

Abstract
mRNA localization is an important posttranscriptional method of targeting proteins to their site of function. The sorting of transcripts to their correct intracellular destination is achieved by a number of mechanisms, including selective degradation or transport by molecular motors along the cytoskeleton. In all cases, this involves mRNA localization signals, or so called zip codes, being recognized by trans-acting cellular factors. In a few cases, primary sequence motifs for RNA localization can be identified, but in general, localization signals operate at the level of secondary (2D) and tertiary (3D) structure. This inevitably means that searching for localization signal motifs is a complex task requiring specialist knowledge of bioinformatics. Furthermore, the publications describing these searching methods tend to be aimed at the bioinformatics community. In this review, we have surveyed the major tools for folding, comparing, and searching potential mRNA localization signals in transcripts or across genomes. Our aim is to provide an overview for biologists, who lack specialist computer and bioinformatics training, of the major RNA bioinformatics tools that are available. The examples provided are focused on mRNA localization signals and RNA stem-loop structures, however these tools can be applied to the study of any RNA signals.
Keywords: RNA Localization, Bioinformatics, Sequence Searching, Secondary structure prediction, Tertiary Structure
Introduction
mRNA localization targets transcripts to particular regions of the cytoplasm, in order to concentrate the synthesis of the proteins they encode at their site of function [1]. Localized transcripts are known in all major eukaryotic model systems and include examples encoding a wide range of types of proteins, from nuclear transcription factors, to membrane proteins and secreted signals. A variety of mechanisms of localization have been demonstrated, but perhaps the predominant mode of transcript sorting within the cell involves transport by molecular motors along the cytoskeleton. RNA cargos are known for all 3 classes of molecular motors, myosin, kinesin and dynein [2]. The process of mRNA localization is no exception. Transcripts are thought to contain localization signals [3], consisting of discrete stem loops structures that associate with a particular combination of RNA binding trans-acting factors, determining the composition of the RNP complexes and site of localization of the transcripts. This can be achieved by recruiting specific molecular motors, influencing the activity of motors [4], dictating a mode of anchoring [5] as well as promoting or preventing degradation. However, in most cases, characterizing the consensuses for RNA binding and/or the signal for localization has proven intractable. Only in rare instances is RNA binding or signals for RNA localization defined at the level of primary sequence. In the vast majority of cases, binding to proteins and signals for RNA localization are defined at the secondary and tertiary structure level. While computational approaches can predict RNA base pairing in relatively small stem loops with reasonable reliability based on the minimum free energy, this may not represent the in vivo folding of the RNA. Such programs only consider the canonical Watson-Crick base pairs (and the G:U wobble pair) and not the thirty or so other non-Watson-Crick bases pairings and triplets, as well as RNA pseudo-knots, G-quartets and other structures. Furthermore, RNA signals are recognized in three dimensions and the surfaces presented to a protein in 3D are very difficult to predict computationally. Finally, there is the added complication that the RNA may change its conformation upon binding to a protein.
This review is intended for readers who have identified a discrete RNA signal of biological significance and are wondering how they can find similar signals in other specific genes, or across multiple genomes. We aim to help the reader decide how to assess whether the signal can be searched on a primary, secondary or tertiary sequence level, and which resources are available for these tasks. We provide a quick guide through the plethora of bioinformatics methods for predicting, comparing and searching for primary, secondary and tertiary RNA structure. Throughout the review, biological examples are highlighted from the field of mRNA localization and RNA stem loop structures, but these methods and the principles described can be applied to any small RNA signal with functions in any aspect of RNA biology. Not all RNA secondary structure prediction methods are described. Instead, we cover the most powerful and commonly used algorithms. The review is divided into three major parts, each dealing with a distinct set of bioinformatics tools required to work with specific kinds of RNA localization signals. The first part addresses searching for similar RNA localizing elements by their primary sequence, and supporting secondary structure information. These types of searches are suited to examples where the localization signals are thought to be highly similar in primary sequence so conventional sequence search algorithms can be employed as well as some more specialized methods such as Stochastic Context Free Grammars (SCFG). The second part describes the majority of cases, involving the comparison of secondary structures of localizing RNA signals. In this case, there is insufficient primary sequence similarity, but sufficient secondary structure consensus for identifying further examples of the signal. The third and final section introduces some software for predicting the tertiary structure of RNAs and highlights the potential of these methods. However, these are so far limited in their utility and do not replace experimental determination of the 3D structures.
1 Primary Sequence
In a few cases, RNA localization signals are defined at the primary sequence level without requiring any secondary structure information. Similar sequences can then be searched for in sequence databases or genomes, using sequence only search methods such as the BLAST [6] suite of tools, hidden Markov models (HMM) or more specialized search programs.
