

Abstract
To optimize the antitumor activity of oncrasin-1, a small molecule RNA polymerase II inhibitor, we evaluated 69 oncrasin-1 analogues for their cytotoxic activity against normal human epithelial cells and K-Ras mutant tumor cells. About 40 of those compounds were as potent as or more potent than oncrasin-1 in tumor cells and had minimal cytotoxic effect on normal cells. Structure-activity relationship analysis revealed that most of the active compounds contained either a hydroxymethyl group or an aldehyde group as a substitute at the 3-position of the indole. Both electron-donating and electron-withdrawing groups in the benzene ring were well tolerated. The hydroxymethyl compounds ranged from equipotent with to 100 times as potent as the corresponding aldehyde compounds. We tested 3 active analogues’ effect on RNA polymerase phosphorylation and found that they all inhibited phosphorylation of the C-terminal domain of RNA polymerase II, suggesting that the active compounds might act through the same mechanisms as oncrasin-1.
Keywords: RNA polymerase II inhibitors, cancer, neoplasm, synthetic lethality, indole
INTRODUCTION
Genetic and epigenetic changes in cancer cells that lead to functional alterations in several key signaling pathways are believed to be the driving force behind oncogenesis and malignant evolution of cancers.1, 2 Those functional changes in signaling pathways have recently been exploited for the development of anticancer therapeutics. Small molecules directly inhibiting critical nodes in oncogenic signaling networks—such as erlotinib and gefitinib against epidermal growth factor receptor,3 imatinib against BCR-Abl fusion protein,4 and sorafenib against Raf kinase and vascular endothelial growth factor receptor 5—have already been successfully used for treatment of various cancers in humans. Small molecules that selectively induce cell death through synthetic lethality in a subset of deregulated oncogene- or mutant tumor suppressor gene-transformed malignant cells but not in their normal counterparts have also been exploited as tumor-selective anticancer agents.6–8 Evidence has demonstrated that functional changes caused by gain-of-function mutations in oncogenes or loss-of-function mutations in tumor suppressor genes may alter cellular signaling or metabolism, rendering the mutant cells more susceptible to a functional change in another gene or rendering the mutant cells susceptible to cell death due to synthetic lethality.9
The advantage of synthetic lethality-mediated anticancer therapy is that it provides lethal selectivity against cancer cells and the opportunity to eliminate malignant cells by indirectly targeting cancer-driving molecules that otherwise cannot be targeted. For example, defects in BRCA1 and BRCA2 genes in some breast and ovary cancer cells make them highly sensitive to small molecule inhibitors of poly (ADP-ribose) polymerase.10, 11 Similarly, oncogene-transformed cells, such as c-Myc-transformed cells, were reported to be more sensitive than their normal counterparts to RNA polymerase II inhibitors, regardless of p53 status, suggesting that RNA polymerase II may serve as a therapeutic target for anticancer therapy.12 Indeed, anticancer agents such as the pan-cyclin-dependent kinase (CDKa) inhibitors flavopiridol and seliciclib elicit their antitumor activity by inhibiting RNA polymerase II function,13–15 leading to suppression of the expression of short-lived antiapoptotic proteins such as Mcl-1, XIAP, Bcl-XL, and survivin.16–18 Taken together, these findings indicate that agents that selectively inhibit RNA polymerase II function will likely be safe and tolerable and may have anticancer efficacy.
We recently reported a novel anticancer agent, designated oncrasin-1, that was identified through synthetic lethality screening on isogenic human ovarian epithelial cells with or without oncogenic Ras expression.8 Molecular characterization revealed that oncrasin-1 induced apoptosis in a subset of cancer cell lines associated with induction of co-aggregation of protein kinase Cι and RNA splicing factors in the nucleus and suppression of phosphorylation of the C-terminal domain (CTD) of the largest subunit of eukaryotic RNA polymerase II.19 CTD phosphorylation is known to be essential for efficient transcription elongation and recruitment of mRNA processing factors, including the capping enzyme and splicing factors required for efficient processing of RNA transcripts.20–26 Inhibiting CTD phosphorylation will ultimately disrupt RNA polymerase II function and promote cell death.16
Compounds identified by library screening often need to be optimized before their possible clinical application can be explored. To optimize oncrasin-1, we have synthesized several oncrasin-1 analogues and derivatives and tested their antitumor activity and selectivity in cultured cell lines. Here we report the structure-activity relationships of these oncrasin-1 analogues and their derivatives.
RESULTS
Synthesis of oncrasin-1 analogues and derivatives
The lead compound, oncrasin-1, was previously reported to induce antitumor activity in a subset of cancer cells harboring K-Ras mutations and to inhibit phosphorylation of the CTD of RNA polymerase II; 8, 19 oncrasin-1 contains an indole structure similar to indole-3-carbinol and a benzyl ring attached to the N atom of the indole. Indole-3-carbinol is a natural constituent of cruciferous vegetables that has been tested for cancer prevention and therapy.27, 28 However, in our initial studies, we found that indole-3-carbinol did not have observable antitumor activity in cancer cells that were highly susceptible to oncrasin-1,8 suggesting that a benzyl group linked to the indole is required for the antitumor activity observed in oncrasin-1. Therefore, most analogues synthesized and tested here contained an indole structure with a benzyl group linked to the N atom of the indole. Occasionally, other molecules were used to replace the benzyl group in the reaction.
The general approaches for the syntheses of oncrasin-1 analogues are presented in Scheme 1. Briefly, indole-3-carboxaldehyde or its analogues were reacted with a benzyl halide under alkali catalysts at room temperature. The products, which were purified on a silica gel or a neutral aluminum C18 column with a CombiFlash Rf-200 system (Teledyne), were either tested for antitumor activity or reduced, oxidized, or otherwise modified to obtain various derivatives as listed in Table 1. According to the substitutes on the 3-position of the indole ring, the derivatives can be subgrouped as carboxaldehyde, methanol, hydroxyalkyl, carboxylic acid, ester, amine, amide, and others. The purity and molecular weight of the final products were determined by high-performance liquid chromatography-mass spectrometry (HPLC/MS/UV230). The chemical structures of the final products were confirmed by nuclear magnetic resonance (NMR) spectrum analyses. For compounds that had purity ≥ 95%, antitumor activity was tested in cultured cells.
Scheme 1.

General approaches for the syntheses of oncrasin-1 analogues. Each synthesis step is described in detail in the Experimental Section.
Table 1.
Analogues and derivatives of oncrasin-1 and their antitumor activities (IC50, μM)
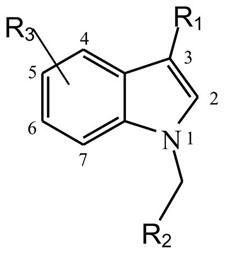 | ||||||
|---|---|---|---|---|---|---|
| Compounds | R1 | R2 | R3 Indole Ring | T29 Cells μM | T29Kt1Cells μM | H460 Cells μM |
| Oncrasin−1 | CHO | 4′-Cl Ph | H | >31.6 | 2.51 | 0.25 |
| 1 | CHO | 2′-F Ph | H | >31.6 | 0.31 | 0.1 |
| 2 | CHO | 3′-F Ph | H | >31.6 | 2.63 | 0.031 |
| 3 | CHO | 3′-Cl Ph | H | >31.6 | 0.37 | 0.037 |
| 4 | CHO | 3′-Br Ph | H | >31.6 | 1.58 | 0.039 |
| 5 | CHO | 4′-Br Ph | H | >31.6 | 0.25 | 0.031 |
| 6 | CHO | 3′-CF3Ph | H | >31.6 | 1.78 | 0.14 |
| 7 | CHO | 4′-CF3Ph | H | >31.6 | >31.6 | 1.58 |
| 8 | CHO | 3′-MePh | H | >31.6 | 6.3 | 0.039 |
| 9 | CHO | 4′-MePh | H | >31.6 | 3.98 | 0.063 |
| 10 | CHO | 3′-NO2Ph | H | >31.6 | 0.50 | 0.039 |
| 11 | CHO | 4′-NO2 Ph | H | >31.6 | 10.0 | 0.50 |
| 12 | CHO | 2′,4′-2Cl Ph | H | >31.6 | 1.58 | 0.1 |
| 13 | CHO | 3′,4′-2Br Ph | H | >31.6 | 0.63 | 0.063 |
| 14 | CHO | Ph | H | >31.6 | 5.0 | 0.045 |
| 15 | CHO | 4′-Cl (CH2)2OPh | H | >31.6 | 4.89 | 0.045 |
| 16 | CHO | 3′-Cl Ph | 5-F | >31.6 | 1.5 | 0.37 |
| 17 | CHO | 4′-Cl Ph | 5-F | >31.6 | 9.09 | 0.29 |
| 18 | CHO | 4′-Cl Ph | 6-F | >31.6 | 9.19 | 0.12 |
| 19 | CHO | 4′-Cl Ph | 5-Cl | >31.6 | 10 | 0.4 |
| 20 | CHO | 4′-Cl Ph | 6-Br | 21 | 32 | 0.09 |
| 21 | CHO | 4′-Cl Ph | 2-O, 6-Cl | 18.6 | 18.2 | 18.2 |
| 22 | CHO | CH (CH3)2 | H | >31.6 | >31.6 | 1.99 |
| 23 | H | Ph | H | >31.6 | >31.6 | >31.6 |
| 24 | H | 4′-Cl Ph | 2-CH2OH, 5-F | 17.4 | 15.1 | 11.5 |
| 25 | CN | 3′-Cl Ph | H | >31.6 | >31.6 | >31.6 |
| 26 | COCH3 | Ph | H | >31.6 | >31.6 | 10 |
| 27 | COCH3 | 2′-Cl Ph | H | >31.6 | >31.6 | >31.6 |
| 28 | COCH3 | 3′-Cl Ph | H | >31.6 | >31.6 | >31.6 |
| 29 | COCH3 | 4′-Cl Ph | H | >31.6 | >31.6 | >31.6 |
| 30 | CH2OH | Ph | H | 14.5 | 0.03 | 0.112 |
| 31 | CH2OH | 3′-F Ph | H | >31.6 | 0.02 | 0.04 |
| 32 | CH2OH | 4′-F Ph | H | 17.0 | 0.03 | 0.118 |
| 33 | CH2OH | 2′-Cl Ph | H | >31.6 | 2.51 | 0.10 |
| 34 | CH2OH | 3′-Cl Ph | H | >31.6 | 0.16 | 0.019 |
| 35 | CH2OH | 4′-Cl Ph | H | >31.6 | 0.079 | 0.016 |
| 36 | CH2OH | 3′-Br Ph | H | >31.6 | 0.032 | 0.028 |
| 37 | CH2OH | 4′-Br Ph | H | >31.6 | 0.39 | 0.031 |
| 38 | CH2OH | 3′-I Ph | H | >31.6 | 0.02 | 0.1 |
| 39 | CH2OH | 3′-CF3 Ph | H | >31.6 | 0.56 | 0.039 |
| 40 | CH2OH | 4′-CF3 Ph | H | >31.6 | 1.25 | 1.25 |
| 41 | CH2OH | 3′-Me Ph | H | >31.6 | 1.00 | 0.031 |
| 42 | CH2OH | 4′-Me Ph | H | >31.6) | 0.15 | 0.031 |
| 43 | CH2OH | 3′-NO2 Ph | H | >31.6 | 0.39 | 0.039 |
| 44 | CH2OH | 4′-NO2 Ph | H | >31.6 | 0.15 | 0.079 |
| 45 | CH2OH | 4′-OMe Ph | H | >31.6 | 0.063 | 0.079 |
| 46 | CH2OH | 2′,4′-2Cl Ph | H | >31.6 | 1.58 | 0.079 |
