. The increased proportion of IL-7R+ cells that is seen during the contraction phase could either be the result of deletion of IL-7R− cells, an intrinsic change in cells from IL-7R− to IL-7R+, or a combination of IL-7R+ cell division and accumulation along with the loss of IL-7R− cells; there is evidence consistent with all of these scenarios. The purging process is enigmatic because cell selection and survival into the memory phase occurs in the apparent absence of virus replication and inflammation. Most evidence indicates that the virus-specific T cells undergo caspase-dependent apoptosis, and the surviving T cells tend to express increased amounts of Bcl2, Bcl-xL, and Bcl6 (Grayson et al., 2006; Grayson et al., 2000; Homann, Teyton, and Oldstone, 2001; Pipkin et al., 2010) compared to effector cells.
Once established, CD4 T cell memory does not require antigen (Swain, Hu, and Huston, 1999) but depends upon cytokines derived from stromal cells. Antiviral CD4+ T cells use IL-7 and, to a lower extent, IL-15 for homeostatic maintenance (Jaleco et al., 2003; Lenz et al., 2004; Purton et al., 2007). In the absence of these cytokines, virus-specific CD4+ T cells decline in number; conversely, their number can be increased above normal when recombinant versions of these cytokines are given to mice (Boyman et al., 2008; Boyman, Surh, and Sprent, 2006; Cho et al., 2007; Rubinstein et al., 2006).
Quiescent memory T cells are responsible for the expedited immunologic protection seen after immunization. As described below, naïve and memory cells differ dramatically in frequency, sensitivity to antigen, cytokine output, and location, which explain why memory cells confer enhanced immunity compared to naïve cells.
Frequency
Immune mice mount recall responses that are detectable sooner than primary responses. The accelerated appearance of the recall response can be explained by the differences in frequency between virus-specific naïve cells (~100 per mouse) that are well below detection by standard assays versus memory cells that can be 500- 1000-fold more abundant. Upon challenge, the naïve cells need to undergo far more rounds of cell division and accumulation before they are detected; memory T cells only need to go through a few rounds; hence, memory responses appear sooner. Most analyses of memory T cells have been done in immune mice, and the results from these mice were compared to those from naive mice. However, there are numerous differences between these two groups of mice, including the virus-specific T cell frequency and the presence of virus-specific antibody and CTL that affect antigen load and APC function. To investigate whether the differentiation changes that occur during the transition of effector cells into memory cells result in quicker cell division rates, LCMV-specific T cells were transferred into naïve mice that were subsequently infected (Whitmire, Eam, and Whitton, 2008) and were allowed to differentiate into memory T cells. These memory T cells were isolated, mixed with naïve cells, labeled with CFSE, and adoptively transferred into the same naïve host. Upon infection, the two donor populations were identified by flow cytometry. The data indicate that when compared in the same host and at similar frequencies, memory T cells and naïve T cells begin cell-division at around the same time (Whitmire, Eam, and Whitton, 2008).
The recall response mediated by memory T cells is typically more robust than the primary response. Once naïve and memory T cells begin dividing, the effector cells derived from the memory T cells accumulate more efficiently than the primary effector cells (MacLeod et al., 2008; Whitmire, Eam, and Whitton, 2008). As a result, there is a rapid rise in the number of secondary effectors. The rapid accumulation of memory cells compared to their naïve counterparts may relate to the efficiency with which memory cells produce cytokines (IFNγ, IL-2) that increase cell division or prevent apoptosis; however, the accumulation could be the result of other epigenetic changes (intrinsically elevated Bcl2 levels). There may also be refinement of virus-specific cells so that only the best responders are selected to survive into the memory phase; these cells may have slight alterations in TCR structure that make them better able to respond compared to the mixed population of naïve T cells that react to the same epitope (Williams, Ravkov, and Bevan, 2008). Other genomic changes may be at play in enhancing the responsiveness of memory cells following infection, including their increased frequency in peripheral tissues (Klonowski et al., 2004; Lefrancois, 2006).
Sensitivity to antigen
Compared to naïve cells, memory T cells are 50- 500-fold more sensitive to antigen (Slifka and Whitton, 2001; Whitmire, Benning, and Whitton, 2006; Williams, Ravkov, and Bevan, 2008). The improvement in sensitivity to low quantities of antigen occurs during the expansion phase and carries on into memory. The molecular mechanisms that permit greater sensitivity have not been worked out: there may be differences in TCR abundance, preformed signaling components associated with the TCR, or increased levels of accessory molecules that improve T cell association with target cells. For example, memory cells express pre-formed costimulatory molecules that can immediately be secreted to the surface of cells to engage APCs (Koguchi et al., 2007).
Effector functions
A major difference between memory T cells and naïve T cells is their ability to rapidly make cytokine (Liu, Whitton, and Slifka, 2004; Rogers, Dubey, and Swain, 2000). When exposed to infection, memory T cells secrete IFNγ within minutes. We utilized an in-vivo method to detect T cells that make IFNγ after infection (Foster et al., 2007; Liu and Whitton, 2005; Whitmire, Eam, and Whitton, 2008). The method involves infecting mice and then treating them with Brefeldin-A to force reactive T cells to sequester cytokine intracellularly. Splenocytes are harvested and immediately surface stained to identify virus-specific cells and stained for intracellular levels of IFNγ. Using this assay, we compared naïve and memory T cells in the same host and found that memory cells respond to infection by producing IFNγ by 6– 12 hours after infection; naïve T cells in the same conditions did not make detectable levels of IFNγ. (Whitmire, Eam, and Whitton, 2008) These data show that memory cells are geared to respond quickly to infection by producing antiviral cytokine. The IFNγ locus is epigenetically modified in memory cells compared to naïve cells (Kersh et al., 2006; Northrop et al., 2006); the histones at this locus are demethylated, thus making the local heterochromatin open and accessible to transcription factors. These data resemble what is found for memory CD8+ T cells: virus-specific CD8+ T cells in immune mice kill target cells within minutes (Barber, Wherry, and Ahmed, 2003; Yates et al., 2007). Hence, a quiescent population of CD8+ memory cells can defend very quickly and during the same periods usually associated only with the innate immune system – thus, memory T cells exert their effects long before their accumulation appears.
Cytokine patterns
As responding T cells differentiate, they increase their ability to produce IFNγ, TNF, and IL-2. It is not clear why there is a difference in kinetics between IFNγ production and IL-2 secretion, but it may relate to the antiviral activity of IFNγ, which is needed immediately. The ability to make these cytokines appears to be sequential: early on many cells make IFNγ but not IL-2, but by the peak of the response, most effector cells make all three cytokines. The early production of IFNγ and TNF serves to diminish virus infection and activate macrophages and dendritic cells. IFNγ production by responding T cells may be directly linked to their enhanced expansion (see below). Early production of IL-2 could be detrimental to expanding effector cells if its impact of on NK cells or Treg cells is greater than that on T cells. The delayed production of IL-2, a growth and survival factor of T cells, may correspond with the transition of effector cells into the memory phase: IL-2 given during the contraction protects antiviral CD8+ and CD4+ T cells from apoptosis and increases memory. The expedited production of IL-2 by memory T cells may play a role in accelerating their accumulation once cell-division commences.
Interferons simultaneously suppress virus replication in target cells and stimulate antiviral T cells that ward off further infection
Numerous viruses encode proteins that either prevent the induction of IFN or block signaling cascades that result from IFN signals. RNA viruses are usually capped to prevent recognition by PRRs and induction of interferon; additionally, members of the picornavirus family (CVB, poliovirus, HAV) prevent encode proteins that block parts of the interferon induction process or block interferon-receptor signals. Other RNA viruses, such as members of the flavivirus produce non-structural proteins that prevent type1 IFN induction as well as IFN signaling. Vaccinia virus encodes decoy receptors for both type 1 and type 2 interferons. The elaborate mechanisms that viruses use to inhibit IFN highlight the key role of these pathways in immune defense.
Numerous pathways are involved in the inflammatory response and result in signals that act directly on T cells. For example, PRRs (NLR, TLR) are involved in initiating the response and lead to IFNαβ production by the infected cell, which may act directly on responding T cells and impact their functions or accumulation. Also, early IFNγ production by NK cells and responding T cells acts upon the IFNγR, which leads to JAK1/2 recruitment followed by STAT1 phosphorylation by JAK1. The STAT1-PO4 forms homodimers that travel to the nucleus and bind to promoter elements called Gamma Activated Sequences, which are near hundreds of genes that are upregulated as a consequence of IFNγR signaling. IFN functions immediately during infection, but the long-term consequence of IFN signaling is the stimulation large numbers of effector and memory T cells. The expansion of the CD4+ T cell population depends on several critical pathways, which likely influences the timing and magnitude of T cell responses. Sequential waves of interferons and costimulatory molecules are expressed after infection, and these interactions may result in distinct effects on T cell differentiation that varies with time (Bertram, Lau, and Watts, 2002).
We have shown that T cells need direct IFNγ signals to fully expand, and they compete for this cytokine as they differentiate into effectors and memory cells (Whitmire et al., 2008; Whitmire, Benning, and Whitton, 2005; Whitmire et al., 2007; Whitmire, Tan, and Whitton, 2005). The receptor for IFNγ, CD119, is expressed ubiquitously; however, the surface level of expression increases selectively on responding virus-specific T cells, consistent with the notion that IFN signals directly into T cells and enhances their ability to respond. To better understand the role of direct IFN signals in virus-specific T cell differentiation, we compared in the same host T cells that were genetically deficient in γR-expression with T cells that express γR (Whitmire et al., 2008; Whitmire, Benning, and Whitton, 2005; Whitmire et al., 2007; Whitmire, Tan, and Whitton, 2005). Under these conditions, the T cells – which are identical in their specificity – were transferred into the same WT host, where they were exposed to the same antigen load and duration. We found that after infection, γR− T cells expanded to lower numbers compared to the T cells that expressed γR, despite being present at equal numbers before infection. Similar results were seen when comparing γR+ versus γR− TCR-transgenic T cells or when endogenous virus-specific T cells from γR+ or γR− mice were compared in the same host. Our findings closely parallel those reported for IFNαβ signals, where T cells that are sensitive to IFNαβ expand in number more vigorously than do T cells that are αβR-deficient (Havenar-Daughton, Kolumam, and Murali-Krishna, 2006; Kolumam et al., 2005), and we confirmed this observation (unpublished data). The findings with αβR-deficient T cells are consistent with the evidence that TLRs enhance T cell memory; TLR signals alter APC antigen presentation capacity and stimulate IFNαβ production from them, which acts directly on responding T cells. Thus, IFNs act on target cells to diminish virus replication and act on responding virus-specific T cells to augment their accumulation.