1.1 Sequence Similarity Searches
In Xenopus laevis, Vg1 RNA is localized to the vegetal cortex of oocytes and has been mapped to a 340nt signal in the 3′UTR [7]. Further investigations found four repeated sequences E1, E2, E3 and E4 [8]. Deletion of the E2 sequences abolish localization, whist the deletion of the E1, E3 and E4 sequences reduce localization efficiency. Two copies of the E2 motif have been shown to be sufficient for the localization of Vg1 and the VM1 motif (UUUCAC) was found to be a critical component [9]. VegT, another vegetally localized Xenopus transcript was identified as having five copies of the E2 motif in a 440nt region of the 3′UTR. The E2 motif was found to be necessary and sufficient for the localization of VegT [10]. Sequence search methods, such as BLAST, could therefore be applied to searching for further RNAs containing multiple copies of the E2 motif. However, due to the short repeated sequence motifs a new program was developed called REPFIND [11], which was used to find further sequences containing clusters of CAC motifs. Interestingly, this led to the discovery of a conserved CAC motif in the majority of RNAs localizing to the vegetal cortex in Xenopus oocytes. In addition, the CAC motifs were found to be conserved across chordates, although their precise functions in localization vary between species [11].
1.2 Statistical Model Sequence Searches
Statistical models for sequence searching are more sensitive than sequence similarity search algorithms such as BLAST. Profile Hidden Markov models (pHMM) are probabilistic models of nucleic acid or protein sequences and are employed by the hmmer [12] and SAM-T98 [13] methods. If there are several similar sequences already known, they can be aligned and transformed into a pHMM, which can be used to search databases for other similar sequences.
Sequence searches can be greatly enhanced by including secondary structure in the search model. These searches use SCFG encoding both sequence and secondary structure into a probabilistic model, also referred to as covariance models, and can be used to find homologous RNAs in sequence databases, with similar sequences and secondary structures. RSEARCH [14] is such a method and is more sensitive than sequence based search methods. Such searches are extremely processor intensive, but with increasing computer power can now be run on genomes of modest size. SCFGs can also be used to predict the structure of a set of closely related RNAs (see the consensus structure prediction section below for more details).
1.3 Databases to Search
Most known localization signals have so far been found in the un-translated regions (UTR) [3,15], so searching databases of UTR sequences is a good starting point. UTRdb [16] is a non-redundant database of eukaryotic 3′UTR and 5′UTRs. Each entry in the database is annotated and links are provided to the genomic and/or protein data. To search for homologous RNAs across genomes, the Ensembl project [17] provides sequence data for a large number of genomes, including all common model organisms. Rfam [18] is a database of families of RNAs and the SCFGs describing them. In addition, Rfam provides a collection of putative RNAs from over 100 genomes, which can be used to search for homologous RNAs.
2 Secondary Structure
Many RNA signals have characteristic stable secondary structures, but lack any recognizable primary sequence features. In such cases, even the most sophisticated primary sequence search methods fail to identify other examples of the same motif. For example, the gurken (GLS) and I factor (ILS) localization signals have similar stable secondary structures, competing for the same cellular machinery required for localization [19]. RNA-binding proteins associate with these signals and then recruit components of the dynein motor complex. The GLS and ILS only share limited sequence similarity, so that primary sequence searches for the GLS fail to find the ILS, and vice versa.
A recent study by Rivas and Eddy for the identification of non-coding RNAs [20], concluded that the stability of RNA secondary structure alone is not sufficient to distinguish them from the folding of random sequences, indicating that there are primary, secondary and/or tertiary structure elements important for the function of these RNAs.
Several reviews have been published in the last few years on the subject of RNA secondary structure prediction [21-24]. However, these have tended to be aimed at bioinformaticians, providing data on the performance of the algorithms rather than their biological application. Reviews have also concentrated on the importance of RNA structure [25], common structural motifs [26] or hairpin structures [27].
2.1 Assumptions for RNA Secondary Structure Prediction
In general, the difficulty of applying secondary structure prediction methods is that they are based around certain assumptions about RNA structure, which may or may not be correct. For example, most secondary structure prediction methods assume Watson-Crick base pairing (including the G:U wobble pair) and ignore non-canonical base pairings, base triples, G-quartets and pseudoknots. The structure of RNA in the predictions is also assumed to be exclusively in the A-form, as described by thermodynamic models. While all current methods assume that RNA folds independently of other RNA and proteins in the cell, the more sophisticated approaches do take into account that RNA structures are dynamic and may form several conformations in vivo.
2.2 Representations of RNA Secondary Structure
The most intuitive method of representing an RNA secondary structure is as an image representing bases as single letter codes. Adjacent bases in the sequence are linked by a single line and base pairings are usually represented by coloured lines or circles (Figure 1A). However, there are other representations such as the connect (ct), dot-bracket and RNA markup language (rnaml) format. The connect format contains columns storing sequence and base pairing indices for the secondary structure. This is the format utilized by Mfold and it is from this format that the structure image and dot-plots are generated (Figure 1B). The dot-bracket notation shows paired bases as matching brackets, and non-paired bases as full stops. RNAfold utilizes this format for its secondary structure predictions and is particularly suited to RNA bioinformatics as it is easily manipulated and stored by computer programs (Figure 1C). The rnaml format is a universal method of encoding RNA secondary structure developed by the RNA structure prediction community [28]. However, the rnaml format is not suitable for reading by humans as it is designed to be read by computer programs.
Fig. 1.