| 47 | CH2OH | 3′,4′-2Cl Ph | H | >31.6 | 0.31 | 0.031 |
| 48 | CH2OH | 3′,4′-2Br Ph | H | >31.6 | 0.10 | 0.05 |
| 49 | CH2OH | H | 5-F | >31.6 | 13.8 | 16.9 |
| 50 | CH2OH | 3′-F Ph | 5-Cl | 10.0 | 0.16 | 0.07 |
| 51 | CH2OH | 4′-Cl Ph | 5-F | 8.12 | 0.08 | 0.03 |
| 52 | CH2OH | 4′-Cl Ph | 6-F | >31.6 | 1.00 | 0.45 |
| 53 | CH2OH | 4′-Cl Ph | 5-Cl | >31.6 | 13.0 | 5.75 |
| 54 | CH2OH | 4′-Cl Ph | 5-OMe | >31.6 | 12.5 | 1.51 |
| 55 | CH2OH | 4′-Me Ph | 5-F | 11.7 | 0.03 | 0.01 |
| 56 | CH2CH2OH | 4′-Cl Ph | H | >31.6 | >31.6 | >31.6 |
| 57 | CH2CH2CH2OH | Ph | H | >31.6 | >31.6 | >31.6 |
| 58 | C(OH)HCH3 | 4′-Cl Ph | H | >31.5 | 19.9 | 25.1 |
| 59 | CH2OCH3 | 4′-Cl Ph | H | >31.6 | 7.94 | 1.99 |
| 60 |
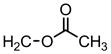
|
4′-Cl Ph | H | >31.6 | 0.063 | 0.039 |
| 61 |
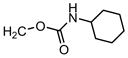
|
4′-Cl Ph | H | >31.6 | 0.2 | 0.02 |
| 62 | COOH | Ph | H | >31.6 | >31.6 | >31.6 |
| 63 | COOH | 4′-Cl Ph | H | >31.6 | >31.6 | >31.6 |
| 64 | COOCH3 | 4′-Cl Ph | H | 17.7 | 25.1 | 31.6 |
| 65 | COOCH3 | 4′-Br Ph | H | 5.00 | 6.30 | 5.00 |
| 66 | CH3 | 4′-Cl Ph | H | >31.6 | >31.6 | >31.6 |
| 67 |
|
4′-Cl Ph | H | 31.6 | 5.01 | 0.10 |
| 68 |
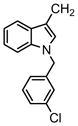
|
3′-Cl Ph | >31.6 | >31.6 | >31.6 | |
| 69 |
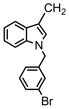
|
3′-Br Ph | >31.6 | >31.6 | >31.6 | |
Antitumor activity and selectivity
Oncrasin-1 was originally identified through synthetic lethality screening on isogenic T29 immortalized normal human ovary epithelial cells and a T29 tumorigenic subclone derived from transformation with mutant K-Ras (T29Kt1).8, 29 Later testing showed that several lung cancer cell lines harboring K-Ras mutations were highly sensitive to oncrasin-1, including H460.8 Therefore, we used T29, T29Kt1, and H460 cell lines to evaluate the cytotoxicity and cancer-cell selectivity of oncrasin-1 analogues and derivatives. Cells were treated with each compound at doses ranging from 10−4.5 M (31.6 μM) to 10−8 M (10 nM) for 3 days, and cell viability was determined by using a sulforhodamine B assay. Cells treated with 1% dimethyl sulfoxide were used as controls. The dose that caused a 50% reduction in the number of surviving cells compared with the number of surviving cells in the control group (the IC50 value) was then calculated for each compound tested.
As shown in Table 1, the IC50 values for 70 compounds (including oncrasin-1) tested on T29, T29Kt1, and H460 cells ranged from 0.01 μM to >31 μM. About 40 compounds showed antiproliferative activity in T29Kt1 and H460 cells with IC50 values at low-micromole (≤5 μM) or nanomole levels (Fig. 1). For most of those approximately 40 active compounds, the IC50 for T29 cells was greater than 31.6 μM, the highest concentration tested, suggesting that those compounds were relatively nontoxic for T29 cells and, like oncrasin-1, were active against both H460 cells and T29Kt1 cells. In a few cases, however, a compound was active against H460 cells but not active against T29Kt1 cells. Most of the active compounds had IC50 values less than or equivalent to those of oncrasin-1, which had IC50 values of >31 μM for T29 cells, 2.51 μM for T29Kt1 cells, and 0.25 μM for H460 cells.
Figure 1.

IC50 values of oncrasin-1 and its analogues in T29, T29Kt1, and H460 cells. All IC50 values of ≥31.6 μM were recorded as 31.6 μM.
Structure-activity relationships
A structure-activity relationship analysis based on IC50 values (Table 1) showed that variations in substitutions of the benzyl group did not have a dramatic impact on the cytotoxicity and selectivity of the oncrasin-1 analogues and derivatives. Both electron-donating and electron-withdrawing groups were well tolerated. Various substitutions of the benzyl group may have changed the IC50 value in H460 and T29Kt1 cells; however, the changes were quantitative rather than qualitative. Replacement of the benzyl group with an isopropyl group (compound 22) resulted in complete or partial loss of activity in H460 and T29Kt1 cells. On the other hand, the linker between the benzyl group and the 1-H indole was not very strict. Although most of the compounds tested had one carbon atom between the indole and the benzene group, a compound with the linker of -(CH2)2-O-(compound 15) retained its activity and selectivity for H460 and T29Kt1 cells. Those results suggested that an aryl substitute at the 1-position of the 1-H indole is preferred for antitumor activity, although the linker between the benzene ring and the 1-H indole is not very strict.
Most of the active compounds contained either an aldehyde group or a hydroxymethyl group as a substitute at the 3-position of the indole. In general, a compound with 3-methanol was more active than its 3-carbaldehyde counterpart. In H460 cells, most hydroxymethyl derivatives (compounds 30–55) were equipotent with to 6 times as potent as the corresponding aldehyde compounds (compounds 1–22), whereas in T29K cells, most hydroxymethyl derivatives were 1 to 100 times as potent as the corresponding aldehyde compounds (Fig. 1). These trends were not dependent on the site or nature (electron donating or withdrawing) of substitution on the benzyl group at the 1-position of indole. Substituting R1 with a ketone (compounds 26–29), a secondary alcohol (compound 58), a methyl (compound 66), or a carboxylic acid (compounds 62 and 63) led to dramatic reduction or complete loss of activity. Moreover, extension of the R1 hydroxymethyl chain by one carbon (compound 56) or two carbons (compound 57) also resulted in dramatic reduction or complete loss of activity in both H460 and T29Kt1 cells, suggesting that the substitute in the indole-3 position is critical for the antitumor activity of this group of compounds. Methylation of the primary alcohol also resulted in a dramatic loss of activity (compound 59). Nevertheless, acylation of the hydroxymethyl group (compound 60) produced a compound equipotent to compound 35.
The effects of substitutes in other positions of the indole varied, depending on the position and substitute. Substitution of the indole at the 5-position or 6-position (compounds 51–55) was well tolerated; 5-F substitution (compounds 51 and 55) produced the best potency.
Stability of hydroxymethyl compounds
We found that hydroxymethyl compounds were not stable under acidic conditions. At pH 2.0, a large portion of those compounds formed dimers within about 30 minutes (Fig. 2). A similar dimer formation at an acidic condition has been reported for indole-3-carbinol,30 which is major part of hydroxymethyl compounds tested here. Nevertheless, hydroxymethyl compounds were stable at pH 4, 6, 7.4 and 9. In contrast, aldehyde compounds were stable at both pH 2.0 and pH 7.4. We synthesized compounds 68 and 69 and evaluated antitumor activity of the dimers. The results showed that dimerization of hydroxymethyl compounds led to complete loss of antitumor activity (compounds 68 and 69). These findings suggested that a specific formulation, such as an enteric-coated formulation, would be required for orally administered hydroxymethyl compounds to be active in vivo.
Figure 2.

Stability of a representative hydroxymethyl analogue at different pH. A) Results of HPLC analyses after incubation of compound 34 at pH 2.0 and pH 7.4 for the indicated times. B) Structure of the tested compound (compound 34) and its dimerized form (compound 68). C) Percentage of compound 34 remaining in solution after incubation at pH 2.0 – pH9 for the indicated times. D) Levels of compound 34 in T29 and T29Kt1 cells overtimes after treatment with 10 μM of compound 34.
We also analyzed whether selective cell killing by an active compound is caused by a difference in bioavailability between sensitive and resistance cell lines. For this purpose, we treated T29 and T29Kt1 cells with 10 μM of compound 34. Because T29Kt1 cells started to die at about 6 h after the treatment, we analyzed cellular content of compound 34 overtime up to 5 h after the treatment. The result showed that, at all time points tested, the levels of compound 34 in T29 cells were twice as that in T29Kt1 cells (Fig. 2D). This result suggested that the resistance observed in T29 cells was not caused by low bioavailability or metabolic instability of the compound in the cells.
Effect on phosphorylation of the CTD of RNA polymerase II
Our initial mechanistic characterization on oncrasin-1-induced antitumor activity revealed that oncrasin-1 suppressed phosphorylation of the CTD of the largest subunit of RNA polymerase II in sensitive cancer cell lines but not in resistant cancer cell lines.19 Phosphorylation of the CTD is essential for efficient mRNA transcription processing, including capping and splicing.20–26 Inhibition of CTD phosphorylation is reported to promote cell death in transformed cells and cancer cells.12, 16
To determine whether active oncrasin-1 derivatives also inhibited CTD phosphorylation, we tested the effect of three active oncrasin-1 analogues (compounds 34, 35, and 50) in two oncrasin-1-sensitive (H157 and H460) and two oncrasin-1-resistant (H1299 and H322) lung cancer cell lines. The effect of these three active compounds on cell viability at 3 days after treatment is shown in Figure 3A. Both H157 and H460 cells were susceptible to the three oncrasin-1 analogues; IC50 values were ≤0.2 μM. In contrast, both H1299 and H322 cells were resistant to the oncrasin-1 analogues; IC50 values were ≥13 μM. Analysis of CTD phosphorylation of cells treated with the oncrasin-1 analogues at a concentration of 1 μM for 12 hours showed that all three compounds dramatically suppressed CTD phosphorylation in H157 and H460 cells but not in H1299 and H322 cells (Fig. 3B). These results demonstrated that the active oncrasin-1 analogues, like oncrasin-1, effectively suppressed CTD phosphorylation in sensitive cancer cell lines but not in resistant cancer cell lines.