Some reports indicate that IFNs are detrimental to T cell memory formation or that there is no impact on memory cell formation (Badovinac, Porter, and Harty, 2004; Dalton et al., 2000; Haring and Harty, 2006; Tewari, Nakayama, and Suresh, 2007). The varied conclusions likely stem from differences in the methods and model systems. For example, analyses comparing T cell memory in separate WT and IFN-deficient mice is problematic when the pathogen load and duration differs. A similar concern holds when a large proportion of APCs or other cell-types (T-reg cells, etc) are IFNγR-deficient, as in a bone-marrow chimera. It is important to consider that the role of IFN likely varies depending on the pathogen: virus infections typically stimulate large amounts of IFN, but bacterial infections stimulate an entirely different set of cytokines: other cytokine networks may dominate in stimulating T cell responses after bacterial infection. The effect of direct IFN signals appears after day 4 post-LCMV infection, when the cells are accumulating exponentially; the role of IFN may not be as apparent in infections or vaccinations where the stimulus does not drive extensive cell division and accumulation. Hence, weak stimuli that drive only a few fold changes in cell number may not appear to be impacted by IFNs.
During a response, restrictive numbers of APC force T cells to compete for antigen; under increasing amounts of competition, smaller and smaller proportions of the cells undergo cell division. We have shown, by varying the frequency of naïve virus-specific TCR-transgenic T cells before infection, that an overabundance of identical T cells is detrimental to antiviral T cell memory. Virus-specific CD8+ T cells and other CD4+ T cell responses show a similar pattern (Blair and Lefrancois, 2007; Foulds and Shen, 2006; Hataye et al., 2006; Kedl et al., 2000; Sarkar et al., 2007; Srinivasan, Foley, and McSorley, 2004; Troy and Shen, 2003; Whitmire et al., 2008; Willis, Kappler, and Marrack, 2006). The negative impact on T cell memory suggests that T cell differentiation may be linked to cell division. As antigen becomes limiting and T cell number increases, the competition for antigen becomes severe with time. T cell production of IFNγ is tightly controlled by contact with antigen (Slifka, Rodriguez, and Whitton, 1999), so the effect of constrained amounts of antigen impacts virus-specific T cell production of IFNγ, thus limiting direct IFNγ signaling into T cells.
We propose a link between T cells that most efficiently elaborate IFNγ secretion and receive IFNγR signals and their competitive fitness to out-expand other T cells and go on to differentiate into memory T cells following acute virus infection (Figure 3). IFNs in particular select cells into the memory pool: competition between cells for limiting amounts of IFN is a deciding factor in determining which cells survive initial expansion and go on to populate the memory pool. It is not yet clear whether the effect of IFNγR signals merely increases the abundance of cells with the potential to differentiate into memory cells or directly stimulates the memory program. However, it is noteworthy that in the absence of direct γR signals, there is a precipitous loss of T cell memory compared to identical cells that express γR. In other words, the initial difference between the two populations at the peak of the response exaggerates as the cells differentiate into the memory phase, suggesting that there is a direct connection between these signals and the memory program. Further studies are required to delineate how direct IFN signals impact memory cell formation and if the long-term effects are set early on or actively deployed during the contraction phase.
Figure 3. Selection of the fittest: virus-specific CD4+ T cells compete for antigen and cytokines, selecting and enriching for high quality memory cells.

Antiviral CD4+ T cells respond to antigen and inflammation by undergoing cell-division, which can be incredibly rapid. As they accumulate in number, effector T cells begin to compete for access to antigen, which constrains cell-division for cells that weakly interact with antigen. In addition, the most vigorously responding T cells produce IFNγ and receive direct IFNγ signals; the best responders receive the greatest amount of IFNγ signaling and continue to accumulate. IFNγ signals also enhance memory cell formation. It is unclear if direct IFNγ signals activate a program of memory cell differentiation or increase the number of memory cell precursors that have the potential to survive.
T cells also appear to compete for limiting amounts of IL-7, IL-15, and IL-2: when these cytokines are delivered as complexes to mice so that they are overabundant across several days, antigen-specific T cells are driven to tremendous number (Boyman et al., 2006; Boyman et al., 2008; Finkelman et al., 1993; Kamimura and Bevan, 2007; Kamimura et al., 2006; Purton, Martin, and Surh, 2008; Rubinstein et al., 2006). Conversely, when T cells are starved of these cytokines, there are severe reductions in the initial expansion and memory levels. It is plausible that these cytokines factor into the memory selection process and support translational efforts to use cytokine-mediated immunotherapy to increase the abundance of vaccine-induced memory T cells.
There are gaps in our understanding of how other pro-inflammatory pathways affect T cell memory formation. Some pathways act directly on responding T cells, others function indirectly through APCs, and some (MyD88, IFNαβ, IFNγ) act on both. NLRs sense bacterial and some DNA or RNA virus infections (Allen et al., 2009; Ichinohe et al., 2009; Kuenzel et al., 2010; Lamkanfi and Kanneganti, 2010; Neerincx et al., 2010). NLRs induce IL-1β production, which signals directly into T cells to enhance their expansion (Ben-Sasson et al., 2009). Consistent with this notion, IL-1R-deficient mice show deficits in lung CD4+ T cell responses following influenza infection (Schmitz et al., 2005). IL-12 and IL-18 are made by activated dendritic cells after virus infection; T cells express receptors for these cytokines and their differentiation into Th1 cells is impacted by these signals (Srinivasan et al., 2007) and may affect the expansion of CD4+ T cells and their differentiation into memory cells (Boelen et al., 2002; Haring and Harty, 2009; Pien et al., 2000; Srinivasan et al., 2007; Tough, Zhang, and Sprent, 2001). Type-III interferons (IFN-λ) are produced during some virus infections and are particularly abundant at skin and mucosal sites. IFN-λ is stimulated by some of the pathways that induce IFNαβ; the receptor for IFN-λ is IL-28Rαβ and is expressed on a limited number of cells, including T cells (Donnelly and Kotenko, 2010). IFN-λ signaling into non-T cells diminishes virus replication in target cells and results in an IFNαβ burst. The effect of IFN-λ signaling into antiviral T cells and their ability to form memory cells remains to be investigated. From the standpoint of improving vaccines against viruses, it would be interesting to learn whether the quality or number of memory cells differs when they differentiate with IFNγ signals versus IL-1β, IFNαβ, IFN-λ, IL-12, or IL-18. DNA viruses and RNA viruses differ in the kinds of innate pathways that are stimulated, and there may be consequent effects on virus-specific T cell memory.
TLR ligands also act on T cells to augment their responses (Marsland et al., 2007). MyD88 conveys signals emanating from TLRs that induce IFN within target cells, but there may be additional functions for this molecule. Intriguing data show that MyD88 signals within T cells influence their accumulation after viral infections (Rahman et al., 2008; Zhao et al., 2009; Zhou et al., 2009). Thus, additional MyD88 functions may overlap with MyD88-mediated induction of IFN signaling to enhance antiviral T cell responses. T cell differentiation is likely determined by a variety of signals, including TCR strength and duration, IFN and other combinations of pro-inflammatory cytokines, and TLR signaling (Nembrini et al., 2006).
Discussion
Antiviral CD4+ T cell responses begin from a small number of cells that undergo tremendous rates of cell division and accumulate under the positive influence of inflammatory cytokines. These early signals enhance their number and further their differentiation into memory cells. Once set, CD4 memory is maintained by homeostatic cytokines and by B cells through unknown mechanisms. Th1 memory cells sustain antiviral CD8 T cell responses and are involved in CD8+ T cell recruitment to peripheral sites; other lineages of antiviral CD4+ T cells can be found after infection, but their longevity and protective role during recall responses need to be studied.
Most studies have examined the influence of innate immune defense on developing T cells, but in an immune individual, the adaptive response occurs concurrently and possibly before innate defenses. How do cytokine-secreting memory T cells affect infected target cells, APC function, and NK cell activity? How do these signals impact ISG functions within infected target cells, given that some proteins (eg, MyD88) may have multiple functions? Conversely, there is evidence that innate immunity influences effective recall responses: in the absence of PKR – a cytoplasmic sensor of viral RNA that stimulates IFN responses – existent memory CD8+ T cells did not effectively eliminate a challenge virus infection (Nakayama et al., 2010). Thus, innate detection of infection and the positive effects of inflammatory cytokines on established T cell memory is an important area for future investigation, as it relates to how best to boost immunity with vaccination.
The role of IFNs, costimulatory molecules, and pro-inflammatory cytokines during chronic virus infection could be very different from that seen during acute infection. The immune system is hyper-activated during HIV infection, such continuous stimulation may induce an endogenous program that downregulates adaptive immunity under these circumstances. During chronic virus infection, virus-specific T cells are either deleted or become dysfunctional over time (Brooks et al., 2005; Fuller et al., 2005; Fuller et al., 2004; Fuller and Zajac, 2003; Mothe et al., 2007; Zajac et al., 1998a; Zajac et al., 1998b). Antiviral CD4+ T cells become non-functional like CD8+ T cells. The molecular mechanisms underlying these effects are still being uncovered; current analyses show PD1L-PD1 (Barber et al., 2006; Bengsch et al., 2010; Blackburn et al., 2009; Freeman et al., 2006; Grosso et al., 2009; Wherry et al., 2007), CD27–CD70 (van Gisbergen et al., 2009), CTLA4-B7, Lag3 (Bengsch et al., 2010; Blackburn et al., 2009), and IL10-IL10R (Brooks et al., 2008; Brooks et al., 2006; Ejrnaes et al., 2006) interactions inhibit reactive cells during chronic infection in mice and in people. During chronic virus infection, reactive T cells may produce IFNγ – possibly lower amounts than seen during acute infection – and the expression of this cytokine might have inhibitory effects during chronic infection: IFNγ induces PD1L on APCs (Muhlbauer et al., 2006; Schreiner et al., 2004), and IFNγ acts on Treg cells to increase their inhibitory activity. Data in other models indicate that IFNγ signals into non-T cells inhibit memory T cell formation (Sercan et al., 2006; Sercan et al., 2010). While the amount and duration of antigen can enhance CD4 T cell responses following acute infection, recent evidence shows that extensive persistence of large amounts of antigen can be detrimental to CD4+ T cell responses (Han et al., 2010), and CD4+ T cells that are removed from such an environment can recover their ability to produce cytokine. Treating chronically infected mice with ribavirin results in a reduction in virus load, and exhausted CD4+ T cells recover some of their functions (Brooks, McGavern, and Oldstone, 2006). Thus, chronic virus infection may diminish CD4+ T cells by induction of immunosuppressive cytokines and inhibitory cell-surface molecules, or by simply over-stimulating them. Future studies need to clarify the roles of type 1 and type 2 IFN on antiviral T cell responses throughout chronic virus infection. Interestingly, persistent LCMV infection of mice does not result in persistently elevated levels of IFNαβ, suggesting that there are other mechanisms at play that diminish innate immune responses – perhaps the inhibited production of IFNαβ is related to the development of exhausted T cells.