Commonly used representations of RNA secondary structure for the Drosophila gurken localization signal. (A) Image format as produced by Mfold. (B) Connect Format, showing columns denoting paired bases and to which base they are paired. (C) Dot-bracket notation, where matching brackets correspond to base pairs, and dots to unpaired bases. This notation is usually accompanied by the RNA sequence to aid interpreting the notation.
2.3 Predicting the Secondary Structure of Individual RNA Sequences
Computational predictions of RNA secondary structure maximize the number of base pairs within a structure, thus minimizing the free energy in the molecule. The complexity in the methods is related to the scoring schemes and the development of more accurate methods of evaluating RNA free energy values. Despite improvements in the methods, the RNA energy models should only be regarded as approximations of the secondary structure. Secondary structure predictions depend on dynamic programming algorithms that start by predicting the optimal substructures of the whole sequence. These are then built up into a matrix, where the dynamic programming algorithm backtracks to find the lowest energy structure for the sequence as a whole. The use of dynamic programming algorithms in the prediction methods always gives the “correct” prediction, but scoring schemes can lead to inaccuracies. When a prediction with minimum free energy (MFE) is made, the structure with the lowest energy (termed optimal) is returned. Some programs also return a number of similar structures with energies close to the lowest energy structure (termed sub-optimal). Sean Eddy has written an excellent review of MFE prediction methods [21].
The most commonly used MFE method is arguably Mfold [29], but there are a wide variety of methods including RNAfold [30] and its sister version RNALfold [31] designed for performing predictions on a genomic scale. Locally stable structures of a defined length are given by folding an entire genome in overlapping windows set at the defined length, progressing along the genome, one nucleotide at a time. Mfold and RNAfold were both developed from the same Zuker-Steigler algorithm, so differences in their prediction accuracies are not significant. Figure 2A shows the MFE based methods in context with other secondary structure prediction methods.
Fig. 2.
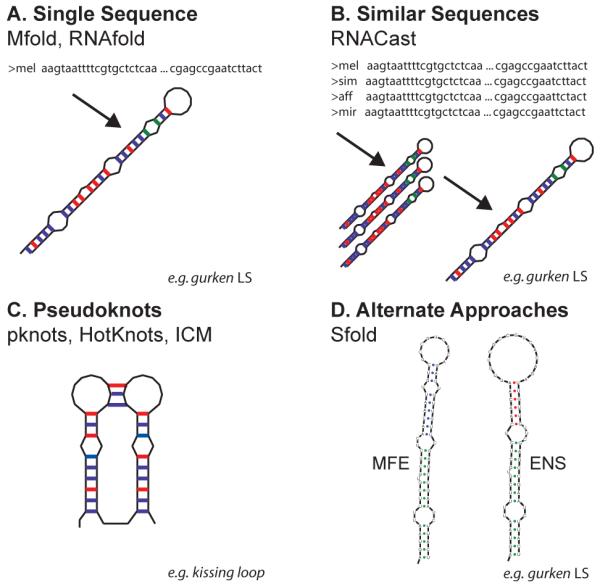
Approaches for predicting RNA secondary structure. (A) For a single sequence (for GLS in the example), the most appropriate methods to use are the MFE based programs (e.g. Mfold, RNAfold). (B) If several similar sequences are available, the most accurate secondary structure predictions are achieved through covariance methods such as RNACast and Pfold. The example shows the GLS from several Drosophilids. (C) Pseudoknot structure prediction, appropriate if experimental evidence points to pseudoknots. Pknots, Hotknots and ICM are all able to predict the secondary structure of pseudoknot-containing structures. The example shown is a kissing loop pseudoknot. (D) Alternate approaches. Programs such as Sfold take into consideration the dynamic nature of RNA and predict ensembles of structures to determine the most likely structures in vivo. ENS denotes an ensemble prediction by Sfold, compared to the MFE prediction: example shows the GLS.
2.4 Accuracy of Secondary Structure Prediction Methods
To calculate the accuracy of a secondary structure prediction method, a reference set of experimentally determined structures is used and are typically from distinct types of RNA (e.g. tRNA, rRNA). Therefore, the choice of data set greatly influences the accuracy of the algorithms, so that a single figure for accuracy can be misleading without providing the reference set used for the test. One of the most important factors in selecting a prediction algorithm is the length of the candidate RNA because some algorithms only perform well with RNAs under certain lengths. Accuracies quoted in the literature vary considerably and reflect the use of different reference sets and techniques for calculating accuracies. Mathews et al.[32] found the accuracy of Mfold to be 73% for a large database of known structures of 700nt or less. Doshi et al. [33] found an accuracy of 41% for Mfold for a reference set of 16S and 23S RNA. Dowell and Eddy [34] report accuracies of 56% (Mfold), 55% (RNAfold), 50% (pknots), and 39% (Pfold) for a reference set of RNase P, SRP and tmRNAs.
