

Abstract
The effects of methoxy-substitution at the 1-, 2-, 3- and 4-positions of 9-aminomethyl-9,10-dihydroanthracene (AMDA) on h5-HT2A receptor affinity were determined. Racemic mixtures of these compounds were found to show the following affinity trend: 3-MeO > 4-MeO > 1-MeO ~ 2-MeO. Comparison of the effects of these substitutions, with the aid of computational molecular modeling techniques, suggest that the various positional and stereochemical isomers of the methoxy-substituted AMDA compounds interact differently with the h5-HT2A receptor. It is predicted that for the compounds with higher affinities, the methoxy oxygen atom is able to interact with hydrogen bond-donating sidechains within alternative h5-HT2A receptor binding sites, whereas the lower-affinity isomers lack this ability.
The chemical features that are responsible for the potency and efficacy of agents that bind to 5-HT2A receptors, as well as the receptor-ligand interactions responsible for the ligands’ observed affinities, have not yet been fully elucidated. Toward that end, we have synthesized and assayed many analogs1 of the selective2 5-HT2A antagonist3 9-aminomethyl-9,10-dihydroanthracene (AMDA, 1). This work in particular builds upon earlier studies1 of AMDA analogs in which it was shown that substituents of widely varying size and polarity may be placed at the 3-position without a large decrease in 5-HT2A affinity. In order to more completely determine the extent of this apparent steric and/or electrostatic tolerance, and in particular to find AMDA analogs that have a significantly reduced affinity for the 5-HT2A receptor, a series of AMDA analogs that are substituted at varying positions around the AMDA core were synthesized and tested. The methoxy group was chosen a general-purpose ‘probe’ functional group because of its synthetic flexibility, small size and amphiphilic nature (i.e. it is able to participate in both polar and non-polar interactions). The results of a systematic study of methoxy-substituted AMDA compounds, and the concomitant effects that these substitutions have upon the compounds’ affinity for the human 5-HT2A receptor are reported here.
Radioligand binding data (5-HT2A receptor affinities) were obtained for each of the target compounds (Table 1). Within the methoxy-substituted AMDA series, the affinities varied by more than 180-fold (4, Ki = 7.5 nM; 3, Ki = 1367 nM). Of all four positional isomers, only 3-methoxy-AMDA (4) showed increased affinity relative to the parent compound AMDA (Ki = 20 nM). 4-Methoxy-AMDA (5, Ki = 124 nM) displayed a modest 6-fold decrease in affinity compared to AMDA, while both 1- and 2-methoxy-AMDA (2, Ki = 1158 nM and 3, Ki = 1367 nM, respectively) were found to have substantially reduced affinity (> 50-fold) compared to AMDA.
Table 1.
Observed binding affinities for AMDA and methoxy-substituted AMDA analogs at the h5-HT2A receptor.
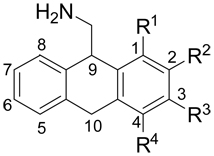 | |||||
|---|---|---|---|---|---|
| Cpd. | R1 | R2 | R3 | R4 | Ki, nMa |
| 1 | -H | -H | -H | -H | 20 |
| 2 | -OCH3 | -H | -H | -H | 1158 |
| 3 | -H | -OCH3 | -H | -H | 1367 |
| 4 | -H | -H | -OCH3 | -H | 7.5 |
| 5 | -H | -H | -H | -OCH3 | 124 |
[3H]Ketanserin labeled cloned 5-HT2A sites. Values represent the mean of computer-derived Ki estimates (using GraphPad Prism) of quadruplicate determinations. Standard errors typically range between 15–25% of the Ki value.
Previous receptor modeling studies1 have identified two possible binding sites for 5-HT2A ligands, as shown schematically in Figure 1. Site 1 is flanked primarily by transmembrane helices TM3, TM5 and TM6, and has been proposed as the binding site for agonists,4, 5 partial agonists6 and antagonists.7, 8 Site 2 is an alternative binding site and is flanked by TM1, TM2, TM3 and TM7. Ketanserin9, a prototypical 5-HT2A antagonist, and similar butyrophenones10 have been proposed to simultaneously bind in both sites.
Figure 1.

Schematic representation of sterically accessible binding sites (Site 1 and Site 2) within the 5-HT2A receptor.