The magnitude of the primary and recall CD4+ T cell response is governed by antigen load and pro-inflammatory cytokines, and the two factors are linked: the antigen load influences how much IFNγ is made by responding T cells and the extent to which they receive direct IFNγ signals. The principal mechanism for priming antiviral CD4+ T cells is through endocytic processes that take-up extracellular protein, degrade it, and load the resulting peptides onto MHCII molecules. However, other mechanisms, including macroautophagy, result in the presentation of peptides from cytosolic proteins (Deretic, 2009; Gannage and Munz, 2009; Lee and Iwasaki, 2008; Munz, 2006; Munz, 2007). CD4+ T cell responses form against HSV and EBV (nuclear antigen 1 EBNA1) proteins that are in the cytoplasm of MHCII+ cells. Certain proteins that have escaped ubiquitin-mediated proteasomal degradation can be captured by autophagosomes and transported to lysosomes. These tend to be long-lived proteins and also ones that form large aggregates, which become encapsulated in autophagosomes that fuse with lysosomes, where the proteins are degraded and loaded onto resident MHCII molecules. In addition to macroautophagy, chaperone-mediated autophagy – the active heat-shock protein mediated transport of proteins directly into the lysosome – results in proteolytic degradation of proteins into peptides that bind to MHCII molecules. Virus proteins aggregate to form structures that efficiently package their genomes, which may induce the autophagosomal process and lead to peptides that are recognized by CD4+ T cells. The extent to which macroautophagy contributes to the overall CD4+ T cell response after virus infection remains to be quantified and compared to the well-established exogenous pathway. Type 1 and Type 2 interferons increase the amount of autophagy within cells, which may enhance the amount of antigen is presented to CD4+ T cells and affect the magnitude of the response. There may also be differences in how proteins are degraded in the late endosome/lysosome pathway versus the autophagosome/lysosome pathway, potentially determining the populations of virus-specific CD4+ T cells that respond. Lysosomes that have fused with autophagosomes may contain different sets of molecules compared to lysosomes that fuse with phagosomes; the co-expression of these other molecules with MHCII molecules on the surface of cells might also influence the developing CD4+ T cell response. Many viruses encode proteins that prevent MHC presentation; this may impact CD4+ T cell responses that depend upon autophagosome-mediated antigen presentation in the same infected cell but not affect CD4+ T cells that recognize antigen produced in the classical phagocytic process or receptor-mediated uptake in non-infected cells. It is unclear what proportion of CD4+ T cells respond to material derived from extracellular protein versus macroautophagy, and how these pathways differ in T cell-dependent protection.
The genomics of antiviral immunity
Nearly all analyses of immune response to vaccination or infection are done in highly inbred mouse models. Pre-clinical studies to evaluate vaccines typically begin with studies in standard inbred mouse models. General response patterns in mice will mimic those in people, but there are exceptions, which need to be studied. Mouse models are useful, because many tools are available to quantify immune responses, which is essential for understanding the underlying processes involved in resolving infection and establishing T cell memory. However, there are several problems with existing mouse models: much of the immune response is dominated by a few genes, different inbred mouse strains have lost some of these genes, and mice vary in their expression of the these genes, which may not match with normal expression patterns. Additionally, many inferences about certain pathways are based on data from gene-ablated mice – such experimental approaches are straightforward and easy to interpret, but rarely do people have absolute deficiencies in those particular genes. Normal individuals vary in alleles of genes and in the amount of expression of those genes, and they differ in non-coding regions of the genome. Critical variation beyond allelic variances at the MHC locus includes differences in intrinsic antiviral pathways and PRRs, including the induction of interferons that amplify differences in subsequent T cell responses. All of these differences with intact genes impact phenotypes seen in people – such natural variation is not replicated in inbred strains of mice. A robust outbred mouse model that factors in the natural variations in components of these networks may better predict which pathways are critical for immunity (Churchill et al., 2004; Yalcin et al., 2010). The quantitative analysis of immune responses following infection or vaccination in outbred populations of mice followed by quantitative associations with certain genetic loci associated with inflammation will play a key role in the future for dissecting how to best develop protective immune responses with vaccines.
Research Highlights.
Memory T cells are a first line of defense against re-infection.
CD4+ T cells directly suppress infection and enhance CD8+ T cell & B cell responses.
T cell differentiation is driven by antigen, interferons, and other pro-inflammatory cytokines.
Acknowledgments
JKW is supported by AI074862 from the NIH (NIAID).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agnellini P, Wiesel M, Schwarz K, Wolint P, Bachmann MF, Oxenius A. Kinetic and mechanistic requirements for helping CD8 T cells. J Immunol. 2008;180(3):1517–25. doi: 10.4049/jimmunol.180.3.1517. [DOI] [PubMed] [Google Scholar]
- Ahn K, Meyer TH, Uebel S, Sempe P, Djaballah H, Yang Y, Peterson PA, Fruh K, Tampe R. Molecular mechanism and species specificity of TAP inhibition by herpes simplex virus ICP47. Embo J. 1996;15(13):3247–55. [PMC free article] [PubMed] [Google Scholar]
- Allen IC, Scull MA, Moore CB, Holl EK, McElvania-TeKippe E, Taxman DJ, Guthrie EH, Pickles RJ, Ting JP. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity. 2009;30(4):556–65. doi: 10.1016/j.immuni.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appay V, Zaunders JJ, Papagno L, Sutton J, Jaramillo A, Waters A, Easterbrook P, Grey P, Smith D, McMichael AJ, Cooper DA, Rowland-Jones SL, Kelleher AD. Characterization of CD4(+) CTLs ex vivo. J Immunol. 2002;168(11):5954–8. doi: 10.4049/jimmunol.168.11.5954. [DOI] [PubMed] [Google Scholar]
- Badovinac VP, Porter BB, Harty JT. CD8+ T cell contraction is controlled by early inflammation. Nat Immunol. 2004;5(8):809–17. doi: 10.1038/ni1098. [DOI] [PubMed] [Google Scholar]
- Barber DL, Wherry EJ, Ahmed R. Cutting edge: rapid in vivo killing by memory CD8 T cells. J Immunol. 2003;171(1):27–31. doi: 10.4049/jimmunol.171.1.27. [DOI] [PubMed] [Google Scholar]
- Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439(7077):682–7. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- Barcy S, Corey L. Herpes simplex inhibits the capacity of lymphoblastoid B cell lines to stimulate CD4+ T cells. J Immunol. 2001;166(10):6242–9. doi: 10.4049/jimmunol.166.10.6242. [DOI] [PubMed] [Google Scholar]
- Battegay M, Moskophidis D, Rahemtulla A, Hengartner H, Mak TW, Zinkernagel RM. Enhanced establishment of a virus carrier state in adult CD4+ T-cell-deficient mice. J Virol. 1994;68(7):4700–4. doi: 10.1128/jvi.68.7.4700-4704.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Sasson SZ, Hu-Li J, Quiel J, Cauchetaux S, Ratner M, Shapira I, Dinarello CA, Paul WE. IL-1 acts directly on CD4 T cells to enhance their antigen-driven expansion and differentiation. Proc Natl Acad Sci U S A. 2009;106(17):7119–24. doi: 10.1073/pnas.0902745106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengsch B, Seigel B, Ruhl M, Timm J, Kuntz M, Blum HE, Pircher H, Thimme R. Coexpression of PD-1, 2B4, CD160 and KLRG1 on exhausted HCV-specific CD8+ T cells is linked to antigen recognition and T cell differentiation. PLoS Pathog. 2010;6(6):e1000947. doi: 10.1371/journal.ppat.1000947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram EM, Lau P, Watts TH. Temporal segregation of 4-1BB versus CD28-mediated costimulation: 4-1BB ligand influences T cell numbers late in the primary response and regulates the size of the T cell memory response following influenza infection. J Immunol. 2002;168(8):3777–85. doi: 10.4049/jimmunol.168.8.3777. [DOI] [PubMed] [Google Scholar]
- Bevan MJ. Cross-priming. Nat Immunol. 2006;7(4):363–5. doi: 10.1038/ni0406-363. [DOI] [PubMed] [Google Scholar]
- Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, Betts MR, Freeman GJ, Vignali DA, Wherry EJ. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol. 2009;10 (1):29–37. doi: 10.1038/ni.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair DA, Lefrancois L. Increased competition for antigen during priming negatively impacts the generation of memory CD4 T cells. Proc Natl Acad Sci U S A. 2007;104(38):15045–50. doi: 10.1073/pnas.0703767104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair GE, Blair-Zajdel ME. Evasion of the immune system by adenoviruses. Curr Top Microbiol Immunol. 2004;273:3–28. doi: 10.1007/978-3-662-05599-1_1. [DOI] [PubMed] [Google Scholar]