Figure 3.

Antitumor activity of three active oncrasin-1 analogues, compounds 34, 35, and 50, in lung cancer cell lines. A) Dose-dependent antitumor activity. Four lung cancer cell lines (H460, H157, H1299, and H322) were treated with various concentrations of the compounds ranging from 0.031 μM to 31 μM. Cell viability was determined 3 days after treatment by using sulforhodamine B assays. Control cells were treated with solvent (dimethyl sulfoxide), and their value was set as 1. The values shown are the means from a quadruplicate assay. B) Western blot analysis. Lung cancer cells were treated with the compounds at a concentration of 1 μM for 24 hours. Phosphorylation of the CTD or the largest subunit of RNA polymerase II was determined with H5 pPol II antibody. β-actin was used as the loading control. C, control; T, treatment.
DISCUSSION AND CONCLUSION
We evaluated 69 oncrasin-1 analogues for their antitumor activity and selectivity in normal and cancerous cell lines. Our results showed that about 40 compounds had antitumor activity similar to or greater than that of oncrasin-1 in H460 lung cancer cells and T29Kt1 ovarian cancer cells. Most of those active compounds also retained their selectivity against tumorigenic H460 and T29Kt1 cells compared with the normal ovarian cell line T29. Our results suggest that these oncrasin-1 analogues and derivatives represent a group of small molecules that have an antitumor spectrum similar to that of oncrasin-1 and possibly molecular mechanisms similar to that of oncrasin-1. Most active compounds contain either a hydroxymethyl group or an aldehyde group at the indole-3-position, suggesting that the active compounds may interact with some cellular nucleophiles to induce antitumor activity. Analysis on intracellular levels of compound 34 in T29 and T29Kt1 cells showed that the resistant cells had relative higher levels of the compound than the sensitive cells did. Whether this is caused by consumption of the compound through conjugation with cellular nucleophiles (glutathione, proteins or nucleic aids) in the sensitive cells, either directly or indirectly through metabolites, remains to be further investigated. It is possible that some metabolic or biochemical differences between the normal and tumor cells may account for the observed selectivity. Nevertheless, a high level of an active compound in resistant cells suggested that the resistance is not associated with a membrane pump mediated compound efflux mechanism.
Our results showed that, like oncrasin-1, all three active compounds inhibited CTD phosphorylation in sensitive cancer cells but not resistant cancer cells, demonstrating that those agents also function as inhibitors of RNA polymerase II. The CTD of the largest subunit of eukaryotic RNA polymerase II contains multiple heptapeptide repeats of the sequence YSPTSPS, 26 in the yeast CTD and 52 in the mammalian CTD. 31 The serine and threonine in the CTD exhibit variable phosphorylation, which is required for efficient transcription elongation and recruitment of mRNA processing factors, including the capping enzyme and splicing factors required for efficient processing of RNA transcripts.24, 26, 32 The CTD is known to be phosphorylated by three major CDKs, CDK7/cyclin H (TFIIH),33, 34 CDK8/cyclin C,35, 36 and CDK9/cyclin T (P-TEFb).37 In addition to the CDKs, there are other protein kinases that also efficiently phosphorylate the CTD, including the DNA-dependent protein kinase,38 the extracellular signal-regulated kinase,39 and the c-abl tyrosine kinase.40, 41 RNA polymerase II enters the assembling transcription complex with its CTD unphosphorylated (IIa form). Phosphorylation of CTD by CDK7 and CDK9 converts polymerase II to its phosphorylated form, enabling efficient RNA elongation and processing.24, 26 How oncrasin-1 and its analogues inhibit phosphorylation of the CTD remains to be determined. Our previous study revealed that the direct inhibitory effects of oncrasin-1 on CDK9 and CDK7 were moderate, suggesting that other protein kinases or indirect mechanisms might contribute to the inhibition of CTD phosphorylation by this group of compounds.
Using small molecule inhibitors of RNA polymerase II as anticancer agents was suggested by Koumenis and Giaccia on the basis of their observation that an RNA polymerase inhibitor, 5,6-dichloro-l-/3-D ribofuranosylbenzimidazole, induced apoptosis more effectively in oncogene-transformed cells than in untransformed cells.12 However, to date, there has been little work in this area because of the potential toxicity of small molecular RNA polymerase II inhibitors. Nevertheless, some recent studies have revealed that certain anticancer agents, including flavopiridol and seliciclib, elicit their antitumor activity by blocking CTD phosphorylation,13–15 providing evidence that inhibiting the CTD function of RNA polymerase II can be exploited as a novel anticancer approach. Our study demonstrates that a group of small molecules could inhibit CTD phosphorylation and induce antitumor activity in a subset of cancer cells without obvious toxic effects on normal cells. Those agents may be a starting point for new anticancer therapeutics.
EXPERIMENTAL SECTION
Chemicals
All reagents were purchased from Sigma-Aldrich or Chembridge in the highest commercially available quality. Of 69 analogous listed in Table 1, nine compounds (compounds 1, 15, 22, 23, 33, 47, 57, 62 and 65) were obtained from ChemBridge Corp (San Diego, CA), one (compound 14) from Sigma-Aldrich (St. Louis, MO), and one (compound 26, NSC118788) from the Developmental Therapeutics Program of the National Cancer Institute. The other 58 compounds were synthesized in our laboratory. Solvents were Reagent grade, HPLC grade, or MS grade depending on whether they were to be used for synthesis or chemical analysis.
Compound synthesis
The compounds and their intermediates were synthesized as illustrated in Scheme 1 and briefly described in the following paragraphs.
Step A: Sodium hydride (1.1 mmol) dispersed in mineral oil was added to a solution of 1H-indole-3-carboxaldehyde or analogues (1.0 mmol) in anhydrous dimethyl sulfoxide at room temperature with stirring. After stirring for 1 hour, benzyl halide (1.2 mmol) was added and the mixture was stirred continuously for 3 to 5 hours. The reaction was stopped by adding ice-water and the mixture was extracted with dichloromethane thrice and brined with 10% NaCl aqueous solution. The organic phase was dried over anhydrous Na2SO4 and filtered, and solvent was removed under reduced pressure. The residue was dissolved in CHCl3, and separated on a silica gel column with CHCl3/MeOH. The products were concentrated using a vacuum evaporator and recrystallized in diethyl ether/n-hexane.
Step B: A product (1.0 mmol) from step A was dissolved in acetone and treated with KMnO4 (1.9 mmol in water). The resulting mixture was stirred for 16 hours at room temperature. The products were decolored with 10% H2O2 and filtered, and the residues were removed. Solvent acetone was reduced to one-third volume under reduced pressure. The resulting solution was acidified with 2N HCl to obtain a precipitate. The product was recrystallized in ethanol.
Step C: A product (1.0 mmol) from step A suspended in alcohol was reduced with an equimolar amount of sodium borohydride on ice bath. The mixture was stirred at room temperature for 1 to 2 hours, and the reaction was stopped by adding acetone. The mixture was poured into ice-water and incubated at 4°C overnight. The precipitate was washed with water thrice and dissolved in chloroform. The organic phase was dried over anhydrous Na2SO4. Finally, the products were purified on a silica gel column with chloroform/MeOH as an eluent.
Step D: A product from step C was modified with aliphatic acid to form an ester product. The product (5 mmol) was dissolved in anhydrous dichloromethane completely by stirring. Aliphatic (C2–C8) carboxylic acid (7.5 mmol), 1,3-dicyclohexylcarbodiimide (5.5 mmol) and 4-(dimethylamine) pyridine (1 mmol) were added into the solution. The mixture was stirred at room temperature for 2 to 3 hours. The product was filtered through paper film, concentrated by evaporation under reduced pressure, redissolved in ethyl acetate, and filtered through Celite®521. The solvent was removed with clean nitrogen gas, and the product was purified on a column of neutral alumina by eluting with dichloromethane/hexane.
Step E: A product from step C (1 mmol) and an isocyanate containing C3–C8 (1.1 mmol) were dissolved in anhydrous dichloromethane. Triethylamine (1.5 mmol) and a catalytic amount of dibutyltin diacetate were added to the solution. After stirring at room temperature for 30 to 50 minutes, solvent was evaporated to cease the reaction. The residue was dissolved in chloroform/methanol (40/1), and the product was separated on a silica gel column. The purified product was concentrated, dissolved in diethyl ether, and crystallized with hexane.
Products purification was performed on silica gels or neutral aluminum C18 columns with a flash chromatograph system (Teledyne Tech Inc., Thousand Oaks, CA). The purity was determined by HPLC (Agilent Technologies 1200 Series) equipped with a C-18 bounded-phase column (Waters, XTerra C18 MS, 3.5μm, 4.5 × 50mm). A gradient elution was performed with acetonitrile and water as a mobile phase and was monitored at 230nm. All compounds used for activity study have purity of ≥ 95%. The chemical entities of compounds were determined by LC/MS/UV230, 1H and/or 13C NMR analyses. Electrospray ionization (ESI) MS spectra were recorded with an Agilent LC/MSD Trap XCT Ultra spectrometer. NMR spectra were recorded with Bruker Avance DRX 300 spectrometer.
1-[(3-flourophenyl) methyl]-1H-indole-3-carboxaldehyde (compound 2)
Compound 2 was prepared from 1H-indole-3-carboxaldehyde and 3-flourobenzyl chloride as described in Step A. 1H NMR (300 MHz, CDCl3) δ 10.10 (m, 1H, H-C=O indole), δ 8.36 (m, 1H, C4-indole), δ 7.78 (m, 1H, C7-indole), δ 7.37-7.26 (s, 2H, C5, C6-indole and 2H, phenyl), δ 7.06-6.87 (m, 1H, C2-indole and 2H, phenyl), δ 5.40 (s, 2H, N-CH2). ESI-MS m/z 254.0 [M+H].
1-[(3-chlorophenyl) methyl]-1H-indole-3-carboxaldehyde (compound 3)
Compound 3 was prepared from 1H-indole-3-carboxaldehyde and 3-chlorobenzyl chloride as described in Step A. 1H NMR (300 MHz, CDCl3) δ 10.05 (m, 1H, H-C=O indole), δ 8.36 (m, 1H, C4-indole), δ 7.75 (m, 1H, C7-indole), δ 7.36-7.20 (s, 2H, C5, C6-indole and 4H, phenyl), δ 7.04 (m, 1H, C2-indole), δ 5.36 (s, 2H, N-CH2). ESI-MS m/z 269.9 [M+H].