2.5 Pseudoknots Structure Prediction
There has been great interest in the development of algorithms to predict the secondary structures of RNAs containing pseudoknots, as these cannot be predicted using standard MFE methods. One such program is Pknots [35], based on standard RNA thermodynamic models, a modified dynamic programming algorithm and thermodynamic models for pseudoknots. However, pknots is only able to process small structures due to the complexity and time requirements of the algorithm. Other programs for predicting pseudoknots include, ILM [36] and HotKnots [37]. Figure 2C shows pseudoknot-capable prediction programs in context of the other classes of prediction software, using a kissing-loop as an example. The KH2 domain of the Fragile-X mental retardation protein (FMRP) competitively binds a kissing loop with brain polyribosomes [38]. It will be interesting to know whether kissing loops have a role in the binding of trans-acting factors to mRNA localization signals.
2.6 Comparative / Consensus Structure Prediction and Motif Finding
Single sequence secondary structure prediction methods can only take into consideration the information held within that one sequence. No inference can be made to the importance of each of the bases in the structure for the function of the RNA. However, if several highly similar sequences are available from related species, then a consensus structure can be determined.
RNAcast [39] (part of the RNAShapes package [40]) takes the approach of calculating the structure for each of a set of sequences independently, then calculates the structure common to all sequences. This does not require any alignment of the sequences unlike Pfold [41], which uses SCFGs to predict the secondary structure of a set of homologous RNAs, assuming a single common structure for the sequences. Sequences must be pre-aligned with programs such as Muscle [42], before Pfold can predict their common structure. More computationally intensive approaches simultaneously predict the MFE structures and perform a structural alignment. Dynalign [43] predicts the structures common to a maximum of two unaligned sequences. Foldalign [44] predicts structures common to two or more sequences and can highlight areas where there is a high degree of sequence similarity. To reduce the computational complexity, both methods implement restrictions such as limiting distances between paired bases and restrictions on the alignments. A further caveat for these methods is the requirement that the sequences be of similar length and adopt similar structures. This is of particular use when validating the prediction of an unknown structure against a known one. Figure 2B shows the consensus methods in context with the other secondary structure prediction methods.
2.7 Statistical Structure Prediction
One of the weaknesses of the MFE methods is that the correct structure is not guaranteed to be the one with the lowest energy, but is likely to be within the sub-optimal structures. A further complication is that RNA is likely to exist in a number of alternative structures in vivo due to the dynamic nature of RNA and the proteins that bind to it. Determining ensembles (families) of secondary structures is one of the focuses of statistical approaches to structure prediction.
Sfold [45] statistically samples representative secondary structures from a Boltzman probability distribution of structures. The statistical sampling provides probabilities for secondary structure motifs. Sfold then clusters the sampled structures by structural similarity, resulting in ensembles of similar structures. The MFE structure is not guaranteed to be within the sample, typically 1000 structures, but can be added artificially to the sample. The output consists of a ranked list, in terms of free energy, of ensembles representing the most likely conformations in vivo. Sfold is particularly suited to determining whether interaction sites remain accessible with variations in the conformation of the structures. The accuracy of the Sfold method was found to be similar to that of the MFE methods (Mfold and RNAfold) [46]. Statistical folding algorithms have been described in more detail in a recent review [47] and are put into context with other prediction methods in Figure 2D.
2.8 2D Structure Comparisons and Searching
RNA signals can be recognized at the structural level, with no known requirement for conservation of sequence. The evolutionary pressure is therefore applied to the maintenance of the structure and not the sequence. In these cases, sequence comparison and alignment methods will fail, and secondary structure comparisons are required. There are two main methods for comparing or calculating the similarity between two independently predicted structures. The first converts structures into a tree or forest representation, where the leaves of the tree are unpaired bases and branches denote paired bases. Once the two structures are in a tree format, the two trees can be compared using tree alignment distance algorithms. Similarities are then quantified and given scores. RNAdistance [30] performs global comparisons of RNA structures represented as trees. However, as the entire lengths of the RNAs are being compared, the algorithm can often become trapped in unimportant fine detail structure comparisons rather than the structures as a whole, leading to anomalous results. In contrast, RNAforester [48] compares structures on a local scale, so can find similarities between two substructures of the RNAs being compared. RNAforester has recently been used in a large-scale classification of a eukaryotic common secondary structure of the internal transcribed spacer 2 (ITS2) [49,50]. ITS2 is a fast evolving, nuclear ribosomal DNA recently suggested as a marker for the taxonomic classification of new species. The conserved core, as shown by RNAforester, consists of four helices forming the loop of a larger stem-loop structure (average length of 212 nt). There are also sequence constraints; a UGGU motif in helix III, a U:U mismatch in helix II. The second method of secondary structure comparison determines whether a structure matches a user defined search motif. RNAMotif [51] converts user defined structure motifs into tree representations. A recursive algorithm then determines whether a sequence, with its structure determined, matches the motif. The smallest structure elements of the motif are searched for first, followed by the larger elements. RNAMotif can be accompanied by a scoring function to quantify the similarity in addition to the pass or fail nature of the motif match. Large numbers of predicted secondary structures can be passed though RNAMotif to determine if any similar secondary structures can be found. The RNAMotif method is extremely flexible and can encode non-canonical base-pairings, triples, quadruplexes and pseudoknots. Another approach is that of ERPIN [52], which performs pattern searching using a set of similar RNAs to produce a template for searching sequence database. The set of RNAs are aligned by sequence, and common secondary structure features determined.