- Blanchard N, Shastri N. Cross-presentation of peptides from intracellular pathogens by MHC class I molecules. Ann N Y Acad Sci. 2010;1183:237–50. doi: 10.1111/j.1749-6632.2009.05135.x. [DOI] [PubMed] [Google Scholar]
- Blattman JN, Antia R, Sourdive DJ, Wang X, Kaech SM, Murali-Krishna K, Altman JD, Ahmed R. Estimating the precursor frequency of naive antigen-specific CD8 T cells. J Exp Med. 2002;195(5):657–64. doi: 10.1084/jem.20001021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocek P, Jr, Foucras G, Paul WE. Interferon gamma enhances both in vitro and in vivo priming of CD4+ T cells for IL-4 production. J Exp Med. 2004;199(12):1619–30. doi: 10.1084/jem.20032014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boelen A, Kwakkel J, Barends M, de Rond L, Dormans J, Kimman T. Effect of lack of Interleukin-4, Interleukin-12, Interleukin-18, or the Interferon-gamma receptor on virus replication, cytokine response, and lung pathology during respiratory syncytial virus infection in mice. J Med Virol. 2002;66(4):552–60. doi: 10.1002/jmv.2180. [DOI] [PubMed] [Google Scholar]
- Borrow P, Martinez-Sobrido L, de La Torre JC. Inhibition of the Type I interferon antiviral response during arenavirus infection. Viruses. 2010;2:2443–2480. doi: 10.3390/v2112443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyman O, Kovar M, Rubinstein MP, Surh CD, Sprent J. Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science. 2006;311(5769):1924–7. doi: 10.1126/science.1122927. [DOI] [PubMed] [Google Scholar]
- Boyman O, Ramsey C, Kim DM, Sprent J, Surh CD. IL-7/anti-IL-7 mAb complexes restore T cell development and induce homeostatic T Cell expansion without lymphopenia. J Immunol. 2008;180(11):7265–75. doi: 10.4049/jimmunol.180.11.7265. [DOI] [PubMed] [Google Scholar]
- Boyman O, Surh CD, Sprent J. Potential use of IL-2/anti-IL-2 antibody immune complexes for the treatment of cancer and autoimmune disease. Expert Opin Biol Ther. 2006;6(12):1323–31. doi: 10.1517/14712598.6.12.1323. [DOI] [PubMed] [Google Scholar]
- Brooks DG, Ha SJ, Elsaesser H, Sharpe AH, Freeman GJ, Oldstone MB. IL-10 and PD-L1 operate through distinct pathways to suppress T-cell activity during persistent viral infection. Proc Natl Acad Sci U S A. 2008;105(51):20428–33. doi: 10.1073/pnas.0811139106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks DG, McGavern DB, Oldstone MB. Reprogramming of antiviral T cells prevents inactivation and restores T cell activity during persistent viral infection. J Clin Invest. 2006;116(6):1675–85. doi: 10.1172/JCI26856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks DG, Teyton L, Oldstone MB, McGavern DB. Intrinsic functional dysregulation of CD4 T cells occurs rapidly following persistent viral infection. J Virol. 2005;79(16):10514–27. doi: 10.1128/JVI.79.16.10514-10527.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks DG, Trifilo MJ, Edelmann KH, Teyton L, McGavern DB, Oldstone MB. Interleukin-10 determines viral clearance or persistence in vivo. Nat Med. 2006;12(11):1301–9. doi: 10.1038/nm1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casrouge A, Beaudoing E, Dalle S, Pannetier C, Kanellopoulos J, Kourilsky P. Size estimate of the alpha beta TCR repertoire of naive mouse splenocytes. J Immunol. 2000;164(11):5782–7. doi: 10.4049/jimmunol.164.11.5782. [DOI] [PubMed] [Google Scholar]
- Cho JH, Boyman O, Kim HO, Hahm B, Rubinstein MP, Ramsey C, Kim DM, Surh CD, Sprent J. An intense form of homeostatic proliferation of naive CD8+ cells driven by IL-2. J Exp Med. 2007;204(8):1787–801. doi: 10.1084/jem.20070740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchill GA, Airey DC, Allayee H, Angel JM, Attie AD, Beatty J, Beavis WD, Belknap JK, Bennett B, Berrettini W, Bleich A, Bogue M, Broman KW, Buck KJ, Buckler E, Burmeister M, Chesler EJ, Cheverud JM, Clapcote S, Cook MN, Cox RD, Crabbe JC, Crusio WE, Darvasi A, Deschepper CF, Doerge RW, Farber CR, Forejt J, Gaile D, Garlow SJ, Geiger H, Gershenfeld H, Gordon T, Gu J, Gu W, de Haan G, Hayes NL, Heller C, Himmelbauer H, Hitzemann R, Hunter K, Hsu HC, Iraqi FA, Ivandic B, Jacob HJ, Jansen RC, Jepsen KJ, Johnson DK, Johnson TE, Kempermann G, Kendziorski C, Kotb M, Kooy RF, Llamas B, Lammert F, Lassalle JM, Lowenstein PR, Lu L, Lusis A, Manly KF, Marcucio R, Matthews D, Medrano JF, Miller DR, Mittleman G, Mock BA, Mogil JS, Montagutelli X, Morahan G, Morris DG, Mott R, Nadeau JH, Nagase H, Nowakowski RS, O’Hara BF, Osadchuk AV, Page GP, Paigen B, Paigen K, Palmer AA, Pan HJ, Peltonen-Palotie L, Peirce J, Pomp D, Pravenec M, Prows DR, Qi Z, Reeves RH, Roder J, Rosen GD, Schadt EE, Schalkwyk LC, Seltzer Z, Shimomura K, Shou S, Sillanpaa MJ, Siracusa LD, Snoeck HW, Spearow JL, Svenson K, Tarantino LM, Threadgill D, Toth LA, Valdar W, de Villena FP, Warden C, Whatley S, Williams RW, Wiltshire T, Yi N, Zhang D, Zhang M, Zou F. The Collaborative Cross, a community resource for the genetic analysis of complex traits. Nat Genet. 2004;36(11):1133–7. doi: 10.1038/ng1104-1133. [DOI] [PubMed] [Google Scholar]
- Cornell CT, Kiosses WB, Harkins S, Whitton JL. Inhibition of protein trafficking by coxsackievirus b3: multiple viral proteins target a single organelle. J Virol. 2006;80(13):6637–47. doi: 10.1128/JVI.02572-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornell CT, Kiosses WB, Harkins S, Whitton JL. Coxsackievirus B3 proteins directionally complement each other to downregulate surface major histocompatibility complex class I. J Virol. 2007;81(13):6785–97. doi: 10.1128/JVI.00198-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotty S, Kersh EN, Cannons J, Schwartzberg PL, Ahmed R. SAP is required for generating long-term humoral immunity. Nature. 2003;421(6920):282–7. doi: 10.1038/nature01318. [DOI] [PubMed] [Google Scholar]
- Dalton DK, Haynes L, Chu CQ, Swain SL, Wittmer S. Interferon gamma eliminates responding CD4 T cells during mycobacterial infection by inducing apoptosis of activated CD4 T cells. J Exp Med. 2000;192(1):117–22. doi: 10.1084/jem.192.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Val M, Hengel H, Hacker H, Hartlaub U, Ruppert T, Lucin P, Koszinowski UH. Cytomegalovirus prevents antigen presentation by blocking the transport of peptide-loaded major histocompatibility complex class I molecules into the medial-Golgi compartment. J Exp Med. 1992;176(3):729–38. doi: 10.1084/jem.176.3.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deretic V. Multiple regulatory and effector roles of autophagy in immunity. Curr Opin Immunol. 2009;21(1):53–62. doi: 10.1016/j.coi.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly RP, Kotenko SV. Interferon-lambda: a new addition to an old family. J Interferon Cytokine Res. 2010;30(8):555–64. doi: 10.1089/jir.2010.0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisfeld AJ, Yee MB, Erazo A, Abendroth A, Kinchington PR. Downregulation of class I major histocompatibility complex surface expression by varicella-zoster virus involves open reading frame 66 protein kinase-dependent and -independent mechanisms. J Virol. 2007;81(17):9034–49. doi: 10.1128/JVI.00711-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ejrnaes M, Filippi CM, Martinic MM, Ling EM, Togher LM, Crotty S, von Herrath MG. Resolution of a chronic viral infection after interleukin-10 receptor blockade. J Exp Med. 2006;203(11):2461–72. doi: 10.1084/jem.20061462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsaesser H, Sauer K, Brooks DG. IL-21 is required to control chronic viral infection. Science. 2009;324(5934):1569–72. doi: 10.1126/science.1174182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkelman FD, Madden KB, Morris SC, Holmes JM, Boiani N, Katona IM, Maliszewski CR. Anti-cytokine antibodies as carrier proteins. Prolongation of in vivo effects of exogenous cytokines by injection of cytokine-anti-cytokine antibody complexes. J Immunol. 1993;151(3):1235–44. [PubMed] [Google Scholar]
- Foster B, Prussin C, Liu F, Whitmire JK, Whitton JL. Detection of intracellular cytokines by flow cytometry. Curr Protoc Immunol. 2007;Chapter 6(Unit 6):24. doi: 10.1002/0471142735.im0624s78. [DOI] [PubMed] [Google Scholar]
- Foulds KE, Shen H. Clonal competition inhibits the proliferation and differentiation of adoptively transferred TCR transgenic CD4 T cells in response to infection. J Immunol. 2006;176(5):3037–43. doi: 10.4049/jimmunol.176.5.3037. [DOI] [PubMed] [Google Scholar]
- Freeman GJ, Wherry EJ, Ahmed R, Sharpe AH. Reinvigorating exhausted HIV-specific T cells via PD-1-PD-1 ligand blockade. J Exp Med. 2006 doi: 10.1084/jem.20061800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frohlich A, Kisielow J, Schmitz I, Freigang S, Shamshiev AT, Weber J, Marsland BJ, Oxenius A, Kopf M. IL-21R on T cells is critical for sustained functionality and control of chronic viral infection. Science. 2009;324(5934):1576–80. doi: 10.1126/science.1172815. [DOI] [PubMed] [Google Scholar]
- Fuller MJ, Hildeman DA, Sabbaj S, Gaddis DE, Tebo AE, Shang L, Goepfert PA, Zajac AJ. Cutting edge: emergence of CD127high functionally competent memory T cells is compromised by high viral loads and inadequate T cell help. J Immunol. 2005;174(10):5926–30. doi: 10.4049/jimmunol.174.10.5926. [DOI] [PubMed] [Google Scholar]