1-[(3-bromophenyl) methyl]-1H-indole-3-carboxaldehyde (compound 4)
Compound 4 was prepared from 1H-indole-3-carboxaldehyde and 3-bromobenzyl bromide as described in Step A. 1H NMR (300 MHz, CDCl3) δ 10.06 (m, 1H, H-C=O indole), δ 8.36 (m, 1H, C4-indole), δ 7.75 (m, 1H, C7-indole), δ 7.48-7.24 (s, 2H, C5, C6-indole and 4H, phenyl), δ 7.08 (m, 1H, C2-indole), δ 5.36 (s, 2H, N-CH2). ESI-MS m/z 314.0 [M+H].
1-[(4-bromophenyl) methyl]-1H-indole-3-carboxaldehyde (compound 5)
Compound 5 was prepared from 1H-indole-3-carboxaldehyde and 4-bromobenzyl bromide as described in Step A. 1H NMR (300 MHz, CDCl3) δ 10.06 (m, 1H, H-C=O indole), δ 8.36 (m, 1H, C4-indole), δ 7.75 (m, 1H, C7-indole), δ 7.49-7.24 (s, 2H, C5, C6-indole and 4H, phenyl), δ 7.08 (m, 1H, C2-indole), δ 5.36 (s, 2H, N-CH2). ESI-MS m/z 314.0 [M+H].
1-[(3-trifluoromethylphenyl) methyl]-1H-indole-3-carboxaldehyde (compound 6)
Compound 6 was prepared from 1H-indole-3-carboxaldehyde an 3-trifluoromethylbenzyl chloride in Step A. 1H NMR (300 MHz, DMSO-d6) δ 9.09 (m, 1H, H-C=O indole), δ 7.39 (m, 1H, C4-indole), δ 6.78 (m, 1H, C7-indole), δ 6.64-6.30 (m, 3H, C2, C5, C6-indole and 4H, phenyl), δ 4.77 (s, 2H, N-CH2). ESI-MS m/z 304.1 [M+H].
1-[(4-trifluoromethylphenyl) methyl]-1H-indole-3-carboxaldehyde (compound 7)
Compound 7 was prepared from 1H-indole-3-carboxaldehyde and 4-trifluoromethylbenzyl chloride as described in Step A. 1H NMR (300 MHz, DMSO-d6) δ 9.09 (m, 1H, H-C=O indole), δ 7.40 (m, 1H, C4-indole), δ 6.77 (m, 1H, C7-indole), δ 6.71-6.30(m, 3H, C2, C5, C6-indole and 4H, phenyl), δ 4.77 (s, 2H, N-CH2). ESI-MS m/z 304.1 [M+H].
1-[(3-methylphenyl) methyl]-1H-indole-3-carboxaldehyde (compound 8)
Compound 8 was prepared from 1H-indole-3-carboxaldehyde and 3-methylbenzyl bromide as described in Step A. 1H NMR (300 MHz, CDCl3) δ 10.03 (m, 1H, H-C=O indole), δ 8.34 (m, 1H, C4-indole), δ 7.73 (m, 1H, C7-indole), δ 7.39-7.14 (m, 2H, C5, C6-indole and 4H, phenyl), δ 7.01 (m, 1H, C2-indole), δ 5.36 (s, 2H, N-CH2), δ 2.35 ( s, 3H, -CH3). ESI-MS m/z 250.0 [M+H].
1-[(4-methylphenyl) methyl]-1H-indole-3-carboxaldehyde (compound 9)
Compound 9 was prepared from 1H-indole-3-carboxaldehyde and 4-methylbenzyl chloride as described in Step A. 1H NMR (300 MHz, CDCl3) δ 10.05 (m, 1H, H-C=O indole), δ 8.35 (m, 1H, C4-indole), δ 7.73 (m, 1H, C7-indole), δ 7.30-6.98 (m, 3H, C2,C5, C6-indole and 4H, phenyl), δ 5.36 (s, 2H, NCH2), δ 2.41(s, 3H, -CH3). ESI-MS m/z 250.0 [M+H].
1-[(3-nitrophenyl) methyl]-1H-indole-3-carboxaldehyde (compound 10)
Compound 10 was prepared from 1H-indole-3-carboxaldehyde and 3-nitrobenzyl chloride as described in Step A. 1H NMR (300 MHz, CDCl3) δ 10.06 (m, 1H, H-C=O indole), δ 8.64 (m, 2H, phenyl), δ 8.37 (m, 1H, C4-indole), δ 7.73-7.66 (m, 1H, C7-indole, 2H, phenyl), δ 7.32-7.10 (s, 3H, C2, C5, C6-indole), δ 5.36 (s, 2H, N-CH2). ESI-MS m/z 281.0 [M+H].
1-[(4-nitrophenyl) methyl]-1H-indole-3-carboxaldehyde (compound 11)
Compound 11 was prepared from 1H-indole-3-carboxaldehyde and 4-nitrobenzyl chloride as described in Step A. 1H NMR (300 MHz, CDCl3) δ 10.06 (m, 1H, H-C=O indole), δ 8.71 (m, 2H, phenyl), δ 8.36 (m, 1H, C4-indole), δ 7.69-7.65 (m, 1H, C7-indole, 2H, phenyl), δ 7.33-7.09 (s, 3H, C2, C5, C6-indole), δ 5.37 (s, 2H, N-CH2). ESI-MS m/z 281.0 [M+H].
1-[(2,4-dichlorophenyl) methyl]-1H-indole-3-carboxaldehyde (compound 12)
Compound 12 was prepared from 1H-indole-3-carboxaldehyde and 2, 4-dichlorobenzyl chloride as described in Step A. 1H NMR (300 MHz, DMSO-d6) δ 9.96 (m, 1H, H-C=O indole), δ 8.35 (m, 1H, C4-indole), δ 8.10 (m, 1H, C7-indole), δ 7.79-7.68 (s, 2H, C5, C6-indole and 2H, phenyl), δ 7.32-7.11 (m, 1H, C2-indole and 1H, phenyl), δ 5.51 (s, 2H, N-CH2). ESI-MS m/z 303.9 [M+H].
1-[(3,4-dibromophenyl) methyl]-1H-indole-3-carboxaldehyde (compound 13)
Compound 13 was prepared from 1H-indole-3-carboxaldehyde and 3, 4-dibromobenzyl chloride as described in Step A. 1H NMR (300 MHz, DMSO-d6) δ 9.96 (m, 1H, H-C=O indole), δ 8.49 (m, 1H, C4-indole), δ 8.12 (m, 1H, C7-indole), δ 7.79-7.71 (s, 2H, C5, C6-indole and 1H, phenyl), δ 7.32-7.17 (m, 1H, C2-indole and 2H, phenyl), δ 5.54 (s, 2H, N-CH2). ESI-MS m/z 391.9 [M+H].
1-[(3-chlorophenyl) methyl]-5-fluoro-1H-indole-3-carboxaldehyde (compound 16)
Compound 16 was prepared from 5-fluoro-1H-indole-3-carboxaldehyde and 3-chlorobenzyl chloride as described in Step A. 1H NMR (300 MHz, CDCl3) δ 10.0 (m, 1H, H-C=O indole), δ 8.03 (m, 1H, C7-indole), δ 7.72 (m, 1H, C4-indole), δ 7.25-7.02 (m, 2H, C2, C6-indole and 4H, phenyl), δ 5.34 (s, 2H, N-CH2). ESI-MS m/z 287.9 [M+H].
1-[(4-chlorophenyl) methyl]-5-fluoro-1H-indole-3-carboxaldehyde (compound 17)
Compound 17 was prepared from 5-fluoro-1H-indole-3-carboxaldehyde and 4-chlorobenzyl chloride as described in Step A. 1H NMR (300 MHz, CDCl3) δ 10.0 (m, 1H, H-C=O indole), δ 8.04 (m, 1H, C7-indole), δ 7.75 (m, 1H, C4-indole), δ 7.27-7.02 (m, 2H, C2,C6-indole and 4H, phenyl), δ 5.36 (s, 2H, N-CH2). ESI-MS m/z 287.9 [M+H].
1-[(4-chlorophenyl) methyl]-6-fluoro-1H-indole-3-carboxaldehyde (compound 18)
Compound 18 was prepared from 6-fluoro-1H-indole-3-carboxaldehyde and 4-chlorobenzyl chloride as described in Step A. 1H NMR (300 MHz, CDCl3) δ 10.0 (m, 1H, H-C=O indole), δ 8.29 (m, 1H, C4-indole), δ 7.85 (m, 1H, C7-indole), δ 7.36-7.27 (m,2H, phenyl), δ 7.13-7.06 (s, 1H, C2-indole and 2H, phenyl), δ 6.95 (m, 1H, C5-indole), δ 5.30 (s, 2H, N-CH2). ESI-MS m/z 287.9 [M+H].
1-[(4-chlorophenyl) methyl]-5-chloro-1H-indole-3-carboxaldehyde (compound 19)
Compound 19 was prepared from 5-chloro-1H-indole-3-carboxaldehyde and 4-chlorobenzyl chloride as described in Step A. 1H NMR (300 MHz, CDCl3) δ 10.0 (m, H-C=O indole), δ 8.35 (m, 1H, C4-indole), δ 7.88 (m, 1H, C7-indole), δ 7.42-7.20( m,1H, C-6 indole and 2H, phenyl), δ 7.15-7.04 (m, 1H, C2-indole and 2H, phenyl), δ 5.34 (s, 2H, N-CH2). ESI-MS m/z 304.0 [M+H].
1-[(4-chlorophenyl) methyl]-6-bromo-1H-indole-3-carboxaldehyde (compound 20)
Compound 20 was prepared from 6-bromo-1H-indole-3-carboxaldehyde and 4-chlorobenzyl chloride as described in Step A. 1H NMR (300 MHz, CDCl3) δ 10.0 (m, H-C=O indole), δ 8.22 (m, 1H, C7-indole), δ 7.92 (m, 1H, C4-indole), δ 7.47-7.32 (m, 2H, C2, C-5 indole and 2H, phenyl), δ 7.10 (m, 2H, phenyl), δ 5.32 (s, 2H, N-CH2). ESI-MS m/z 349.1 [M+H].
1-[(4-chlorophenyl)methyl]-6-chloro-2-oxoindoline-3-carboxaldehyde (compound 21)
Compound 21 was prepared from 6-chloro-2-oxoindole-3-carboxaldehyde and 4-chlorobenzyl bromide as described in Step A. 1H NMR (300 MHz, DMSO-d6) δ 10.39 (m, H-C=O indole), δ 7.81 (m, 1H, C5-indole), δ 7.51-7.39 (m,2H, C4, C7-indole and 2H, phenyl), δ 6.94 (m, 2H, phenyl), δ 5.40 (s, 2H, N-CH2, 1H, C2-indole). ESI-MS m/z 319.5 [M+H].
1-[(4-chlorophenyl) methyl]-5-fluoro-1H-indole-2-methanol (compound 24)
Compound 24 was prepared from 5-fluoro-1H-indole-2-carboxaldehyde and 4-chlorobenzyl chloride and then reduced as described in the Step C. 1H NMR (300 MHz, CDCl3) δ 7.24 (m, 2H, phenyl), δ 7.13 (m, 1H, C7-indole), δ 6.96-6.91 (m, 2H, C4, C6-indole, 2H, phenyl), δ 6.58 (m, 1H, C3-indole), δ 5.44 (s, 2H, N-CH2), δ 4.78 (s, 2H, HO-CH2-). ESI-MS m/z 271.8 [M+H-H2O].