If the interaction sites of the RNA are known then this can be incorporated into the prediction of secondary structure. RNAhybrid [53] models the hybridization of short RNA sequences to longer target sequences, and is particularly useful in the fields of miRNA and siRNA searching. Other successful methods for searching for miRNAs have been developed by the Bartel lab [54,55].
3 Tertiary Structure
Currently the best methods of determining the tertiary structures of RNAs are NMR and X-ray crystallography. However, considerable time and resources are required. The computational modeling of tertiary structures of RNA is a field very much in its infancy, but there have been significant advances allowing tertiary structures to be modeled, investigated and searched for. RNA tertiary structure modeling is a more straightforward problem than protein structure prediction, due to the strong constraints provided by base pairing. Therefore, RNA structure predictions are tractable with reasonable computer power. Tertiary structure models produced with currently available software may lack atomic resolution of experimentally determined structures, but can give valuable insight into the topology, the RNA and the nature of its binding to proteins. Tertiary structure modeling is very useful for modeling mutations in an RNA whose wild type structure has been determined experimentally. Experimentally determined RNA structures can be found in the Nucleic Acid Database (NDB) [56] and are further organized by structure and function in the Structural Classification of RNA database (SCOR) [57].
3.1 Ab initio Structure Prediction and Molecular Dynamics
Ab initio folding methods, are based on the physical properties of RNA molecules rather than on previously determined structures (as is the case with threading approaches). Typically force fields are used to describe the atoms within the RNA molecule, and then the interactions of the atoms are simulated over a defined period of time to model the structure. In a recent study, the structure prediction of a UUUU tetraloop was investigated and compared to NMR models [58]. The computational modeling predicted by the Amber molecular dynamics program [59] was found to be in good overall agreement with the NMR model.
3.2 Threading Modeling / Partial Experimental Data
Threading is where a structural model for an RNA is built up one base at a time, by comparing its sequence to a database of experimentally determined structures. Threading methods rely heavily on structure databases and will improve as more structures are determined. MC-SYM [60] is an example of a threading program and has great potential for the refinement of threading based methods and model building.
3.3 Tertiary Structure Searching
Ash1 mRNA from S. cerevisiae has been shown to have four different localization signals [61-63]; three in coding regions (E1, E2A, E2B) and one in the 3′UTR (E3). Each of the four elements is able to localize the Ash1 mRNA to the bud of the yeast cells. Initially, no obvious consensus sequence was identified. However, a consensus of two cytosine bases was identified by their orientation in the three dimensional structure of the localization signals [64]. A stem of four or five bases is flanked by two internal loops each containing a consensus cytosine on opposite strands. One of the cytosines forms a further CGA motif within the structure, however this is not absolutely required. These cytosines are separated by six nucleotides despite the variations in the secondary structure. MC-SEARCH was then used to determine whether the four signals can form similar tertiary structures with the cytosines orientated in the same manner. MC-SEARCH was able to search existing 3D RNA databases using secondary structure definitions of the four localization signals. The search revealed numerous structures containing the four cytosine motifs, and that they consistently span 28Å, indicating that different secondary structures can still form similar tertiary structures. RNAMotif was then used to search for further localization signals in S. cerevisiae. Two mRNAs containing the motif and were found to localize to the yeast bud. Figure 3D shows this search strategy in context with the other search methods described in this review.
Fig. 3.

Search strategies for finding similar RNAs based on sequence and/or structure. Example applications are given for each of the methods. (A) If there are sequence constraints. (B) Finding homologous RNAs with sequence and secondary structure similarity. (C) Low sequence similarity with secondary structure similarity. (D) Tertiary structure constraints, where there are no obvious sequence or secondary structure similarities.
4 Conclusion
While current bioinformatics methods are most successful when based upon previously identified localization signals, genome wide searches may also aid in the elucidation of a sequence and/or secondary structure consensus for localization. One of the most obvious weaknesses of these kinds of approaches is that the folding algorithms do not take into account the binding of other RNAs and proteins. The development of folding methods that include interacting molecules will have a big impact on the field of RNA localization through the structural prediction of localization signals. Furthermore, improvements in tertiary structure prediction in the coming years are likely to have the most significant impact on the success of such searches. Tertiary structure prediction of localization signals and the structures of mutations in the signals will provide invaluable insight into the nature of the recognition of the RNA by the localization machinery.
Acknowledgments
ID is supported by a Wellcome Trust Senior Research Fellowship (067413). RSH is supported by a Medical Research Council Career Establishment Grant and an eDIKT grant from SFC to ID. We thank David Finnegan, Georgia Vendra, Eve Hartswood and David Ish-Horowicz for discussions.