- Fuller MJ, Khanolkar A, Tebo AE, Zajac AJ. Maintenance, loss, and resurgence of T cell responses during acute, protracted, and chronic viral infections. J Immunol. 2004;172(7):4204–14. doi: 10.4049/jimmunol.172.7.4204. [DOI] [PubMed] [Google Scholar]
- Fuller MJ, Zajac AJ. Ablation of CD8 and CD4 T cell responses by high viral loads. J Immunol. 2003;170(1):477–86. doi: 10.4049/jimmunol.170.1.477. [DOI] [PubMed] [Google Scholar]
- Gannage M, Munz C. Autophagy in MHC class II presentation of endogenous antigens. Curr Top Microbiol Immunol. 2009;335:123–40. doi: 10.1007/978-3-642-00302-8_6. [DOI] [PubMed] [Google Scholar]
- Grayson JM, Weant AE, Holbrook BC, Hildeman D. Role of Bim in regulating CD8+ T-cell responses during chronic viral infection. J Virol. 2006;80(17):8627–38. doi: 10.1128/JVI.00855-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grayson JM, Zajac AJ, Altman JD, Ahmed R. Cutting edge: increased expression of Bcl-2 in antigen-specific memory CD8+ T cells. J Immunol. 2000;164(8):3950–4. doi: 10.4049/jimmunol.164.8.3950. [DOI] [PubMed] [Google Scholar]
- Grosso JF, Goldberg MV, Getnet D, Bruno TC, Yen HR, Pyle KJ, Hipkiss E, Vignali DA, Pardoll DM, Drake CG. Functionally distinct LAG-3 and PD-1 subsets on activated and chronically stimulated CD8 T cells. J Immunol. 2009;182(11):6659–69. doi: 10.4049/jimmunol.0804211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton SE, Wolkers MC, Schoenberger SP, Jameson SC. The generation of protective memory-like CD8+ T cells during homeostatic proliferation requires CD4+ T cells. Nat Immunol. 2006;7(5):475–81. doi: 10.1038/ni1326. [DOI] [PubMed] [Google Scholar]
- Hammarlund E, Lewis MW, Hansen SG, Strelow LI, Nelson JA, Sexton GJ, Hanifin JM, Slifka MK. Duration of antiviral immunity after smallpox vaccination. Nat Med. 2003;9(9):1131–1137. doi: 10.1038/nm917. [DOI] [PubMed] [Google Scholar]
- Han S, Asoyan A, Rabenstein H, Nakano N, Obst R. Role of antigen persistence and dose for CD4+ T-cell exhaustion and recovery. Proc Natl Acad Sci U S A. 2010;107(47):20453–8. doi: 10.1073/pnas.1008437107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hand TW, Morre M, Kaech SM. Expression of IL-7 receptor alpha is necessary but not sufficient for the formation of memory CD8 T cells during viral infection. Proc Natl Acad Sci U S A. 2007;104(28):11730–5. doi: 10.1073/pnas.0705007104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haring JS, Harty JT. Aberrant Contraction of Antigen-Specific CD4 T Cells After Infection in the Absence of IFN-{gamma} or its Receptor. Infect Immun. 2006 doi: 10.1128/IAI.00847-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haring JS, Harty JT. Interleukin-18-related genes are induced during the contraction phase but do not play major roles in regulating the dynamics or function of the T-cell response to Listeria monocytogenes infection. Infect Immun. 2009;77(5):1894–903. doi: 10.1128/IAI.01315-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington LE, Janowski KM, Oliver JR, Zajac AJ, Weaver CT. Memory CD4 T cells emerge from effector T-cell progenitors. Nature. 2008;452(7185):356–60. doi: 10.1038/nature06672. [DOI] [PubMed] [Google Scholar]
- Harrington LE, Most Rv R, Whitton JL, Ahmed R. Recombinant vaccinia virus-induced T-cell immunity: quantitation of the response to the virus vector and the foreign epitope. J Virol. 2002;76(7):3329–37. doi: 10.1128/JVI.76.7.3329-3337.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hataye J, Moon JJ, Khoruts A, Reilly C, Jenkins MK. Naive and Memory CD4+ T Cell Survival Controlled by Clonal Abundance. Science. 2006 doi: 10.1126/science.1124228. [DOI] [PubMed] [Google Scholar]
- Havenar-Daughton C, Kolumam GA, Murali-Krishna K. Cutting Edge: The direct action of type I IFN on CD4 T cells is critical for sustaining clonal expansion in response to a viral but not a bacterial infection. J Immunol. 2006;176(6):3315–9. doi: 10.4049/jimmunol.176.6.3315. [DOI] [PubMed] [Google Scholar]
- Hegazy AN, Peine M, Helmstetter C, Panse I, Frohlich A, Bergthaler A, Flatz L, Pinschewer DD, Radbruch A, Lohning M. Interferons direct Th2 cell reprogramming to generate a stable GATA-3(+)T-bet(+) cell subset with combined Th2 and Th1 cell functions. Immunity. 2010;32(1):116–28. doi: 10.1016/j.immuni.2009.12.004. [DOI] [PubMed] [Google Scholar]
- Hegde NR, Tomazin RA, Wisner TW, Dunn C, Boname JM, Lewinsohn DM, Johnson DC. Inhibition of HLA-DR assembly, transport, and loading by human cytomegalovirus glycoprotein US3: a novel mechanism for evading major histocompatibility complex class II antigen presentation. J Virol. 2002;76(21):10929–41. doi: 10.1128/JVI.76.21.10929-10941.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heise MT, Connick M, Virgin HWt. Murine cytomegalovirus inhibits interferon gamma-induced antigen presentation to CD4 T cells by macrophages via regulation of expression of major histocompatibility complex class II-associated genes. J Exp Med. 1998;187(7):1037–46. doi: 10.1084/jem.187.7.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homann D, Teyton L, Oldstone MB. Differential regulation of antiviral T-cell immunity results in stable CD8+ but declining CD4+ T-cell memory. Nat Med. 2001;7(8):913–9. doi: 10.1038/90950. [DOI] [PubMed] [Google Scholar]
- Homann D, Tishon A, Berger DP, Weigle WO, von Herrath MG, Oldstone MB. Evidence for an underlying CD4 helper and CD8 T-cell defect in B-cell-deficient mice: failure to clear persistent virus infection after adoptive immunotherapy with virus-specific memory cells from muMT/muMT mice. J Virol. 1998;72(11):9208–16. doi: 10.1128/jvi.72.11.9208-9216.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Huston G, Duso D, Lepak N, Roman E, Swain SL. CD4(+) T cell effectors can become memory cells with high efficiency and without further division. Nat Immunol. 2001;2(8):705–10. doi: 10.1038/90643. [DOI] [PubMed] [Google Scholar]
- Ichinohe T, Lee HK, Ogura Y, Flavell R, Iwasaki A. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J Exp Med. 2009;206(1):79–87. doi: 10.1084/jem.20081667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson SE, Mason GM, Wills MR. Human cytomegalovirus immunity and immune evasion. Virus Res. 2010 doi: 10.1016/j.virusres.2010.10.031. [DOI] [PubMed] [Google Scholar]
- Jacob J, Baltimore D. Modelling T-cell memory by genetic marking of memory T cells in vivo. Nature. 1999;399(6736):593–7. doi: 10.1038/21208. [DOI] [PubMed] [Google Scholar]
- Jaleco S, Swainson L, Dardalhon V, Burjanadze M, Kinet S, Taylor N. Homeostasis of naive and memory CD4+ T cells: IL-2 and IL-7 differentially regulate the balance between proliferation and Fas-mediated apoptosis. J Immunol. 2003;171(1):61–8. doi: 10.4049/jimmunol.171.1.61. [DOI] [PubMed] [Google Scholar]
- Janssen EM, Lemmens EE, Wolfe T, Christen U, von Herrath MG, Schoenberger SP. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature. 2003;421(6925):852–6. doi: 10.1038/nature01441. [DOI] [PubMed] [Google Scholar]
- Jellison ER, Kim SK, Welsh RM. Cutting edge: MHC class II-restricted killing in vivo during viral infection. J Immunol. 2005;174(2):614–8. doi: 10.4049/jimmunol.174.2.614. [DOI] [PubMed] [Google Scholar]
- Johnston RJ, Poholek AC, DiToro D, Yusuf I, Eto D, Barnett B, Dent AL, Craft J, Crotty S. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science. 2009;325(5943):1006–10. doi: 10.1126/science.1175870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, Gapin L, Kaech SM. Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity. 2007;27(2):281–95. doi: 10.1016/j.immuni.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaech SM, Tan JT, Wherry EJ, Konieczny BT, Surh CD, Ahmed R. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat Immunol. 2003;4(12):1191–8. doi: 10.1038/ni1009. [DOI] [PubMed] [Google Scholar]
- Kamimura D, Bevan MJ. Naive CD8+ T cells differentiate into protective memory-like cells after IL-2 anti IL-2 complex treatment in vivo. J Exp Med. 2007;204(8):1803–12. doi: 10.1084/jem.20070543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamimura D, Sawa Y, Sato M, Agung E, Hirano T, Murakami M. IL-2 in vivo activities and antitumor efficacy enhanced by an anti-IL-2 mAb. J Immunol. 2006;177(1):306–14. doi: 10.4049/jimmunol.177.1.306. [DOI] [PubMed] [Google Scholar]
- Kanai K, Satoh Y, Yamanaka H, Kawaguchi A, Horie K, Sugata K, Hoshikawa Y, Sata T, Sairenji T. The vIL-10 gene of the Epstein-Barr virus (EBV) is conserved in a stable manner except for a few point mutations in various EBV isolates. Virus Genes. 2007;35(3):563–9. doi: 10.1007/s11262-007-0153-5. [DOI] [PubMed] [Google Scholar]
- Kedl RM, Rees WA, Hildeman DA, Schaefer B, Mitchell T, Kappler J, Marrack P. T cells compete for access to antigen-bearing antigen-presenting cells. J Exp Med. 2000;192(8):1105–13. doi: 10.1084/jem.192.8.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemball CC, Harkins S, Whitmire JK, Flynn CT, Feuer R, Whitton JL. Coxsackievirus B3 inhibits antigen presentation in vivo, exerting a profound and selective effect on the MHC class I pathway. PLoS Pathog. 2009;5(10):e1000618. doi: 10.1371/journal.ppat.1000618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemball CC, Harkins S, Whitton JL. Enumeration and functional evaluation of virus-specific CD4+ and CD8+ T cells in lymphoid and peripheral sites of coxsackievirus B3 infection. J Virol. 2008;82(9):4331–42. doi: 10.1128/JVI.02639-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersh EN, Fitzpatrick DR, Murali-Krishna K, Shires J, Speck SH, Boss JM, Ahmed R. Rapid demethylation of the IFN-gamma gene occurs in memory but not naive CD8 T cells. J Immunol. 2006;176(7):4083–93. doi: 10.4049/jimmunol.176.7.4083. [DOI] [PubMed] [Google Scholar]