1-[(3-chlorophenyl) methyl]-1H-indole-3-carbonitrile (compound 25)
Compound 25 was prepared from 1H-indole-3-carbonitrile and 3-chlorobenzyl chloride as described in Step A. 1H NMR (300 MHz, DMSO-d6), δ 8.50 (m, 1H, C4-indole), δ 7.66 (m, 2H, indole), δ 7.41-7.25 (m, 1H, indole and 4H, phenyl), δ 7.04 (m, 1H, C2-indole), δ 5.54 (s, 2H, N-CH2), δ 5.32 (s, 2H, CNCH2). ESI-MS m/z 281.1 [M+H].
1-[(2-chlorophenyl) methyl]-1H-indole-3-ethanone (compound 27)
Compound 27 was prepared from 1H-indole-3-ethanone and 2-chlorobenzyl chloride as described in Step A. δ 8.45 (m, 1H, C2-indole), δ 7.73 (m, 1H, C4-indole), δ 7.32-7.18 (s, 3H, indole and 4H, phenyl), δ 5.35 (s, 2H, N-CH2), δ 2.87 (s, 3H, O=C-CH3). ESI-MS m/z 284.1 [M+H].
1-[3-chlorophenyl) methyl]-1H-indole-3-ethanone (compound 28)
Compound 28 was prepared from 1H-indole-3-ethanone and 3-chlorobenzyl chloride as described in Step A. δ 8.43 (m, 1H, C2-indole), δ 7.74 (m, 1H, C4-indole), δ 7.30-7.15 (m, 3H, -indole and 4H, phenyl), δ 5.36 (s, 2H, N-CH2), δ 2.87 (s, 3H, O=C-CH3). ESI-MS m/z 284.1 [M+H].
1-[4-chlorophenyl) methyl]-1H-indole-3-ethanone (compound 29)
Compound 29 was prepared from 1H-indole-3-ethanone and 4-chlorobenzyl chloride as described in Step A. δ 8.41 (m, 1H, C2-indole), δ 7.76 (m, 1H, C4-indole), δ 7.32-7.28 (m, 3H, indole, 2H, phenyl), δ 7.11-6.85 (m, 2H, phenyl), δ 5.36 (s, 2H, N-CH2), δ 2.56 (s, 3H, O=C-CH3). 13C NMR (CDCl3, 75HMz), δ193.1 (C=O), 136.3 (indole C-9), 134.7 (indole C-2), 134.3 (phenyl C-1), 134.1 (phenyl C-4), 129.2 (phenyl C-2 and C-6), 128.2 (phenyl C-3 and C-5), 126.4 (indole C-8), 123.6 (indole C-5), 122.8 (indole C-6), 122.7 (indole C-4), 117.7 (indole C-3), 110.0 (indole C-7), 50.1 (N-CH2-), 27.7 (CH3). ESI-MS m/z 284.1 [M+H].
1-(phenylmethyl)-1H-indole-3-methanol (compound 30)
Compound 30 was prepared from compound 14 as described in Step C. 1H NMR (300 MHz, CDCl3) δ 7.65 (m, 1H, C4-indole), δ 7.32-7.03 (s, 3H, indole and 5H, phenyl), δ 6.78 (m, 1H, C-2 indole), δ 5.27 (s, 2H, N-CH2), δ 4.28 (s, 2H, CH2-OH). ESI-MS m/z 220.1 [M+H-H2O].
1-(3-fluorophenylmethyl)-1H-indole-3-methanol (compound 31)
Compound 31 was prepared from compound 2 as described Step C. 1H NMR (300 MHz, CDCl3) δ 7.79 (m, 1H, C4-indole), δ 7.25-7.10 (s, 3H, indole and 1H, phenyl), δ 6.92 (m, 3H, phenyl), δ 6.81 (m, 1H, C-2 indole) δ 5.31 (s, 2H, N-CH2), δ 4.91 (s, 2H, CH2-OH). ESI-MS m/z 238.1 [M+H-H2O].
1-(4-fluorophenylmethyl)-1H-indole-3-methanol (compound 32)
Compound 32 was prepared from 1H-indole-3-carboxaldehyde and 4-flourobenzyl chloride as described in Steps A and C. 1H NMR (300 MHz, CDCl3) δ 7.88 (m, 1H, C4-indole), δ 7.23-7.01 (s, 3H, indole and 4H, phenyl), δ 6.83 (m, 1H, C-2 indole), δ 5.27 (s, 2H, N-CH2), δ 4.89 (s, 2H, CH2-OH). ESI-MS m/z 238.1 [M+H-H2O].
1-[(3-chlorophenyl) methyl]-1H-indole-3-methanol (compound 34)
Compound 34 was prepared from Compound 16 according to Step C. 1H NMR (300 MHz, CDCl3) δ 7.78 (m, 1H, C4-indole), δ 7.23-7.15 (s, 3H, indole and 4H, phenyl), δ 7.00 (m, 1H, C-2 indole), δ 5.28 (s, 2H, N-CH2), δ 4.91 (s, 2H, CH2-OH). 13C NMR (CDCl3, 75HMz), 141.2 (phenyl C-1), 136.7 (indole C-9), 133.6 (phenyl C-3),130.8(phenyl C-5), 127.8 (phenyl C-2, indole C-8), 127.3 (phenyl C-6, indole C-2), 126.3 (phenyl C-4), 121.8 (indole C-5), 119.8 (indole C-6), 119.2 (indole C-4), 116.5 (indole C-3), 110.2 (indole C-7), 55.7 (-CH2OH), 48.8 (N-CH2). ESI-MS m/z 254.1 [M+H-H2O].
1-[(4-chlorophenyl) methyl]-1H-indole-3-methanol (compound 35)
Compound 35 was prepared from Oncrasin-1 according to Step C. 1H NMR (300 MHz, CDCl3) δ 7.77 (m, 1H, C4-indole), δ 7.30-6.81 (s, 4H, indole and 4H, phenyl), δ 5.27 (s, 2H, N-CH2), δ 4.90 (s, 2H, CH2-OH). 13C NMR (CDCl3, 75HMz), 136.7 (indole C-9), 135.7 (phenyl C-1), 133.5 (phenyl C-4), 128.9 (phenyl C-2 and C-6), 128.2 (phenyl C-3 and C-5), 127.2 (indole C-8), 126.7 (indole C-2), 122.4 (indole C-5), 119.9 (indole C-6), 119.3 (indole C-3), 115.7 (indole C-4), 109.7 (indole C-7), 57.2 (-CH2OH), 49.4 (N-CH2-). ESI-MS m/z 254.1 [M+H-H2O].
1-[(3-bromophenyl) methyl]-1H-indole-3-methanol (compound 36)
Compound was prepared from compound 4 according to Step C. 1H NMR (300 MHz, CDCl3) δ 7.77 (m, 1H, C4-indole), δ 7.43-7.63 (m, 2H, phenyl), δ 7.24-7.14 (s, 3H, indole, 2H, phenyl), δ 7.03 (m, 1H, C-2 indole), δ 5.27 (s, 2H, N-CH2), δ 4.91 (s, 2H, CH2-OH). ESI-MS m/z 315.9 [M+H-H2O].
1-[(4-bromophenyl) methyl]-1H-indole-3-methanol (compound 37)
Compound 37 was prepared from Compound 5 according to Step C. 1H NMR (300 MHz, CDCl3) δ 7.70 (m, 1H, C4-indole), δ 7.43 (m, 2H, phenyl), δ 7.20-7.06 (m, 3H, indole), δ 6.98-6.89 (m, 1H, C-2 indole and 2H, phenyl), δ 5.23 (s, 2H, N-CH2), δ 4.26 (s, 2H, CH2-OH). ESI-MS m/z 315.9 [M+H-H2O].
1-[(3-iodiophenyl) methyl]-1H-indole-3-methanol (compound 38)
Compound 38 was prepared from 1H-indole-3-carboxaldehyde and 3-iodiobenzyl bromide according to Steps A and C. 1H NMR (300 MHz, CDCl3) δ 7.77 (m, 1H, C4-indole), δ 7.60-7.47 (m, 2H, phenyl), δ 7.20-7.04 (m, 3H,C-5,C-6, C-7-indole, 2H Phenyl), δ 5.24 (s, 2H, N-CH2), δ 4.91 (s, 2H, CH2-OH). ESI-MS m/z 346.0 [M+H-H2O].
1-[(3-fluoromethylphenyl) methyl]-1H-indole-3-methanol (compound 39)
Compound 39 was prepared from compound 6 according to Step C. 1H NMR (300 MHz, CDCl3) δ 7.70 (m, 1H, C4-indole), δ 7.65-7.40 (m, 3H, indole, 4H, phenyl), δ 6.91 (m, 1H, C-2 indole), δ 5.34 (s, 2H, NCH2), δ 4.81 (s, 2H, CH2-OH). ESI-MS m/z 288.0 [M+H-H2O].
1-[(4-fluoromethylphenyl) methyl]-1H-indole-3-methanol (compound 40)
Compound 40 was prepared from compound 7 according to Step C. 1H NMR (300 MHz, CDCl3) δ 7.74 (m, 1H, C4-indole), δ 7.02 (m, 2H, C-2 phenyl), δ 7.36-7.14 (m, 4H, indole, 2H, phenyl), δ 5.37 (s, 2H, NCH2), δ 4.88 (s, 2H, CH2-OH). ESI-MS m/z 288.0 [M+H-H2O].
1-[(3-methylphenyl) methyl]-1H-indole-3-methanol (compound 41)
Compound 41 was prepared from compound 8 according to Step C. 1H NMR (300 MHz, CDCl3) δ 7.76 (m, 1H, C4-indole), δ 7.31-7.10 ( s, 3H, C5, C6, C7-indole, 4H Phenyl), δ 6.97 (m, 1H, C2-indole), δ 5.24 (s, 2H, N-CH2), δ 4.87 (s, 2H, CH2-OH), δ 2.29 (s, 3H, Ph-CH3). ESI-MS m/z 233.9 [M+H-H2O].
1-[(4-methylphenyl) methyl]-1H-indole-3-methanol (compound 42)
Compound 42 was prepared from compound 9 according to Step C. 1H NMR (300 MHz, CDCl3) δ 7.75 (m, 1H, C4-indole), δ 7.32-7.06 (m, 4H, indole, 4H Phenyl), δ 5.26 (s, 2H, N-CH2), δ 4.89 (s, 2H, CH2-OH), δ 2.33 (s, 3H, Ph-CH3). ESI-MS m/z 233.9 [M+H-H2O].