References
- [1].Palacios IM, St Johnston D. Getting the message across: the intracellular localization of mRNAs in higher eukaryotes. Annu Rev Cell Dev Biol. 2001;17:569–614. doi: 10.1146/annurev.cellbio.17.1.569. [DOI] [PubMed] [Google Scholar]
- [2].Tekotte H, Davis I. Intracellular mRNA localization: motors move messages. Trends Genet. 2002;18:636–42. doi: 10.1016/s0168-9525(02)02819-6. [DOI] [PubMed] [Google Scholar]
- [3].Van de Bor V, Davis I. mRNA localisation gets more complex. Curr Opin Cell Biol. 2004;16:300–7. doi: 10.1016/j.ceb.2004.03.008. [DOI] [PubMed] [Google Scholar]
- [4].Bullock SL, Nicol A, Gross SP, Zicha D. Guidance of bidirectional motor complexes by mRNA cargoes through control of dynein number and activity. Curr Biol. 2006;16:1447–52. doi: 10.1016/j.cub.2006.05.055. [DOI] [PubMed] [Google Scholar]
- [5].Delanoue R, Davis I. Dynein anchors its mRNA cargo after apical transport in the Drosophila blastoderm embryo. Cell. 2005;122:97–106. doi: 10.1016/j.cell.2005.04.033. [DOI] [PubMed] [Google Scholar]
- [6].Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- [7].Mowry KL, Melton DA. Vegetal messenger RNA localization directed by a 340-nt RNA sequence element in Xenopus oocytes. Science. 1992;255:991–4. doi: 10.1126/science.1546297. [DOI] [PubMed] [Google Scholar]
- [8].Deshler JO, Highett MI, Schnapp BJ. Localization of Xenopus Vg1 mRNA by Vera protein and the endoplasmic reticulum. Science. 1997;276:1128–31. doi: 10.1126/science.276.5315.1128. [DOI] [PubMed] [Google Scholar]
- [9].Gautreau D, Cote CA, Mowry KL. Two copies of a subelement from the Vg1 RNA localization sequence are sufficient to direct vegetal localization in Xenopus oocytes. Development. 1997;124:5013–20. doi: 10.1242/dev.124.24.5013. [DOI] [PubMed] [Google Scholar]
- [10].Kwon S, Abramson T, Munro TP, John CM, Kohrmann M, Schnapp BJ. UUCAC- and vera-dependent localization of VegT RNA in Xenopus oocytes. Curr Biol. 2002;12:558–64. doi: 10.1016/s0960-9822(02)00740-6. [DOI] [PubMed] [Google Scholar]
- [11].Betley JN, Frith MC, Graber JH, Choo S, Deshler JO. A ubiquitous and conserved signal for RNA localization in chordates. Curr Biol. 2002;12:1756–61. doi: 10.1016/s0960-9822(02)01220-4. [DOI] [PubMed] [Google Scholar]
- [12].Eddy SR. Profile hidden Markov models. Bioinformatics. 1998;14:755–63. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- [13].Karplus K, Barrett C, Hughey R. Hidden Markov models for detecting remote protein homologies. Bioinformatics. 1998;14:846–56. doi: 10.1093/bioinformatics/14.10.846. [DOI] [PubMed] [Google Scholar]
- [14].Klein RJ, Eddy SR. RSEARCH: finding homologs of single structured RNA sequences. BMC Bioinformatics. 2003;4:44. doi: 10.1186/1471-2105-4-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].St Johnston D. Moving messages: the intracellular localization of mRNAs. Nat Rev Mol Cell Biol. 2005;6:363–75. doi: 10.1038/nrm1643. [DOI] [PubMed] [Google Scholar]
- [16].Mignone F, Grillo G, Licciulli F, Iacono M, Liuni S, Kersey PJ, et al. UTRdb and UTRsite: a collection of sequences and regulatory motifs of the untranslated regions of eukaryotic mRNAs. Nucleic Acids Res. 2005;33:D141–6. doi: 10.1093/nar/gki021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Birney E, Andrews TD, Bevan P, Caccamo M, Chen Y, Clarke L, et al. An overview of Ensembl. Genome Res. 2004;14:925–8. doi: 10.1101/gr.1860604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Griffiths-Jones S, Bateman A, Marshall M, Khanna A, Eddy SR. Rfam: an RNA family database. Nucleic Acids Res. 2003;31:439–41. doi: 10.1093/nar/gkg006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Van De Bor V, Hartswood E, Jones C, Finnegan D, Davis I. gurken and the I factor retrotransposon RNAs share common localization signals and machinery. Dev Cell. 2005;9:51–62. doi: 10.1016/j.devcel.2005.04.012. [DOI] [PubMed] [Google Scholar]
- [20].Rivas E, Eddy SR. Secondary structure alone is generally not statistically significant for the detection of noncoding RNAs. Bioinformatics. 2000;16:583–605. doi: 10.1093/bioinformatics/16.7.583. [DOI] [PubMed] [Google Scholar]
- [21].Eddy SR. How do RNA folding algorithms work? Nat Biotechnol. 2004;22:1457–8. doi: 10.1038/nbt1104-1457. [DOI] [PubMed] [Google Scholar]
- [22].Mathews DH. Revolutions in RNA secondary structure prediction. J Mol Biol. 2006;359:526–32. doi: 10.1016/j.jmb.2006.01.067. [DOI] [PubMed] [Google Scholar]