- Khanolkar A, Fuller MJ, Zajac AJ. CD4 T cell-dependent CD8 T cell maturation. J Immunol. 2004;172(5):2834–44. doi: 10.4049/jimmunol.172.5.2834. [DOI] [PubMed] [Google Scholar]
- Klonowski KD, Williams KJ, Marzo AL, Blair DA, Lingenheld EG, Lefrancois L. Dynamics of blood-borne CD8 memory T cell migration in vivo. Immunity. 2004;20(5):551–62. doi: 10.1016/s1074-7613(04)00103-7. [DOI] [PubMed] [Google Scholar]
- Knappe A, Hor S, Wittmann S, Fickenscher H. Induction of a novel cellular homolog of interleukin-10, AK155, by transformation of T lymphocytes with herpesvirus saimiri. J Virol. 2000;74(8):3881–7. doi: 10.1128/jvi.74.8.3881-3887.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koguchi Y, Thauland TJ, Slifka MK, Parker DC. Preformed CD40 ligand exists in secretory lysosomes in effector and memory CD4+ T cells and is quickly expressed on the cell surface in an antigen-specific manner. Blood. 2007;110(7):2520–7. doi: 10.1182/blood-2007-03-081299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolumam GA, Thomas S, Thompson LJ, Sprent J, Murali-Krishna K. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J Exp Med. 2005;202(5):637–50. doi: 10.1084/jem.20050821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondrack RM, Harbertson J, Tan JT, McBreen ME, Surh CD, Bradley LM. Interleukin 7 regulates the survival and generation of memory CD4 cells. J Exp Med. 2003;198(12):1797–806. doi: 10.1084/jem.20030735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotenko SV, Saccani S, Izotova LS, Mirochnitchenko OV, Pestka S. Human cytomegalovirus harbors its own unique IL-10 homolog (cmvIL-10) Proc Natl Acad Sci U S A. 2000;97(4):1695–700. doi: 10.1073/pnas.97.4.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuenzel S, Till A, Winkler M, Hasler R, Lipinski S, Jung S, Grotzinger J, Fickenscher H, Schreiber S, Rosenstiel P. The nucleotide-binding oligomerization domain-like receptor NLRC5 is involved in IFN-dependent antiviral immune responses. J Immunol. 2010;184(4):1990–2000. doi: 10.4049/jimmunol.0900557. [DOI] [PubMed] [Google Scholar]
- Kumaraguru U, Banerjee K, Rouse BT. In vivo rescue of defective memory CD8+ T cells by cognate helper T cells. J Leukoc Biol. 2005;78(4):879–87. doi: 10.1189/jlb.0105007. [DOI] [PubMed] [Google Scholar]
- Lamkanfi M, Kanneganti TD. Nlrp3: an immune sensor of cellular stress and infection. Int J Biochem Cell Biol. 2010;42(6):792–5. doi: 10.1016/j.biocel.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane TE, Liu MT, Chen BP, Asensio VC, Samawi RM, Paoletti AD, Campbell IL, Kunkel SL, Fox HS, Buchmeier MJ. A central role for CD4(+) T cells and RANTES in virus-induced central nervous system inflammation and demyelination. J Virol. 2000;74(3):1415–24. doi: 10.1128/jvi.74.3.1415-1424.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Iwasaki A. Autophagy and antiviral immunity. Curr Opin Immunol. 2008;20(1):23–9. doi: 10.1016/j.coi.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefrancois L. Development, trafficking, and function of memory T-cell subsets. Immunol Rev. 2006;211:93–103. doi: 10.1111/j.0105-2896.2006.00393.x. [DOI] [PubMed] [Google Scholar]
- Leist TP, Kohler M, Zinkernagel RM. Impaired generation of anti-viral cytotoxicity against lymphocytic choriomeningitis and vaccinia virus in mice treated with CD4-specific monoclonal antibody. Scand J Immunol. 1989;30(6):679–86. doi: 10.1111/j.1365-3083.1989.tb02476.x. [DOI] [PubMed] [Google Scholar]
- Lemmermann NA, Gergely K, Bohm V, Deegen P, Daubner T, Reddehase MJ. Immune evasion proteins of murine cytomegalovirus preferentially affect cell surface display of recently generated peptide presentation complexes. J Virol. 2010;84(3):1221–36. doi: 10.1128/JVI.02087-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenz DC, Kurz SK, Lemmens E, Schoenberger SP, Sprent J, Oldstone MB, Homann D. IL-7 regulates basal homeostatic proliferation of antiviral CD4+T cell memory. Proc Natl Acad Sci U S A. 2004;101(25):9357–62. doi: 10.1073/pnas.0400640101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewandowski GA, Lo D, Bloom FE. Interference with major histocompatibility complex class II-restricted antigen presentation in the brain by herpes simplex virus type 1: a possible mechanism of evasion of the immune response. Proc Natl Acad Sci U S A. 1993;90(5):2005–9. doi: 10.1073/pnas.90.5.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Huston G, Swain SL. IL-7 promotes the transition of CD4 effectors to persistent memory cells. J Exp Med. 2003;198(12):1807–15. doi: 10.1084/jem.20030725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Whitton JL. Cutting Edge: Re-evaluating the In Vivo Cytokine Responses of CD8+ T Cells during Primary and Secondary Viral Infections. J Immunol. 2005;174(10):5936–40. doi: 10.4049/jimmunol.174.10.5936. [DOI] [PubMed] [Google Scholar]
- Liu F, Whitton JL, Slifka MK. The rapidity with which virus-specific CD8+ T cells initiate IFN-gamma synthesis increases markedly over the course of infection and correlates with immunodominance. J Immunol. 2004;173(1):456–62. doi: 10.4049/jimmunol.173.1.456. [DOI] [PubMed] [Google Scholar]
- MacLeod MK, McKee A, Crawford F, White J, Kappler J, Marrack P. CD4 memory T cells divide poorly in response to antigen because of their cytokine profile. Proc Natl Acad Sci U S A. 2008;105(38):14521–6. doi: 10.1073/pnas.0807449105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall DR, Turner SJ, Belz GT, Wingo S, Andreansky S, Sangster MY, Riberdy JM, Liu T, Tan M, Doherty PC. Measuring the diaspora for virus-specific CD8+ T cells. Proc Natl Acad Sci U S A. 2001;98(11):6313–8. doi: 10.1073/pnas.101132698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsland BJ, Nembrini C, Grun K, Reissmann R, Kurrer M, Leipner C, Kopf M. TLR ligands act directly upon T cells to restore proliferation in the absence of protein kinase C-theta signaling and promote autoimmune myocarditis. J Immunol. 2007;178(6):3466–73. doi: 10.4049/jimmunol.178.6.3466. [DOI] [PubMed] [Google Scholar]
- Marzo AL, Vezys V, Williams K, Tough DF, Lefrancois L. Tissue-level regulation of Th1 and Th2 primary and memory CD4 T cells in response to Listeria infection. J Immunol. 2002;168(9):4504–10. doi: 10.4049/jimmunol.168.9.4504. [DOI] [PubMed] [Google Scholar]
- Masopust D, Vezys V, Marzo AL, Lefrancois L. Preferential localization of effector memory cells in nonlymphoid tissue. Science. 2001;291(5512):2413–7. doi: 10.1126/science.1058867. [DOI] [PubMed] [Google Scholar]
- Matloubian M, Concepcion RJ, Ahmed R. CD4+ T cells are required to sustain CD8+ cytotoxic T-cell responses during chronic viral infection. J Virol. 1994;68(12):8056–63. doi: 10.1128/jvi.68.12.8056-8063.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHeyzer-Williams LJ, McHeyzer-Williams MG. Developmentally distinct Th cells control plasma cell production in vivo. Immunity. 2004;20(2):231–42. doi: 10.1016/s1074-7613(04)00028-7. [DOI] [PubMed] [Google Scholar]
- McHeyzer-Williams LJ, Pelletier N, Mark L, Fazilleau N, McHeyzer-Williams MG. Follicular helper T cells as cognate regulators of B cell immunity. Curr Opin Immunol. 2009;21(3):266–73. doi: 10.1016/j.coi.2009.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melief CJ, Schoenberger SP. Enhancement of proliferation and downregulation of TRAIL expression on CD8+ T cells by IL-21. Eur J Immunol. 2010;40(11):2990–2. doi: 10.1002/eji.201041076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon JJ, Chu HH, Pepper M, McSorley SJ, Jameson SC, Kedl RM, Jenkins MK. Naive CD4(+) T cell frequency varies for different epitopes and predicts repertoire diversity and response magnitude. Immunity. 2007;27(2):203–13. doi: 10.1016/j.immuni.2007.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mothe BR, Stewart BS, Oseroff C, Bui HH, Stogiera S, Garcia Z, Dow C, Rodriguez-Carreno MP, Kotturi M, Pasquetto V, Botten J, Crotty S, Janssen E, Buchmeier MJ, Sette A. Chronic lymphocytic choriomeningitis virus infection actively down-regulates CD4+ T cell responses directed against a broad range of epitopes. J Immunol. 2007;179(2):1058–67. doi: 10.4049/jimmunol.179.2.1058. [DOI] [PubMed] [Google Scholar]
- Mueller SN, Jones CM, Stock AT, Suter M, Heath WR, Carbone FR. CD4+ T cells can protect APC from CTL-mediated elimination. J Immunol. 2006;176(12):7379–84. doi: 10.4049/jimmunol.176.12.7379. [DOI] [PubMed] [Google Scholar]
- Muhlbauer M, Fleck M, Schutz C, Weiss T, Froh M, Blank C, Scholmerich J, Hellerbrand C. PD-L1 is induced in hepatocytes by viral infection and by interferon-alpha and -gamma and mediates T cell apoptosis. J Hepatol. 2006;45(4):520–8. doi: 10.1016/j.jhep.2006.05.007. [DOI] [PubMed] [Google Scholar]