1-[(3-nitrophenyl) methyl]-1H-indole-3-methanol (compound 43)
Compound 43 was prepared from compound 10 according to Step C. 1H NMR (300 MHz, CDCl3) δ 8.14-8.06 (m, 2H, phenyl), δ 7.77 (m, 2H, phenyl), δ 7.49-7.02 (s, 5H, indole),δ 5.42 (s, 2H, N-CH2), δ 4.90 (s, 2H, CH2-OH). ESI-MS m/z 265.1 [M+H-H2O].
1-[(4-nitrophenyl) methyl]-1H-indole-3-methanol (compound 44)
Compound 44 was prepared from compound 11 according to Step C. 1H NMR (300 MHz, CDCl3) δ 8.12–8.19 (m, 2H, phenyl), δ 7.79 (m, 2H, phenyl), δ 7.23-6.93 (m, 5H, indole), δ 5.42 (s, 2H, N-CH2), δ 4.93 (s, 2H, CH2-OH). ESI-MS m/z 265.1 [M+H-H2O].
1-[(4-methoxyphenyl) methyl]-1H-indole-3-methanol (compound 45)
Compound 45 was prepared from 1-[(4-methoxyphenyl) methyl]-1H-indole-3-carboxaldehyde according to Step C procedure. 1H NMR (300 MHz, CDCl3) δ 7.76 (m, 1H, C-4 indole), δ 7.34-7.29 (m, 3H, C-5, C-6, C-7 indole and 3H, phenyl), δ 7.23-6.93 (m, 1H, C-2 indole and 1H, phenyl), δ 5.42 (s, 2H, NCH2), δ 4.89 (s, 2H, CH2-OH), δ 3.79 (s, 3H, CH3-O). ESI-MS m/z 250.1 [M+H-H2O].
1-[(2,4-dichlorophenyl) methyl]-1H-indole-3-methanol (compound 46)
Compound 46 was prepared from compound 12 according to Step C. 1H NMR (300 MHz, CDCl3) δ 7.80 (m, 1H, C-4 indole), δ 7.47 (m, 1H, C-7 indole), δ 7.33-6.69 (m, 2H, C-5,C-6, indole and 3H, phenyl), δ 6.58 (m, 1H, C-2 indole), δ 5.36 (s, 2H, N-CH2), δ 4.92 (s, 2H, CH2-OH). ESI-MS m/z 288.0 [M+H-H2O].
1-[(3,4-dibromophenyl) methyl]-1H-indole-3-methanol (compound 48)
Compound 48 was prepared from compound 13 according to Step C. 1H NMR (300 MHz, CDCl3) δ 7.75 (m, 1H, C4-indole), δ 7.50-6.88 (m, 3H, C-5,C-6, C-7-indole and 3H Phenyl), δ 5.24 (s, 2H, N-CH2), δ 4.91 (s, 2H, CH2-OH). ESI-MS m/z 375.9 [M+H-H2O].
5-fluoro-1-methyl-1H-indole-3-methanol (compound 49)
Compound 49 was prepared from 5-fluoro-1-methyl-1H-indole-3-carboxaldehyde according to Step C. 1H NMR (300 MHz, CDCl3) δ 76.97-6.77 (m, 4H, C2, C4, C6, C7-indole), δ 4.19 (s, 2H, CH2-OH), δ 3.53 (s, 2H, N-CH3). ESI-MS m/z 162.0 [M+H-H2O].
1-[(3-fluorophenyl) methyl]-5-chloro-1H-indole-3-methanol (compound 50)
Compound 50 was prepared from 5-chloro-1H-indole-3-carboxaldehyde and 3-flourobenzyl chloride according to Steps A and C. 1H NMR (300 MHz, CDCl3) δ 7.42-7.21 (m, 2H, C4, C6-indole), δ 7.16-7.09 (m, 1H, C7-indole, 2H, Phenyl), δ 6.98-6.77 (m, 1H,C-2-indole and 2H, phenyl), δ 5.26 (s, 2H, N-CH2), δ 4.89 (s, 2H, CH2-OH). 13C NMR (CDCl3, 75HMz), δ162.1 (phenyl C-3), 139.5 (phenyl C-1), 135.1 (indole C-9), 130.4 (phenyl C-5), 129.0 (indole C-8), 127.6 (indole C-5), 125.7 (indole C-2), 125.0 (phenyl C-6), 122.2 (indole C-4), 119.0 (indole C-6), 115.4 (phenyl C-2), 113.6 (phenyl C-4), 113.4 (indole C-7), 112.6 (indole C-3), 57.0 (-CH2OH), 49.7 (N-CH2-). ESI-MS m/z 272.0 [M+H-H2O].
1-[(4-chlorophenyl) methyl]-5-fluoro-1H-indole-3-methanol (compound 51)
Compound 51 was prepared from compound 17 according to Step C. 1H NMR (300 MHz, CDCl3) δ 7.71 (m, 1H, C7-indole), δ 7.51 (m, 2H, Phenyl), δ 7.15-6.94 (m, 3H,C-2,C-4, C-6 indole and 2H, phenyl), δ 5.23 (s, 2H, N-CH2), δ 4.86 (s, 2H, CH2-OH). ESI-MS m/z 272.0 [M+H-H2O].
1-[(4-chlorophenyl) methyl]-6-fluoro-1H-indole-3-methanol (compound 52)
Compound 52 was prepared from compound 18 according to Step C. 1H NMR (300 MHz, CDCl3) δ 7.67 (m, 1H, C4-indole), δ 7.28 (m, 2H, Phenyl), δ 7.10-7.05 (m, 2H,C-5, C-7 indole and 2H, phenyl), δ 6.85 (m, 1H, C2-indole), δ 5.27 (s, 2H, N-CH2), δ 4.07 (s, 2H, CH2-OH). ESI-MS m/z 272.0 [M+HH2O].
1-[(4-chlorophenyl) methyl]-5-chloro-1H-indole-3-methanol (compound 53)
Compound 53 was prepared from compound 19 according to Step C. 1H NMR (300 MHz, CDCl3) δ 7.72 (m, 1H, C4-indole), δ 7.51 (m, 1H, C7-indole), δ 7.15-6.90 ( m, 3H, C-2, C-4, C-5 indole and 4H, phenyl), δ 5.23 (s, 2H, N-CH2), δ 4.74 (s, 2H, CH2-OH). ESI-MS m/z 288.0 [M+H-H2O].
1-[(4-chlorophenyl) methyl]-5-methoxy-1H-indole-3-methanol (compound 54)
Compound 54 was prepared from 5-methoxy-1H-indole-3-carboxaldehyde and 4-chlorobenzyl chloride according to Steps A and C. 1H NMR (300 MHz, CDCl3) δ 7.17-7.01 (m, 4H, phenyl), δ 6.95-6.72 (m, 4H, indole), δ 5.19 (s, 2H, N-CH2), δ 4.85 (s, 2H, CH2-OH), 3.80 (s, 3H, CH3O-). ESI-MS m/z 284.1 [M+H-H2O].
1-[(4-methylphenyl) methyl]-5-fluoro-1H-indole-3-methanol (compound 55)
Compound 55 was prepared from 5-fluoro-1H-indole-3-carboxaldehyde and 4-methylbenzyl chloride according to Steps A and C. 1H NMR (300 MHz, CDCl3) δ 7.28 (m, 1H, C7-indole), δ 7.18-7.02 (m, 2H, C4, C6-indole and 4H, phenyl), δ 6.94 (m, 1H, C2-indole), δ 5.23 (s, 2H, N-CH2), δ 4.83 (s, 2H, CH2-OH), δ 2.19 (s, 3H, CH3-Ph). ESI-MS m/z 251.8 [M+H-H2O].
2-[1-[(4-chlorophenyl) methyl]-1H-indole-3-]ethanol (compound 56)
Compound 56 was prepared from 2-(3-hydroxyethyl) indole and 4-chlorobenzyl chloride as described in Step A. 1H NMR (300 MHz, CDCl3) δ 7.65 (m, 1H, C4-indole), δ 7.21-7.14 ( m, 3H, indole and 2H, phenyl), δ 7.07-6.93 (m, 1H, C2-indole and 2H, phenyl), δ 5.27 (s, 2H, N-CH2), δ 3.93 (s, 2H, CH2-CH2-OH), δ 3.06 (s, 2H, CH2-CH2-OH). ESI-MS m/z 268.1 [M+H-H2O].
1-[1-[(4-chlorophenyl) methyl]-1H-indole-3-] ethanol (compound 58)
Compound 58 was prepared from compound 29 according to Step C. 1H NMR (300 MHz, CDCl3) δ 7.65 (m, 1H, C4-indole), δ 7.39-7.10 (m, 3H, indole and 2H, phenyl), δ 7.08-6.96 (m, 1H, C2-indole and 2H, phenyl), δ 5.35 (s, 2H, N-CH2), δ 4.89 (m, 1H, CH3-CH-OH), δ 1.47 (s, 3H, CH3-CH-OH). ESI-MS m/z 268.1 [M+H-H2O].
1-[(4-chlorophenyl) methyl]-1H-indole-3-methoxymethane (compound 59)
Compound 59 was prepared from 3-methoxymethyl-1H-indole and 4-chlorobenzyl chloride as described in Step A. 1H NMR (300 MHz, CDCl3) δ 7.72 (m, 1H, C4-indole), δ 7.36-7.16 (m, 3H, indole and 4H, phenyl), δ 6.93 (m, 1H, C2-indole), δ 5.28 (s, 2H, N-CH2), 4.68 (s, 2H, -CH2-OCH3), 1.58 (s, 3H, -CH2-OCH3). ESI-MS m/z 285.9 [M+H].
1-[(4-chlorophenyl) methyl]-1H-indole-3-methyl acetate (compound 60)
Compound 60 was prepared from compound 35 and acetic anhydride according to Step D. 1H NMR (300 MHz, CDCl3) δ 7.75 (m, 1H, C4-indole), δ 7.25-7.17(s, 3H, indole and 4H, phenyl), δ 6.92 (m, 1H, C2-indole), δ 5.36 (s, 2H, -CH2-OOCH3 ), δ 5.29 (s, 2H, N-CH2), δ 2.09 (s, 3H, -CH2-OOCH3). ESI-MS m/z 313.9 [M+H].
1-[(4-chlorophenyl) methyl]-1H-indole-3-methyl cyclohexylcarbamate (compound 61)
Compound 61 was prepared from compound 35 and cyclohexyl isocyanate according to Step E. 1H NMR (300 MHz, CDCl3) δ 7.76 (m, 1H, C4-indole), δ 7.30-7.15 (s, 3H, indole and 4H, phenyl), δ 7.05 (m, 1H, C2-indole), δ 5.33 (s, 2H,-N-CH2), δ 5.27 (s, 2H, indole-C3-CH2-O-), δ 3.60 (m, 1H, C1-cyclohexyl), δ 1.39-1.11 (m, 10H, cyclohexyl group), did not see -NH-. ESI-MS m/z 397.1 [M+H].