- [23].Reeder J, Hochsmann M, Rehmsmeier M, Voss B, Giegerich R. Beyond Mfold: recent advances in RNA bioinformatics. J Biotechnol. 2006;124:41–55. doi: 10.1016/j.jbiotec.2006.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Mathews DH, Turner DH. Prediction of RNA secondary structure by free energy minimization. Curr Opin Struct Biol. 2006;16:270–8. doi: 10.1016/j.sbi.2006.05.010. [DOI] [PubMed] [Google Scholar]
- [25].Holbrook SR. RNA structure: the long and the short of it. Curr Opin Struct Biol. 2005;15:302–8. doi: 10.1016/j.sbi.2005.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Hendrix DK, Brenner SE, Holbrook SR. RNA structural motifs: building blocks of a modular biomolecule. Q Rev Biophys. 2005;38:221–43. doi: 10.1017/S0033583506004215. [DOI] [PubMed] [Google Scholar]
- [27].Svoboda P, Cara AD. Hairpin RNA: a secondary structure of primary importance. Cell Mol Life Sci. 2006;63:901–8. doi: 10.1007/s00018-005-5558-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Waugh A, Gendron P, Altman R, Brown JW, Case D, Gautheret D, et al. RNAML: a standard syntax for exchanging RNA information. Rna. 2002;8:707–17. doi: 10.1017/s1355838202028017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Zuker M. Calculating nucleic acid secondary structure. Curr Opin Struct Biol. 2000;10:303–10. doi: 10.1016/s0959-440x(00)00088-9. [DOI] [PubMed] [Google Scholar]
- [30].Hofacker IL. Vienna RNA secondary structure server. Nucleic Acids Res. 2003;31:3429–31. doi: 10.1093/nar/gkg599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Hofacker IL, Priwitzer B, Stadler PF. Prediction of locally stable RNA secondary structures for genome-wide surveys. Bioinformatics. 2004;20:186–90. doi: 10.1093/bioinformatics/btg388. [DOI] [PubMed] [Google Scholar]
- [32].Mathews DH, Disney MD, Childs JL, Schroeder SJ, Zuker M, Turner DH. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc Natl Acad Sci U S A. 2004;101:7287–92. doi: 10.1073/pnas.0401799101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Doshi KJ, Cannone JJ, Cobaugh CW, Gutell RR. Evaluation of the suitability of free-energy minimization using nearest-neighbor energy parameters for RNA secondary structure prediction. BMC Bioinformatics. 2004;5:105. doi: 10.1186/1471-2105-5-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Dowell RD, Eddy SR. Evaluation of several lightweight stochastic context-free grammars for RNA secondary structure prediction. BMC Bioinformatics. 2004;5:71. doi: 10.1186/1471-2105-5-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Rivas E, Eddy SR. A dynamic programming algorithm for RNA structure prediction including pseudoknots. J Mol Biol. 1999;285:2053–68. doi: 10.1006/jmbi.1998.2436. [DOI] [PubMed] [Google Scholar]
- [36].Ruan J, Stormo GD, Zhang W. An iterated loop matching approach to the prediction of RNA secondary structures with pseudoknots. Bioinformatics. 2004;20:58–66. doi: 10.1093/bioinformatics/btg373. [DOI] [PubMed] [Google Scholar]
- [37].Ren J, Rastegari B, Condon A, Hoos HH. HotKnots: heuristic prediction of RNA secondary structures including pseudoknots. Rna. 2005;11:1494–504. doi: 10.1261/rna.7284905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Darnell JC, Fraser CE, Mostovetsky O, Stefani G, Jones TA, Eddy SR, et al. Kissing complex RNAs mediate interaction between the Fragile-X mental retardation protein KH2 domain and brain polyribosomes. Genes Dev. 2005;19:903–18. doi: 10.1101/gad.1276805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Reeder J, Giegerich R. Consensus shapes: an alternative to the Sankoff algorithm for RNA consensus structure prediction. Bioinformatics. 2005;21:3516–23. doi: 10.1093/bioinformatics/bti577. [DOI] [PubMed] [Google Scholar]
- [40].Steffen P, Voss B, Rehmsmeier M, Reeder J, Giegerich R. RNAshapes: an integrated RNA analysis package based on abstract shapes. Bioinformatics. 2006;22:500–3. doi: 10.1093/bioinformatics/btk010. [DOI] [PubMed] [Google Scholar]
- [41].Knudsen B, Hein J. Pfold: RNA secondary structure prediction using stochastic context-free grammars. Nucleic Acids Res. 2003;31:3423–8. doi: 10.1093/nar/gkg614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Mathews DH. Predicting a set of minimal free energy RNA secondary structures common to two sequences. Bioinformatics. 2005;21:2246–53. doi: 10.1093/bioinformatics/bti349. [DOI] [PubMed] [Google Scholar]