- Munz C. Autophagy and antigen presentation. Cell Microbiol. 2006;8(6):891–8. doi: 10.1111/j.1462-5822.2006.00714.x. [DOI] [PubMed] [Google Scholar]
- Munz C. Viral evasion of autophagy. Cell Host Microbe. 2007;1(1):9–11. doi: 10.1016/j.chom.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Nakanishi Y, Lu B, Gerard C, Iwasaki A. CD8(+) T lymphocyte mobilization to virus-infected tissue requires CD4(+) T-cell help. Nature. 2009;462(7272):510–3. doi: 10.1038/nature08511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama Y, Plisch EH, Sullivan J, Thomas C, Czuprynski CJ, Williams BR, Suresh M. Role of PKR and Type I IFNs in viral control during primary and secondary infection. PLoS Pathog. 2010;6(6):e1000966. doi: 10.1371/journal.ppat.1000966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neerincx A, Lautz K, Menning M, Kremmer E, Zigrino P, Hosel M, Buning H, Schwarzenbacher R, Kufer TA. A role for the human nucleotide-binding domain, leucine-rich repeat-containing family member NLRC5 in antiviral responses. J Biol Chem. 2010;285(34):26223–32. doi: 10.1074/jbc.M110.109736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nembrini C, Abel B, Kopf M, Marsland BJ. Strong TCR signaling, TLR ligands, and cytokine redundancies ensure robust development of type 1 effector T cells. J Immunol. 2006;176(12):7180–8. doi: 10.4049/jimmunol.176.12.7180. [DOI] [PubMed] [Google Scholar]
- Neumann J, Eis-Hubinger AM, Koch N. Herpes simplex virus type 1 targets the MHC class II processing pathway for immune evasion. J Immunol. 2003;171(6):3075–83. doi: 10.4049/jimmunol.171.6.3075. [DOI] [PubMed] [Google Scholar]
- Northrop JK, Thomas RM, Wells AD, Shen H. Epigenetic remodeling of the IL-2 and IFN-gamma loci in memory CD8 T cells is influenced by CD4 T cells. J Immunol. 2006;177(2):1062–9. doi: 10.4049/jimmunol.177.2.1062. [DOI] [PubMed] [Google Scholar]
- Obar JJ, Khanna KM, Lefrancois L. Endogenous naive CD8+ T cell precursor frequency regulates primary and memory responses to infection. Immunity. 2008;28(6):859–69. doi: 10.1016/j.immuni.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obst R, van Santen HM, Mathis D, Benoist C. Antigen persistence is required throughout the expansion phase of a CD4(+) T cell response. J Exp Med. 2005;201(10):1555–65. doi: 10.1084/jem.20042521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opferman JT, Ober BT, Ashton-Rickardt PG. Linear differentiation of cytotoxic effectors into memory T lymphocytes. Science. 1999;283(5408):1745–8. doi: 10.1126/science.283.5408.1745. [DOI] [PubMed] [Google Scholar]
- Orr MT, Edelmann KH, Vieira J, Corey L, Raulet DH, Wilson CB. Inhibition of MHC class I is a virulence factor in herpes simplex virus infection of mice. PLoS Pathog. 2005;1(1):e7. doi: 10.1371/journal.ppat.0010007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pien GC, Satoskar AR, Takeda K, Akira S, Biron CA. Cutting edge: selective IL-18 requirements for induction of compartmental IFN-gamma responses during viral infection. J Immunol. 2000;165(9):4787–91. doi: 10.4049/jimmunol.165.9.4787. [DOI] [PubMed] [Google Scholar]
- Pipkin ME, Sacks JA, Cruz-Guilloty F, Lichtenheld MG, Bevan MJ, Rao A. Interleukin-2 and inflammation induce distinct transcriptional programs that promote the differentiation of effector cytolytic T cells. Immunity. 2010;32(1):79–90. doi: 10.1016/j.immuni.2009.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purton JF, Martin CE, Surh CD. Enhancing T cell memory: IL-7 as an adjuvant to boost memory T-cell generation. Immunol Cell Biol. 2008;86(5):385–6. doi: 10.1038/icb.2008.30. [DOI] [PubMed] [Google Scholar]
- Purton JF, Tan JT, Rubinstein MP, Kim DM, Sprent J, Surh CD. Antiviral CD4+ memory T cells are IL-15 dependent. J Exp Med. 2007;204(4):951–61. doi: 10.1084/jem.20061805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman AH, Cui W, Larosa DF, Taylor DK, Zhang J, Goldstein DR, Wherry EJ, Kaech SM, Turka LA. MyD88 plays a critical T cell-intrinsic role in supporting CD8 T cell expansion during acute lymphocytic choriomeningitis virus infection. J Immunol. 2008;181(6):3804–10. doi: 10.4049/jimmunol.181.6.3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravkov EV, Williams MA. The magnitude of CD4+ T cell recall responses is controlled by the duration of the secondary stimulus. J Immunol. 2009;183(4):2382–9. doi: 10.4049/jimmunol.0900319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock KL, Farfan-Arribas DJ, Shen L. Proteases in MHC class I presentation and cross-presentation. J Immunol. 2010;184(1):9–15. doi: 10.4049/jimmunol.0903399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers PR, Dubey C, Swain SL. Qualitative changes accompany memory T cell generation: faster, more effective responses at lower doses of antigen. J Immunol. 2000;164(5):2338–46. doi: 10.4049/jimmunol.164.5.2338. [DOI] [PubMed] [Google Scholar]
- Roman E, Miller E, Harmsen A, Wiley J, Von Andrian UH, Huston G, Swain SL. CD4 effector T cell subsets in the response to influenza: heterogeneity, migration, and function. J Exp Med. 2002;196(7):957–68. doi: 10.1084/jem.20021052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinstein MP, Kovar M, Purton JF, Cho JH, Boyman O, Surh CD, Sprent J. Converting IL-15 to a superagonist by binding to soluble IL-15R{alpha} Proc Natl Acad Sci U S A. 2006;103(24):9166–71. doi: 10.1073/pnas.0600240103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S, Teichgraber V, Kalia V, Polley A, Masopust D, Harrington LE, Ahmed R, Wherry EJ. Strength of stimulus and clonal competition impact the rate of memory CD8 T cell differentiation. J Immunol. 2007;179(10):6704–14. doi: 10.4049/jimmunol.179.10.6704. [DOI] [PubMed] [Google Scholar]
- Schmitz N, Kurrer M, Bachmann MF, Kopf M. Interleukin-1 is responsible for acute lung immunopathology but increases survival of respiratory influenza virus infection. J Virol. 2005;79(10):6441–8. doi: 10.1128/JVI.79.10.6441-6448.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiner B, Mitsdoerffer M, Kieseier BC, Chen L, Hartung HP, Weller M, Wiendl H. Interferon-beta enhances monocyte and dendritic cell expression of B7-H1 (PD-L1), a strong inhibitor of autologous T-cell activation: relevance for the immune modulatory effect in multiple sclerosis. J Neuroimmunol. 2004;155(1–2):172–82. doi: 10.1016/j.jneuroim.2004.06.013. [DOI] [PubMed] [Google Scholar]
- Seddon B, Tomlinson P, Zamoyska R. Interleukin 7 and T cell receptor signals regulate homeostasis of CD4 memory cells. Nat Immunol. 2003;4(7):680–6. doi: 10.1038/ni946. [DOI] [PubMed] [Google Scholar]
- Sercan O, Hammerling GJ, Arnold B, Schuler T. Cutting Edge: Innate Immune Cells Contribute to the IFN-{gamma}-Dependent Regulation of Antigen-Specific CD8+ T Cell Homeostasis. J Immunol. 2006;176(2):735–9. doi: 10.4049/jimmunol.176.2.735. [DOI] [PubMed] [Google Scholar]
- Sercan O, Stoycheva D, Hammerling GJ, Arnold B, Schuler T. IFN-gamma receptor signaling regulates memory CD8+ T cell differentiation. J Immunol. 2010;184(6):2855–62. doi: 10.4049/jimmunol.0902708. [DOI] [PubMed] [Google Scholar]
- Sevilla N, Kunz S, Holz A, Lewicki H, Homann D, Yamada H, Campbell KP, de La Torre JC, Oldstone MB. Immunosuppression and resultant viral persistence by specific viral targeting of dendritic cells. J Exp Med. 2000;192(9):1249–60. doi: 10.1084/jem.192.9.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevilla N, Kunz S, McGavern D, Oldstone MB. Infection of dendritic cells by lymphocytic choriomeningitis virus. Curr Top Microbiol Immunol. 2003;276:125–44. doi: 10.1007/978-3-662-06508-2_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shedlock DJ, Shen H. Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science. 2003;300(5617):337–9. doi: 10.1126/science.1082305. [DOI] [PubMed] [Google Scholar]
- Shiow LR, Rosen DB, Brdickova N, Xu Y, An J, Lanier LL, Cyster JG, Matloubian M. CD69 acts downstream of interferon-alpha/beta to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature. 2006;440(7083):540–4. doi: 10.1038/nature04606. [DOI] [PubMed] [Google Scholar]
- Sievers E, Neumann J, Raftery M, SchOnrich G, Eis-Hubinger AM, Koch N. Glycoprotein B from strain 17 of herpes simplex virus type I contains an invariant chain homologous sequence that binds to MHC class II molecules. Immunology. 2002;107(1):129–35. doi: 10.1046/j.1365-2567.2002.01472.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slifka MK, Rodriguez F, Whitton JL. Rapid on/off cycling of cytokine production by virus-specific CD8+ T cells. Nature. 1999;401(6748):76–9. doi: 10.1038/43454. [DOI] [PubMed] [Google Scholar]
- Slifka MK, Whitton JL. Functional avidity maturation of CD8(+) T cells without selection of higher affinity TCR. Nat Immunol. 2001;2(8):711–7. doi: 10.1038/90650. [DOI] [PubMed] [Google Scholar]
- Srinivasan A, Foley J, McSorley SJ. Massive number of antigen-specific CD4 T cells during vaccination with live attenuated Salmonella causes interclonal competition. J Immunol. 2004;172(11):6884–93. doi: 10.4049/jimmunol.172.11.6884. [DOI] [PubMed] [Google Scholar]
- Srinivasan A, Salazar-Gonzalez RM, Jarcho M, Sandau MM, Lefrancois L, McSorley SJ. Innate immune activation of CD4 T cells in salmonella-infected mice is dependent on IL-18. J Immunol. 2007;178(10):6342–9. doi: 10.4049/jimmunol.178.10.6342. [DOI] [PubMed] [Google Scholar]