1-[(4-chlorophenyl) methyl]-1H-indole-3-carboxylic acid (compound 63)
Compound 63 was prepared from Oncrasin-1 according to Step B. 1H NMR (300 MHz, CDCl3) δ 8.85 (s, 1H, C2-indole), δ 8.17 (m, 1H, C4-indole), δ 7.95 (m, 1H, C7-indole), δ 7.44-7.24 (s, 2H, C5, C6-indole and 4H, phenyl), δ 5.78 (s, 2H, N-CH2), did not see -COOH. ESI-MS m/z 286.0 [M+H].
Methyl [1-[(4-chlorophenyl) methyl]]-1H-indole-3-carboxylate (compound 64)
Compound 64 was prepared from methyl indole-3-carboxylate and 4-chlorobenzyl chloride according to Step A. 1H NMR (300 MHz, CDCl3) δ 8.23 (m, 1H, C4-indole), δ 7.85 (m, 1H, C2-indole), δ 7.35-7.29 (s, 3H, indole and 2H, phenyl), δ 7.09 (m, 2H, phenyl), 5.34 (s, 2H, N-CH2), δ 3.95(s, 3H, COOCH3). 13C NMR (CDCl3, 75HMz), δ 165.3 (OC=O), 136.6 (indole C-9), 134.4 (indole C-2), 134.3 (phenyl C-1), 134.0 (phenyl C-4), 129.2 (phenyl C-2 and C-6), 128.3 (phenyl C-3 and C-5), 126.8 (indole C-8), 123.1(indole C-5), 122.1(indole C-6) 121.9 (indole C-4), 110.1 (indole C-7), 107.8 (indole C-3), 51.0 (N-CH2-), 50.0 (OCH3). ESI-MS m/z 299.9 [M+H].
1-[(4-chlorophenyl) methyl]-1H-indole-3-methane (compound 66)
Compound 66 was prepared from 3-methyl indole and 4-chlorobenzyl chloride according to procedure of Step A. 1H NMR (300 MHz, CDCl3) δ 7.61 ( m, 1H, C4-indole), δ 7.20-7.02 (m, 3H, indole and 4H, phenyl), δ 6.90 (m, 1H, C2-indole), 5.25 (s, 2H, N-CH2), δ 2.35 ( s, 3H, indole-3-CH3). ESI-MS m/z 256.0 [M+H].
Methyl 2-[1-[(4-chlorophenyl) methyl]-1H-indole-3-methylamino] acetate (compound 67)
Compound 67 was prepared from 1-[(4-chlorophenyl) methyl]-1H-indole-3-carboxaldehyde and glycinmethylester hydrochloride according to Step A. 1H NMR (300 MHz, CDCl3) δ 7.73 (m, 2H, C4, C7-indole), δ 7.22-7.02 (m, 2H, indole and 4H, phenyl), δ 6.90 (m, 1H, C2-indole), 5.25 (s, 2H, N-CH2), δ 4.01 (s, 2H, CH2-NH-CH2-COOCH3), δ 3.73 (s, 3H, CH2-NH-CH2-COOCH3), δ 3.51 (s, 2H, CH2-NH-CH2-COOCH3). ESI-MS m/z 341.8 [M+H-H].
bis[1-[(3-chlorophenyl) methyl]-1H-indole-3-]methane (compound 68)
Compound 68 was prepared from 3,3′-methyleneindole and 4-chlorobenzyl chloride according to Step A. 1H NMR (300 MHz, CDCl3) δ 7.65 (m, 2H, C4, C4′--indole), δ 7.23-7.17 (s, 6H, C5, C6, C7, C5′, C6′, C7′-indole), δ 7.15-7.10 (m, 6H, phenyl), δ 6.93-6.34 (m, 2H, C2, C2′-indole, 2H phenyl), 5.29 (s, 4H, 2 N-CH2), δ 4.29 ( s, 2H, bridge-CH2-). 13C NMR (CDCl3, 75HMz), δ 140.0 (indole 2C-9), 136.7 (phenyl 2C-1), 134.6 (phenyl 2C-3), 130.0 (phenyl 2C-5), 128.3 (phenyl 2C-2), 127.7 (indole 2C-8), 126.7 (phenyl 2C-6), 126.3 (indole 2C-2), 124.7 (phenyl 2C-4), 121.9 (indole 2C-5), 119.5 (indole 2C-6), 119.1 (indole 2C-4), 115.1 (indole 2C-3), 109.4 (indole 2C-7), 49.3 (2 NCH2-), 21.2 (-CH2-). ESI-MS m/z 495.0 [M+H].
bis[1-[(3-bromophenyl) methyl]-1H-indole-3-]methane (compound 69)
Compound 69 was prepared from 3,3′-methyleneindole and 4-bromobenzyl bromide according to Step A. 1H NMR (300 MHz, CDCl3) δ 7.65 ( m, 2H, C4, C4′-indole), δ 7.38 (m, 2H, C7, C7′-indole), δ 7.19-7.09 (m, 4H, C5, C6, C5′, C6′-indole and 4H, phenyl), δ 6.97 (m, 2H, phenyl), δ 6.83 (C2, C2′-indole), δ 5.23 (s, 4H, 2 N-CH2), δ 4.29 (s, 2H, bridge-CH2-). ESI-MS m/z 583.0 [M+H].
Cell culture
H460, H157, H1299, and H322 human lung cancer cells were provided by Dr. John Minna at The University of Texas Southwestern Medical School (Dallas, TX). T29 immortalized normal human ovarian epithelial cells and K-Ras-transformed T29Kt1 cells were provided by Dr. Jinsong Liu in our institution. All cell lines were propagated routinely in a monolayer culture in RPMI-1640 medium or in Dulbecco’s modified Eagle’s medium supplemented with 10% heat-inactivated fetal calf serum, 100 units/ml penicillin, and 100 μg/ml streptomycin. All cells were maintained in the presence of 5% CO2 at 37°C.
Cell viability assay
The inhibitory effects of oncrasin-1 and other agents on cell growth were determined by using the sulforhodamine B assay, as described previously. 8 Each experiment was performed in quadruplicate and repeated at least three times. The IC50 value was determined by using the CurveExpert Version 1.3 program.
Western blot analysis
Western blot analysis was performed as we have previously described. 42 Phosphorylated CTD-specific antibody (H5, Covance, Princeton, NJ) and antibody for β-actin (Sigma) were diluted 1:1000 before use for Western blot analysis.
Levels of compound 34 in cells after treatment
T29 and T29Kt1 cells cultured in ϕ15 cm plates to 80–90% confluence were treated with 10μM of compound 34. Cells were harvested at different time points by trypsinization. After washed once with phosphate buffered saline, cells were lyzed with 0.5ml of cell lysis buffer M-PER (Thermo Scientific). After homogenizing in a Bullet Blender (Next Advance Inc., Averill Park, NY) and a brief spin, the supernatant was collected. Protein concentration was determined with BCA protein assay reagent (Pierce, Rockford, IL). 400μl of supernatant was mixed with 800 μl of acetonitrile to precipitate proteins. After a brief spin at 4°C, the clean extracts were concentrated by nitrogen gas under pressure to 400μl before used for HPLC analysis as described above to determine the levels of compound 34. 1-[(4-chlorophenyl) methyl]-4-benzyloxy-1H-indole-3-methanol was used as internal standard. The levels of compound 34 were normalized with protein amount in the samples.
Acknowledgments
We thank Dr. Joel Morris at the National Institute of Health for helpful suggestions and Stephanie Deming for editorial review of this manuscript. This work was supported in part by Cancer Center Support Grant CA-016672 to The University of Texas MD Anderson Cancer Center, which supports the Lung Program, the Nuclear Magnetic Resonance Facility, and the DNA Analysis Facility; National Cancer Institute grants R01 CA 092487 and R01 CA 124951 (B. Fang); the Rapid Access to Interventional Development Program; INNOFUND of China (09C26213301228) and National Natural Science Foundation of China No.30973563 (X Wei).
Footnotes
Abbreviations: CDK, cyclin-dependent kinase; CTD, C-terminal domain; HPLC, high-performance liquid chromatography; IC50, dose that causes a 50% reduction in the number of surviving cells compared with the number of surviving cells in the control group; MS, mass spectrometry; NMR, nuclear magnetic resonance.