Each compound in Table 1 (with explicit consideration of stereoisomers) was docked into two graphical receptor models representing the agonist-selected Site 1 and the antagonist-selected Site 2 (see Experimental Methods). The overall quality of the docked poses was determined using the ChemScore fitness function with visual inspection of the docked solutions, and the top-ranking solution for each isomer was selected (see Supplemental Table S1). In each case, GOLD was able to place the ligand into both Site 1 and Site 2. However, productive hydrogen bonds were formed only for the more potent 3-methoxy-AMDA ((R)-4 and (S)-4) and 4-methoxy-AMDA ((S)-5).11 For (R)-4, the methoxy oxygen H-bonds with S1593.36, for (S)-4, the H-bond donor is S1312.61, and for (S)-5, H-bonding occurs with S2395.43 (see Figure 2). In each of these solutions, the ammonium ion interacts with D1553.32 and with other lipophilic/aromatic residues surrounding the AMDA core (see Supplemental Table S2). The current models are consistent with earlier mutagenesis and molecular modeling studies.12 The fact that both isomers of 4 are able to H-bond with the receptor and both receive a relatively high ChemScore is consistent with its low Ki value. Although there are several hydrogen bond donating sites within the agonist-selected Site 1 (S1593.36, T1603.37, S2395.43, S2425.46, W3366.48, N3436.55) and antagonist-selected Site 2 (S771.35, T811.39, S1312.61, W1513.28, S1593.36, S3737.46), our results suggest that 1-methoxy AMDA (2) and 2-methoxy-AMDA (3) are not able to position themselves within these sites in a way that allows the methoxy group to beneficially interact with them. This in turn is consistent with the significantly reduced affinity of 2 and 3 for h5-HT2A. The binding affinity of the methoxy-AMDA compounds can thus be directly correlated with their ability to H-bond with residues in Site 1 or Site 2.
Figure 2.

Proposed binding modes of the three MeO-AMDA isomers that exhibit substantial hydrogen-binding interactions with residues within the binding crevice of the h5-HT2A receptor. The receptor backbone for models representing Site 1 (agonist-selected) are indicated with a green ribbon; those representing Site 2 (antagonist-selected) are indicated with an orange ribbon. The sidechains of 5-HT2A residues within 5 Å of the ligand are displayed as capped sticks and the ligand is rendered as a ball-and-stick model. a. (R)-3-methoxy-AMDA ((R)-4) in Site 2. b. (S)-3-methoxy-AMDA ((S)-4) in Site 2. c. (S)-4-methoxy-AMDA ((S)-5) in Site 1.
It should be noted that an alternative explanation for the reduced activity of 2 is the possibility of internal H-bond formation. This would reduce the effectiveness of the amine and the methoxy group to act as an H-bond donor and acceptor, respectively, and could potentially impose a ligand conformation that reduces the complementarity of the ligand to its binding site.
AMDA (1) was synthesized as previously described.3 The synthesis of 1-methoxy- (2) and 2-methoxy-9-aminomethyl-9,10-dihydroanthracene (3) (Scheme 1) was brought about by the 1,4-addition of 2-[(methoxymethoxy)-methyl]phenylmagnesium bromide13 to the nitrostyrenes 6a and 6b14 to give substituted 1,1-diaryl-2-nitroethanes 7a and 7b. The MOM-protected benzyl alcohols 7a and 7b were deprotected using HCl to give the nitro alcohols 8a and 8b, which were reduced to their respective amines to give amino alcohols 9a and 9b. Cyclodehydration of amino alcohols 9a and 9b using freshly prepared PPE in CHCl3 gave the 1- and 2-methoxy substituted AMDAs 2 and 3 respectively. Synthesis of 3-methoxy- (4) and 4-methoxy-9-aminomethyl-9,10-dihydroanthracenes (5) (Scheme 2) was brought about by a standard Suzuki cross-coupling reaction between commercially available 3-methoxybenzyl bromide 10a or 2-methoxybenzyl bromide 10b15 and 2-formylphenylboronic acid to yield 2-(3-methoxybenzyl)benzaldehyde 11a and 2-(2-methoxybenzyl)benzaldehyde 11b. Cyanosilylation of the aldehydes 11a and 11b using TMSCN gave the corresponding cyano trimethylsilyl ethers 12a and 12b, which were reduced with LiAlH4 to give the respective amino alcohols 13a and 13b. Cyclodehydration of amino alcohols 13a and 13b using CH3SO3H and PPA respectively gave the 3-methoxy- and 4-methoxy-substituted AMDAs 4 and 5.
Scheme 1.

Reagents and conditions: (a) 1-bromo-2-((methoxymethoxy)methyl)benzene, Mg, I2, THF, 65 → 0 °C, 12 h; (b) HCl, MeOH, 65 °C, 5 h; (c) H2, (50 psi), MeOH, 10% Pd/C, 25 °C, 36 h; (d) PPE, CHCl3, reflux, 3 h.
Scheme 2.