- [44].Gorodkin J, Heyer LJ, Stormo GD. Finding the most significant common sequence and structure motifs in a set of RNA sequences. Nucleic Acids Res. 1997;25:3724–32. doi: 10.1093/nar/25.18.3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ding Y, Lawrence CE. A statistical sampling algorithm for RNA secondary structure prediction. Nucleic Acids Res. 2003;31:7280–301. doi: 10.1093/nar/gkg938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Gardner PP, Giegerich R. A comprehensive comparison of comparative RNA structure prediction approaches. BMC Bioinformatics. 2004;5:140. doi: 10.1186/1471-2105-5-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Ding Y. Statistical and Bayesian approaches to RNA secondary structure prediction. Rna. 2006;12:323–31. doi: 10.1261/rna.2274106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Hochsmann M, Toller T, Giegerich R, Kurtz S. Local similarity in RNA secondary structures; Proc IEEE Comput Soc Bioinform Conf; 2003; pp. 159–68. 2. [PubMed] [Google Scholar]
- [49].Schultz J, Maisel S, Gerlach D, Muller T, Wolf M. A common core of secondary structure of the internal transcribed spacer 2 (ITS2) throughout the Eukaryota. Rna. 2005;11:361–4. doi: 10.1261/rna.7204505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Schultz J, Muller T, Achtziger M, Seibel PN, Dandekar T, Wolf M. The internal transcribed spacer 2 database--a web server for (not only) low level phylogenetic analyses. Nucleic Acids Res. 2006;34:W704–7. doi: 10.1093/nar/gkl129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Macke TJ, Ecker DJ, Gutell RR, Gautheret D, Case DA, Sampath R. RNAMotif, an RNA secondary structure definition and search algorithm. Nucleic Acids Res. 2001;29:4724–35. doi: 10.1093/nar/29.22.4724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Lambert A, Fontaine JF, Legendre M, Leclerc F, Permal E, Major F, et al. The ERPIN server: an interface to profile-based RNA motif identification. Nucleic Acids Res. 2004;32:W160–5. doi: 10.1093/nar/gkh418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Rehmsmeier M, Steffen P, Hochsmann M, Giegerich R. Fast and effective prediction of microRNA/target duplexes. Rna. 2004;10:1507–17. doi: 10.1261/rna.5248604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Jones-Rhoades MW, Bartel DP. Computational identification of plant microRNAs and their targets, including a stress-induced miRNA. Mol Cell. 2004;14:787–99. doi: 10.1016/j.molcel.2004.05.027. [DOI] [PubMed] [Google Scholar]
- [55].Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003;115:787–98. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- [56].Berman HM, Olson WK, Beveridge DL, Westbrook J, Gelbin A, Demeny T, et al. The nucleic acid database. A comprehensive relational database of three-dimensional structures of nucleic acids. Biophys J. 1992;63:751–9. doi: 10.1016/S0006-3495(92)81649-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Tamura M, Hendrix DK, Klosterman PS, Schimmelman NR, Brenner SE, Holbrook SR. SCOR: Structural Classification of RNA, version 2.0. Nucleic Acids Res. 2004;32:D182–4. doi: 10.1093/nar/gkh080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Koplin J, Mu Y, Richter C, Schwalbe H, Stock G. Structure and dynamics of an RNA tetraloop: a joint molecular dynamics and NMR study. Structure. 2005;13:1255–67. doi: 10.1016/j.str.2005.05.015. [DOI] [PubMed] [Google Scholar]
- [59].Case DA, Cheatham TE, 3rd, Darden T, Gohlke H, Luo R, Merz KM, Jr., et al. The Amber biomolecular simulation programs. J Comput Chem. 2005;26:1668–88. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Gendron P, Lemieux S, Major F. Quantitative analysis of nucleic acid three-dimensional structures. J Mol Biol. 2001;308:919–36. doi: 10.1006/jmbi.2001.4626. [DOI] [PubMed] [Google Scholar]
- [61].Chartrand P, Meng XH, Huttelmaier S, Donato D, Singer RH. Asymmetric sorting of ash1p in yeast results from inhibition of translation by localization elements in the mRNA. Mol Cell. 2002;10:1319–30. doi: 10.1016/s1097-2765(02)00694-9. [DOI] [PubMed] [Google Scholar]
- [62].Chartrand P, Meng XH, Singer RH, Long RM. Structural elements required for the localization of ASH1 mRNA and of a green fluorescent protein reporter particle in vivo. Curr Biol. 1999;9:333–6. doi: 10.1016/s0960-9822(99)80144-4. [DOI] [PubMed] [Google Scholar]
- [63].Gonzalez I, Buonomo SB, Nasmyth K, von Ahsen U. ASH1 mRNA localization in yeast involves multiple secondary structural elements and Ash1 protein translation. Curr Biol. 1999;9:337–40. doi: 10.1016/s0960-9822(99)80145-6. [DOI] [PubMed] [Google Scholar]
- [64].Olivier C, Poirier G, Gendron P, Boisgontier A, Major F, Chartrand P. Identification of a conserved RNA motif essential for She2p recognition and mRNA localization to the yeast bud. Mol Cell Biol. 2005;25:4752–66. doi: 10.1128/MCB.25.11.4752-4766.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]