- Stohlman SA, Bergmann CC, Lin MT, Cua DJ, Hinton DR. CTL effector function within the central nervous system requires CD4+ T cells. J Immunol. 1998;160(6):2896–904. [PubMed] [Google Scholar]
- Sun JC, Bevan MJ. Defective CD8 T cell memory following acute infection without CD4 T cell help. Science. 2003;300(5617):339–42. doi: 10.1126/science.1083317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun JC, Williams MA, Bevan MJ. CD4+ T cells are required for the maintenance, not programming, of memory CD8+ T cells after acute infection. Nat Immunol. 2004;5(9):927–33. doi: 10.1038/ni1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swain SL, Hu H, Huston G. Class II-independent generation of CD4 memory T cells from effectors. Science. 1999;286(5443):1381–3. doi: 10.1126/science.286.5443.1381. [DOI] [PubMed] [Google Scholar]
- Teijaro JR, Verhoeven D, Page CA, Turner D, Farber DL. Memory CD4 T cells direct protective responses to influenza virus in the lungs through helper-independent mechanisms. J Virol. 2010;84(18):9217–26. doi: 10.1128/JVI.01069-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temme S, Eis-Hubinger AM, McLellan AD, Koch N. The herpes simplex virus-1 encoded glycoprotein B diverts HLA-DR into the exosome pathway. J Immunol. 2010;184(1):236–43. doi: 10.4049/jimmunol.0902192. [DOI] [PubMed] [Google Scholar]
- Tewari K, Nakayama Y, Suresh M. Role of direct effects of IFN-gamma on T cells in the regulation of CD8 T cell homeostasis. J Immunol. 2007;179(4):2115–25. doi: 10.4049/jimmunol.179.4.2115. [DOI] [PubMed] [Google Scholar]
- Tough DF, Zhang X, Sprent J. An IFN-gamma-dependent pathway controls stimulation of memory phenotype CD8+ T cell turnover in vivo by IL-12, IL-18, and IFN-gamma. J Immunol. 2001;166(10):6007–11. doi: 10.4049/jimmunol.166.10.6007. [DOI] [PubMed] [Google Scholar]
- Troy AE, Shen H. Cutting edge: homeostatic proliferation of peripheral T lymphocytes is regulated by clonal competition. J Immunol. 2003;170(2):672–6. doi: 10.4049/jimmunol.170.2.672. [DOI] [PubMed] [Google Scholar]
- van Gisbergen KP, van Olffen RW, van Beek J, van der Sluijs KF, Arens R, Nolte MA, van Lier RA. Protective CD8 T cell memory is impaired during chronic CD70-driven costimulation. J Immunol. 2009;182(9):5352–62. doi: 10.4049/jimmunol.0802809. [DOI] [PubMed] [Google Scholar]
- Varga SM, Welsh RM. Stability of virus-specific CD4+ T cell frequencies from acute infection into long term memory. J Immunol. 1998;161(1):367–74. [PubMed] [Google Scholar]
- Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, Kalia V, Subramaniam S, Blattman JN, Barber DL, Ahmed R. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. 2007;27(4):670–84. doi: 10.1016/j.immuni.2007.09.006. [DOI] [PubMed] [Google Scholar]
- Whitmire JK, Asano MS, Murali-Krishna K, Suresh M, Ahmed R. Long-term CD4 Th1 and Th2 memory following acute lymphocytic choriomeningitis virus infection. J Virol. 1998;72(10):8281–8. doi: 10.1128/jvi.72.10.8281-8288.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitmire JK, Benning N, Eam B, Whitton JL. Increasing the CD4+ T cell precursor frequency leads to competition for IFN-gamma thereby degrading memory cell quantity and quality. J Immunol. 2008;180(10):6777–85. doi: 10.4049/jimmunol.180.10.6777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitmire JK, Benning N, Whitton JL. Cutting edge: early IFN-gamma signaling directly enhances primary antiviral CD4+ T cell responses. J Immunol. 2005;175(9):5624–8. doi: 10.4049/jimmunol.175.9.5624. [DOI] [PubMed] [Google Scholar]
- Whitmire JK, Benning N, Whitton JL. Precursor frequency, nonlinear proliferation, and functional maturation of virus-specific CD4+ T cells. J Immunol. 2006;176(5):3028–36. doi: 10.4049/jimmunol.176.5.3028. [DOI] [PubMed] [Google Scholar]
- Whitmire JK, Eam B, Benning N, Whitton JL. Direct Interferon-{gamma} Signaling Dramatically Enhances CD4+ and CD8+ T Cell Memory. J Immunol. 2007;179(2):1190–7. doi: 10.4049/jimmunol.179.2.1190. [DOI] [PubMed] [Google Scholar]
- Whitmire JK, Eam B, Whitton JL. Tentative T cells: memory cells are quick to respond, but slow to divide. PLoS Pathog. 2008;4(4):e1000041. doi: 10.1371/journal.ppat.1000041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitmire JK, Tan JT, Whitton JL. Interferon-{gamma} acts directly on CD8+ T cells to increase their abundance during virus infection. J Exp Med. 2005;201(7):1053–9. doi: 10.1084/jem.20041463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins C, Gale M., Jr Recognition of viruses by cytoplasmic sensors. Curr Opin Immunol. 2010;22(1):41–7. doi: 10.1016/j.coi.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams MA, Bevan MJ. Effector and memory CTL differentiation. Annu Rev Immunol. 2007;25:171–92. doi: 10.1146/annurev.immunol.25.022106.141548. [DOI] [PubMed] [Google Scholar]
- Williams MA, Ravkov EV, Bevan MJ. Rapid culling of the CD4+ T cell repertoire in the transition from effector to memory. Immunity. 2008;28(4):533–45. doi: 10.1016/j.immuni.2008.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis RA, Kappler JW, Marrack PC. CD8 T cell competition for dendritic cells in vivo is an early event in activation. Proc Natl Acad Sci U S A. 2006;103(32):12063–8. doi: 10.1073/pnas.0605130103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windheim M, Hilgendorf A, Burgert HG. Immune evasion by adenovirus E3 proteins: exploitation of intracellular trafficking pathways. Curr Top Microbiol Immunol. 2004;273:29–85. doi: 10.1007/978-3-662-05599-1_2. [DOI] [PubMed] [Google Scholar]
- Xu RH, Remakus S, Ma X, Roscoe F, Sigal LJ. Direct presentation is sufficient for an efficient anti-viral CD8+ T cell response. PLoS Pathog. 2010;6(2):e1000768. doi: 10.1371/journal.ppat.1000768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yalcin B, Nicod J, Bhomra A, Davidson S, Cleak J, Farinelli L, Osteras M, Whitley A, Yuan W, Gan X, Goodson M, Klenerman P, Satpathy A, Mathis D, Benoist C, Adams DJ, Mott R, Flint J. Commercially available outbred mice for genome-wide association studies. PLoS Genet. 2010;6(9) doi: 10.1371/journal.pgen.1001085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates A, Graw F, Barber DL, Ahmed R, Regoes RR, Antia R. Revisiting estimates of CTL killing rates in vivo. PLoS One. 2007;2(12):e1301. doi: 10.1371/journal.pone.0001301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yewdell JW. Designing CD8+ T cell vaccines: it’s not rocket science (yet) Curr Opin Immunol. 2010;22(3):402–10. doi: 10.1016/j.coi.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yewdell JW, Haeryfar SM. Understanding presentation of viral antigens to CD8+ T cells in vivo: the key to rational vaccine design. Annu Rev Immunol. 2005;23:651–82. doi: 10.1146/annurev.immunol.23.021704.115702. [DOI] [PubMed] [Google Scholar]
- Yi JS, Du M, Zajac AJ. A vital role for interleukin-21 in the control of a chronic viral infection. Science. 2009;324(5934):1572–6. doi: 10.1126/science.1175194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi JS, Ingram JT, Zajac AJ. IL-21 deficiency influences CD8 T cell quality and recall responses following an acute viral infection. J Immunol. 2010;185(8):4835–45. doi: 10.4049/jimmunol.1001032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusuf I, Kageyama R, Monticelli L, Johnston RJ, Ditoro D, Hansen K, Barnett B, Crotty S. Germinal center T follicular helper cell IL-4 production is dependent on signaling lymphocytic activation molecule receptor (CD150) J Immunol. 2010;185(1):190–202. doi: 10.4049/jimmunol.0903505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zajac AJ, Blattman JN, Murali-Krishna K, Sourdive DJ, Suresh M, Altman JD, Ahmed R. Viral immune evasion due to persistence of activated T cells without effector function. J Exp Med. 1998a;188(12):2205–13. doi: 10.1084/jem.188.12.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zajac AJ, Murali-Krishna K, Blattman JN, Ahmed R. Therapeutic vaccination against chronic viral infection: the importance of cooperation between CD4+ and CD8+ T cells. Curr Opin Immunol. 1998b;10(4):444–9. doi: 10.1016/s0952-7915(98)80119-2. [DOI] [PubMed] [Google Scholar]
- Zhao Y, De Trez C, Flynn R, Ware CF, Croft M, Salek-Ardakani S. The adaptor molecule MyD88 directly promotes CD8 T cell responses to vaccinia virus. J Immunol. 2009;182(10):6278–86. doi: 10.4049/jimmunol.0803682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S, Kurt-Jones EA, Cerny AM, Chan M, Bronson RT, Finberg RW. MyD88 intrinsically regulates CD4 T-cell responses. J Virol. 2009;83(4):1625–34. doi: 10.1128/JVI.01770-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuniga EI, Liou LY, Mack L, Mendoza M, Oldstone MB. Persistent virus infection inhibits type I interferon production by plasmacytoid dendritic cells to facilitate opportunistic infections. Cell Host Microbe. 2008;4(4):374–86. doi: 10.1016/j.chom.2008.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuniga MC, Wang H, Barry M, McFadden G. Endosomal/lysosomal retention and degradation of major histocompatibility complex class I molecules is induced by myxoma virus. Virology. 1999;261(2):180–92. doi: 10.1006/viro.1999.9840. [DOI] [PubMed] [Google Scholar]