Reagents and conditions: (a) 2-formylphenylboronic acid, Pd(PPh3)4, Na2CO3, toluene:ethanol (9:1), 100 °C, 3 h; (b) TMSCN, ZnI2, CH2Cl2, 65 °C, 4 h; (c) LiAlH4, THF, reflux, 12h; (d) CH3SO3H, 25 °C, 12h; PPA, 60 °C, 6 h.
The molecular modeling methodology used to generate the h5-HT2A receptor-ligand complex models will be described briefly here, and is discussed in detail elsewhere.12 To begin, the h5-HT2A receptor sequence was aligned using ClustalX 1.8316 with a profile of several related class A GPCRs17 that included bovine rhodopsin. The result was an unambiguous alignment (in the TM helical regions) of the h5-HT2A sequence with that of bovine rhodopsin. Manual modifications were made to the alignment in the region of the second extracellular loop to properly align the cysteine residues of the disulfide linkage. This alignment, along with a file containing the atomic coordinates of bovine rhodopsin (‘A’ chain of PDB code 1U19), was used as input to the MODELLER software package18, 19 to generate a population of 100 h5-HT2A homology models, each with a different conformation. For the template rhodopsin structure, all residues within 12 Å of the bound retinal ligand were mutated to alanine to encourage MODELLER to produce structurally diverse receptor conformations. The N- and C-termini were truncated. The third intracellular loop was modeled simply as a poly-Gly chain whose backbone coordinates were taken from the structure of rhodopsin. Each receptor in the population was subsequently energy-minimized without constraint in SYBYL 7.2 (Tripos Inc., St. Louis, MO) using the Tripos Force Field (TFF) with Gasteiger-Hückel charges, a distance-dependent dielectric constant of 4, a non-bonded interaction cutoff = 8 Å, and were terminated at an energy gradient of 0.05 kcal/(mol·Å). The automated docking program GOLD20, 21 version 3.01 (Cambridge Crystallographic Data Centre, Cambridge, UK) was then used to dock a potent 5-HT2A agonist (1-(2,5-dimethoxy-4-bromophenyl)-2-aminopropane; DOB) and a potent antagonist (ketanserin) into each of the 100 receptor models. Based on the fitness scores and quality of the docked poses, one receptor model was selected to represent the agonist binding site (Site 1 in Figure 1) and a second model was selected to represent an additional antagonist binding site (Site 2 in Figure 1). After a small amount of conformational refinement and checks for stereochemical integrity, the two receptor models were saved and used for subsequent docking exercises.
Ligand molecules were sketched in using SYBYL and energy-minimized using the same method and parameters as were used for the receptor models. Basic amines were protonated to form ammonium ions. Ligand chirality was treated explicitly, with each isomer sketched, energy-minimized and saved as a separate structure file. GOLD was then used to dock each ligand structure into each of the two selected receptor models. The parameter set defined by the “standard default settings” option was used in conjunction with a protein H-bond constraint (default settings) that biased the docked solutions in favor of those in which the ligand ammonium ion interacted with the conserved D1553.32. Ten genetic algorithm (GA) runs were performed for each ligand at both Site 1 and Site 2. Short molecular dynamics (MD) simulations were then carried out to enable the receptor-ligand complex to sample alternative locally-accessible low-energy states in order to improve the binding free energy, and to simultaneously increase the degree of receptor-ligand complementarity. The MD simulations were run at 300K for 100 ps using the TFF with assigned Gasteiger-Hückel charges, a distance-dependent dielectric constant = 4.0 and a non-bonded interaction cutoff of 8.0 Å. To maintain the structural integrity of the receptor-ligand complexes during the MD run, an aggregate was defined that constrained the atoms of all residues greater than 8.0 Å from the GOLD-docked solution to their starting coordinates. The receptor-ligand complexes were then subjected to a final energy minimization step using the same parameters as shown above. All molecular modeling was performed on MIPS R14K- and R16K-based IRIX 6.5 Silicon Graphics Fuel and Tezro workstations.
Binding assays and data analysis were performed through the NIMH Psychoactive Drug Screening Program (PDSP). The 5HT2A competitive binding assay employs [3H]ketanserin (a 5-HT2A antagonist) as the radioligand. Binding data were analyzed using Prism (GraphPad Software, Inc., San Diego, CA). Details of the binding assay protocol may be found at the PDSP home page, http://pdsp.med.unc.edu.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by United States Public Health Service Grant R01-MH57969 (RBW), R01-MH57635 (BLR), K02-MH01366 (BLR) and R01-MH61887 (BLR), U19-MH82441 (BLR) and the NIMH Psychoactive Drug Screening Program (BLR).