References
- 1.Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, Sougnez C, Greulich H, Muzny DM, Morgan MB, Fulton L, Fulton RS, Zhang Q, Wendl MC, Lawrence MS, Larson DE, Chen K, Dooling DJ, Sabo A, Hawes AC, Shen H, Jhangiani SN, Lewis LR, Hall O, Zhu Y, Mathew T, Ren Y, Yao J, Scherer SE, Clerc K, Metcalf GA, Ng B, Milosavljevic A, Gonzalez-Garay ML, Osborne JR, Meyer R, Shi X, Tang Y, Koboldt DC, Lin L, Abbott R, Miner TL, Pohl C, Fewell G, Haipek C, Schmidt H, Dunford-Shore BH, Kraja A, Crosby SD, Sawyer CS, Vickery T, Sander S, Robinson J, Winckler W, Baldwin J, Chirieac LR, Dutt A, Fennell T, Hanna M, Johnson BE, Onofrio RC, Thomas RK, Tonon G, Weir BA, Zhao X, Ziaugra L, Zody MC, Giordano T, Orringer MB, Roth JA, Spitz MR, Wistuba II, Ozenberger B, Good PJ, Chang AC, Beer DG, Watson MA, Ladanyi M, Broderick S, Yoshizawa A, Travis WD, Pao W, Province MA, Weinstock GM, Varmus HE, Gabriel SB, Lander ES, Gibbs RA, Meyerson M, Wilson RK. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–1075. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, Silliman N, Szabo S, Dezso Z, Ustyanksky V, Nikolskaya T, Nikolsky Y, Karchin R, Wilson PA, Kaminker JS, Zhang Z, Croshaw R, Willis J, Dawson D, Shipitsin M, Willson JK, Sukumar S, Polyak K, Park BH, Pethiyagoda CL, Pant PV, Ballinger DG, Sparks AB, Hartigan J, Smith DR, Suh E, Papadopoulos N, Buckhaults P, Markowitz SD, Parmigiani G, Kinzler KW, Velculescu VE, Vogelstein B. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 3.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, Haber DA. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 4.Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, Zimmermann J, Lydon NB. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nature Med. 1996;2:561–566. doi: 10.1038/nm0596-561. [DOI] [PubMed] [Google Scholar]
- 5.Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, Chen C, Zhang X, Vincent P, McHugh M, Cao Y, Shujath J, Gawlak S, Eveleigh D, Rowley B, Liu L, Adnane L, Lynch M, Auclair D, Taylor I, Gedrich R, Voznesensky A, Riedl B, Post LE, Bollag G, Trail PA. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64:7099–7109. doi: 10.1158/0008-5472.CAN-04-1443. [DOI] [PubMed] [Google Scholar]
- 6.Torrance CJ, Agrawal V, Vogelstein B, Kinzler KW. Use of isogenic human cancer cells for high-throughput screening and drug discovery. Nature Biotech. 2001;19:940–945. doi: 10.1038/nbt1001-940. [DOI] [PubMed] [Google Scholar]
- 7.Dolma S, Lessnick SL, Hahn WC, Stockwell BR. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell. 2003;3:285–296. doi: 10.1016/s1535-6108(03)00050-3. [DOI] [PubMed] [Google Scholar]
- 8.Guo W, Wu S, Liu J, Fang B. Identification of a small molecule with synthetic lethality for K-ras and protein kinase C iota. Cancer Res. 2008;68:7403–7408. doi: 10.1158/0008-5472.CAN-08-1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaelin WG., Jr The concept of synthetic lethality in the context of anticancer therapy. Nature Rev Cancer. 2005;5:689–698. doi: 10.1038/nrc1691. [DOI] [PubMed] [Google Scholar]
- 10.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase.[Erratum appears in Nature. 2007 May 17;447(7142):346] Nature. 2005;434:913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 11.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC, Ashworth A. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 12.Koumenis C, Giaccia A. Transformed cells require continuous activity of RNA polymerase II to resist oncogene-induced apoptosis. Mol Cell Biol. 1997;17:7306–7316. doi: 10.1128/mcb.17.12.7306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baumli S, Lolli G, Lowe ED, Troiani S, Rusconi L, Bullock AN, Debreczeni JE, Knapp S, Johnson LN. The structure of P-TEFb (CDK9/cyclin T1), its complex with flavopiridol and regulation by phosphorylation. EMBO J. 2008;27:1907–1918. doi: 10.1038/emboj.2008.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chao SH, Price DH. Flavopiridol inactivates P-TEFb and blocks most RNA polymerase II transcription in vivo. J Biol Chem. 2001;276:31793–31799. doi: 10.1074/jbc.M102306200. [DOI] [PubMed] [Google Scholar]
- 15.Maccallum DE, Melville J, Frame S, Watt K, Anderson S, Gianella-Borradori A, Lane DP, Green SR. Seliciclib (CYC202, R-Roscovitine) induces cell death in multiple myeloma cells by inhibition of RNA polymerase II-dependent transcription and down-regulation of Mcl-1. Cancer Res. 2005;65:5399–5407. doi: 10.1158/0008-5472.CAN-05-0233. [DOI] [PubMed] [Google Scholar]
- 16.Chen R, Keating MJ, Gandhi V, Plunkett W. Transcription inhibition by flavopiridol: mechanism of chronic lymphocytic leukemia cell death. Blood. 2005;106:2513–2519. doi: 10.1182/blood-2005-04-1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wittmann S, Bali P, Donapaty S, Nimmanapalli R, Guo F, Yamaguchi H, Huang M, Jove R, Wang HG, Bhalla K. Flavopiridol down-regulates antiapoptotic proteins and sensitizes human breast cancer cells to epothilone B-induced apoptosis. Cancer Res. 2003;63:93–99. [PubMed] [Google Scholar]
- 18.Raje N, Kumar S, Hideshima T, Roccaro A, Ishitsuka K, Yasui H, Shiraishi N, Chauhan D, Munshi NC, Green SR, Anderson KC. Seliciclib (CYC202 or R-roscovitine), a small-molecule cyclin-dependent kinase inhibitor, mediates activity via downregulation of Mcl-1 in multiple myeloma. Blood. 2005;106:1042–1047. doi: 10.1182/blood-2005-01-0320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guo W, Wu S, Wang L, Wang R, Wei L, Liu J, Fang B. Interruption of RNA processing machinery by a small compound 1-[(4-chlorophenyl) methyl]-1H-indole-3-carboxaldehyde (oncrasin-1) Mol Cancer Ther. 2009;8:441–448. doi: 10.1158/1535-7163.MCT-08-0839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Archambault J, Chambers RS, Kobor MS, Ho Y, Cartier M, Bolotin D, Andrews B, Kane CM, Greenblatt J. An essential component of a C-terminal domain phosphatase that interacts with transcription factor IIF in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 1997;94:14300–14305. doi: 10.1073/pnas.94.26.14300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bird G, Zorio DA, Bentley DL. RNA polymerase II carboxy-terminal domain phosphorylation is required for cotranscriptional pre-mRNA splicing and 3′-end formation. Mol Cell Biol. 2004;24:8963–8969. doi: 10.1128/MCB.24.20.8963-8969.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hirose Y, Tacke R, Manley JL. Phosphorylated RNA polymerase II stimulates pre-mRNA splicing. Genes Dev. 1999;13:1234–1239. doi: 10.1101/gad.13.10.1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim E, Du L, Bregman DB, Warren SL. Splicing factors associate with hyperphosphorylated RNA polymerase II in the absence of pre-mRNA. J Cell Biol. 1997;136:19–28. doi: 10.1083/jcb.136.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McCracken S, Fong N, Yankulov K, Ballantyne S, Pan G, Greenblatt J, Patterson SD, Wickens M, Bentley DL. The C-terminal domain of RNA polymerase II couples mRNA processing to transcription. Nature. 1997;385:357–361. doi: 10.1038/385357a0. [DOI] [PubMed] [Google Scholar]
- 25.Millhouse S, Manley JL. The C-terminal domain of RNA polymerase II functions as a phosphorylation-dependent splicing activator in a heterologous protein. Mol Cell Biol. 2005;25:533–544. doi: 10.1128/MCB.25.2.533-544.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Misteli T, Spector DL. RNA polymerase II targets pre-mRNA splicing factors to transcription sites in vivo. Mol Cell. 1999;3:697–705. doi: 10.1016/s1097-2765(01)80002-2. [DOI] [PubMed] [Google Scholar]
- 27.Chinni SR, Li Y, Upadhyay S, Koppolu PK, Sarkar FH. Indole-3-carbinol (I3C) induced cell growth inhibition, G1 cell cycle arrest and apoptosis in prostate cancer cells. Oncogene. 2001;20:2927–2936. doi: 10.1038/sj.onc.1204365. [DOI] [PubMed] [Google Scholar]
- 28.Garcia HH, Brar GA, Nguyen DH, Bjeldanes LF, Firestone GL. Indole-3-carbinol (I3C) inhibits cyclin-dependent kinase-2 function in human breast cancer cells by regulating the size distribution, associated cyclin E forms, and subcellular localization of the CDK2 protein complex. J Biol Chem. 2005;280:8756–8764. doi: 10.1074/jbc.M407957200. [DOI] [PubMed] [Google Scholar]
- 29.Liu J, Yang G, Thompson-Lanza JA, Glassman A, Hayes K, Patterson A, Marquez RT, Auersperg N, Yu Y, Hahn WC, Mills GB, Bast RC., Jr A genetically defined model for human ovarian cancer. Cancer Res. 2004;64:1655–1663. doi: 10.1158/0008-5472.can-03-3380. [DOI] [PubMed] [Google Scholar]
- 30.Anderton MJ, Manson MM, Verschoyle RD, Gescher A, Lamb JH, Farmer PB, Steward WP, Williams ML. Pharmacokinetics and tissue disposition of indole-3-carbinol and its acid condensation products after oral administration to mice. Clinical Cancer Res. 2004;10:5233–5241. doi: 10.1158/1078-0432.CCR-04-0163. [DOI] [PubMed] [Google Scholar]
- 31.Oelgeschlager T. Regulation of RNA polymerase II activity by CTD phosphorylation and cell cycle control. J Cell Physiol. 2002;190:160–169. doi: 10.1002/jcp.10058. [DOI] [PubMed] [Google Scholar]
- 32.Mortillaro MJ, Blencowe BJ, Wei X, Nakayasu H, Du L, Warren SL, Sharp PA, Berezney R. A hyperphosphorylated form of the large subunit of RNA polymerase II is associated with splicing complexes and the nuclear matrix. Proc Natl Acad Sci U S A. 1996;93:8253–8257. doi: 10.1073/pnas.93.16.8253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Larochelle S, Chen J, Knights R, Pandur J, Morcillo P, Erdjument-Bromage H, Tempst P, Suter B, Fisher RP. T-loop phosphorylation stabilizes the CDK7-cyclin H-MAT1 complex in vivo and regulates its CTD kinase activity. EMBO J. 2001;20:3749–3759. doi: 10.1093/emboj/20.14.3749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shiekhattar R, Mermelstein F, Fisher RP, Drapkin R, Dynlacht B, Wessling HC, Morgan DO, Reinberg D. Cdk-activating kinase complex is a component of human transcription factor TFIIH. Nature. 1995;374:283–287. doi: 10.1038/374283a0. [DOI] [PubMed] [Google Scholar]
- 35.Akoulitchev S, Chuikov S, Reinberg D. TFIIH is negatively regulated by cdk8-containing mediator complexes. Nature. 2000;407:102–106. doi: 10.1038/35024111. [DOI] [PubMed] [Google Scholar]
- 36.Leclerc V, Tassan JP, O’Farrell PH, Nigg EA, Leopold P. Drosophila Cdk8, a kinase partner of cyclin C that interacts with the large subunit of RNA polymerase II. Mol Biol Cell. 1996;7:505–513. doi: 10.1091/mbc.7.4.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim YK, Bourgeois CF, Isel C, Churcher MJ, Karn J. Phosphorylation of the RNA polymerase II carboxyl-terminal domain by CDK9 is directly responsible for human immunodeficiency virus type 1 Tat-activated transcriptional elongation. Mol Cell Biol. 2002;22:4622–4637. doi: 10.1128/MCB.22.13.4622-4637.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peterson SR, Jesch SA, Chamberlin TN, Dvir A, Rabindran SK, Wu C, Dynan WS. Stimulation of the DNA-dependent protein kinase by RNA polymerase II transcriptional activator proteins. Journal of Biological Chemistry. 1995;270:1449–1454. doi: 10.1074/jbc.270.3.1449. [DOI] [PubMed] [Google Scholar]
- 39.Bonnet F, Vigneron M, Bensaude O, Dubois MF. Transcription-independent phosphorylation of the RNA polymerase II C-terminal domain (CTD) involves ERK kinases (MEK1/2) Nucleic Acids Res. 1999;27:4399–4404. doi: 10.1093/nar/27.22.4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Baskaran R, Chiang GG, Wang JY. Identification of a binding site in c-Ab1 tyrosine kinase for the C-terminal repeated domain of RNA polymerase II. Mol Cell Biol. 1996;16:3361–3369. doi: 10.1128/mcb.16.7.3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baskaran R, Dahmus ME, Wang JY. Tyrosine phosphorylation of mammalian RNA polymerase II carboxyl-terminal domain. Proc Natl Acad Sci U S A. 1993;90:11167–11171. doi: 10.1073/pnas.90.23.11167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu S, Zhu H, Gu J, Zhang L, Teraishi F, Davis JJ, Jacob DA, Fang B. Induction of Apoptosis and Down-regulation of Bcl-XL in Cancer Cells by a novel small molecule 2[[3-(2,3-dichlorophenoxy)propyl]amino] ethanol (2,3-DCPE) Cancer Res. 2003;64:1110–1113. doi: 10.1158/0008-5472.can-03-2790. [DOI] [PubMed] [Google Scholar]