REFERENCES AND NOTES
- 1.Westkaemper RB, Glennon RA. Curr. Top. Med. Chem. 2002;2:575. doi: 10.2174/1568026023393741. [DOI] [PubMed] [Google Scholar]
- 2.Runyon SP, Savage JE, Taroua M, Roth BL, Glennon RA, Westkaemper RB. Bioorganic and Medicinal Chemistry Letters. 2001;11:655. doi: 10.1016/s0960-894x(01)00023-3. [DOI] [PubMed] [Google Scholar]
- 3.Westkaemper RB, Runyon SP, Bondarev ML, Savage JE, Roth BL, Glennon RA. European Journal of Pharmacology. 1999;380:R5. doi: 10.1016/s0014-2999(99)00525-7. [DOI] [PubMed] [Google Scholar]
- 4.Braden MR, Parrish JC, Naylor JC, Nichols DE. Molecular Pharmacology. 2006;70:1956. doi: 10.1124/mol.106.028720. [DOI] [PubMed] [Google Scholar]
- 5.Shapiro DA, Kristiansen K, Kroeze WK, Roth BL. Molecular Pharmacology. 2000;58:877. doi: 10.1124/mol.58.5.877. [DOI] [PubMed] [Google Scholar]
- 6.Chambers JJ, Nichols DE. J. Comput.-Aided Mol. Des. 2002;16:511. doi: 10.1023/a:1021275430021. [DOI] [PubMed] [Google Scholar]
- 7.Muntasir HA, Rashid M, Komiyama T, Kawakami J, Nagatomo T. Journal of Pharmacological Sciences. 2006;102:55. doi: 10.1254/jphs.fp0060171. [DOI] [PubMed] [Google Scholar]
- 8.Westkaemper RB, Runyon SP, Savage JE, Roth BL, Glennon RA. Bioorganic and Medicinal Chemistry Letters. 2001;11:563. doi: 10.1016/s0960-894x(01)00010-5. [DOI] [PubMed] [Google Scholar]
- 9.Dezi C, Brea J, Alvarado M, Raviña E, Masaguer CF, Loza MI, Sanz F, Pastor M. Journal of Medicinal Chemistry. 2007;50:3242. doi: 10.1021/jm070277a. [DOI] [PubMed] [Google Scholar]
- 10.Brea J, Castro M, Loza MI, Masaguer CF, Raviña E, Dezi C, Pastor M, Sanz F, Cabrero-Castel A, Galán-Rodríguez B, Fernández-Espejo E, Maldonado R, Robledo P. Neuropharmacology. 2006;51:251. doi: 10.1016/j.neuropharm.2006.03.021. [DOI] [PubMed] [Google Scholar]
- 11.Individual amino acid residues of the receptor are identified by the traditional residue identifier indicating the residue’s position in the primary amino acid sequence, followed by the general Ballesteros-Weinstein GPCR residue identifier as a superscript. See Ballesteros JA, Weinstein H. Methods Neurosci. 1995;25:366.
- 12.Runyon SP, Savage JE, Mosier PD, Roth BL, Glennon RA, Westkaemper RB. Journal of Medicinal Chemistry. submitted. [Google Scholar]
- 13.Ashwood MS, Bell LA, Houghton PG, Wright SHB. Synthesis. 1988;1988:379. [Google Scholar]
- 14.Furniss BS, Hannaford AJ, Smith PWG, Tatchell AR. Vogel’s Textbook of Practical Organic Chemistry. Harlow: Longman; 1989. [Google Scholar]
- 15.Dijks FA, Grove SJA, Carlyle IC, Thorn SN, Rae DR, Ruigt GSF, Leysen D. Patent WO/1999/18941. International Patent. 1999
- 16.Chenna R, Sugawara H, Koike T, Lopez R, Gibson TJ, Higgins DG, Thompson JD. Nucleic Acids Res. 2003;31:3497. doi: 10.1093/nar/gkg500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bissantz C, Bernard P, Hilbert M, Rognan D. Proteins. 2003;50:5. doi: 10.1002/prot.10237. [DOI] [PubMed] [Google Scholar]
- 18.Fiser A, Šali A. In: Methods in Enzymology: Macromolecular Crystallography: Part D. Carter CWJ, Sweet RM, editors. Vol. 374. 2003. p. 461. [Google Scholar]
- 19.Šali A, Blundell TL. Journal of Molecular Biology. 1993;234:779. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 20.Jones G, Willett P, Glen RC. Journal of Molecular Biology. 1995;245:43. doi: 10.1016/s0022-2836(95)80037-9. [DOI] [PubMed] [Google Scholar]
- 21.Jones G, Willett P, Glen RC, Leach AR, Taylor R. Journal of Molecular Biology. 1997;267:727. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.