

Abstract
Rare, inactivating mutations in the BRCA1 gene appear to play a limited role in prostate cancer. To our knowledge, however, no study has comprehensively assessed the role of other BRCA1 sequence variations, e.g., missense mutations, in prostate cancer. In a study of 817 men with and without prostate cancer from 323 familial and early-onset prostate cancer families, we used family-based association tests and conditional logistic regression to investigate the association between prostate cancer and single nucleotide polymorphisms (SNPs) tagging common haplotype variation in a 200 kb-region surrounding (and including) the BRCA1 gene. We also used the Genotype-IBD Sharing Test (GIST) to determine whether our most strongly associated SNP could account for prostate cancer linkage to chromosome 17q21 in a sample of 154 families from our previous genome-wide linkage study. The strongest evidence for prostate cancer association was for a glutamine-to-arginine substitution at codon 356 (Gln356Arg) in exon 11 of the BRCA1 gene. The minor (Arg) allele was preferentially transmitted to affected men (p=0.005 for a dominant model), with an estimated odds ratio of 2.25 (95% confidence interval = 1.21 to 4.20). Notably, BRCA1 Gln356Arg is not in strong linkage disequilibrium with other BRCA1 coding SNPs or any known HapMap SNP on chromosome 17. In addition, GIST results suggest that Gln356Arg accounts (in part) for our prior evidence of prostate cancer linkage to chromosome 17q21 (p=0.022). Thus, we have identified a common, non-synonymous substitution in the BRCA1 gene that is associated with and linked to prostate cancer.
Keywords: prostate, cancer, genetics, BRCA1, risk
Introduction
Germ-line, loss-of-function mutations in the BRCA1 gene on chromosome 17q21 substantially increase the lifetime risk of developing breast and ovarian cancer. BRCA1 mutations may also confer an increased risk of other cancers. Specifically, some studies (although not all(1;2)) have suggested an increased risk of prostate cancer among male carriers of deleterious BRCA1 mutations in breast and ovarian cancer families(3–5). For example, Ford et al.(3) reported a significant excess of prostate cancer cases in breast and ovarian families with evidence of linkage to BRCA1. Similarly, based on families with a history of breast and/or ovarian cancer and with at least one individual known to carry a pathogenic mutation in the BRCA1 gene, Thompson et al.(5) reported an approximately two-fold increased relative risk of prostate cancer in BRCA1 mutation carriers compared to non-carriers, although the effect was restricted to men who were young at their time of diagnosis (less than 65 years of age).
In a previous genome-wide scan (GWS) based on 175 families from the University of Michigan Prostate Cancer Genetics Project (PCGP), our strongest signal for prostate cancer linkage was on chromosome 17q21 (maximum logarithm of odds (LOD) scores (MLSs) of 2.36 in all families and 3.28 in the subset of families with 4 or more affected men)(6). Similarly, in a subsequent, combined genome-wide linkage analyses of 1,233 families from ten(7) independent studies of hereditary prostate cancer (including the 175 families from our PCGP study) chromosome 17q21 was among the top five regions with evidence of prostate cancer linkage. Notably, the strongest linkage signal for hereditary prostate cancer in the PCGP study is within 5 cM of the BRCA1 gene (on chromosome 17q21), suggesting the presence of a susceptibility locus near, if not within, the BRCA1 gene region.
Since the majority of identified BRCA1 mutations are nonsense or frameshift mutations that result in a truncated protein product, most studies to date have focused exclusively on the relationship between obvious loss-of-function mutations (e.g., protein truncating mutations) in BRCA1 and prostate cancer. Screening of hereditary prostate cancer families (including our own(8)), however, suggests that these rare, highly penetrant mutations are unlikely to account for a sizeable fraction of hereditary prostate cancer cases(9–11). Still, to our knowledge, no large, comprehensive study has definitively assessed the role of other sequence variations in BRCA1, e.g., missense mutations, as a contributor to prostate cancer risk. To more comprehensively survey genetic variation in the BRCA1 gene in relation to prostate cancer risk, we genotyped a set of SNPs tagging haplotype variation in a 200 kb-region surrounding (and including) the BRCA1 gene and assessed their association with prostate cancer in a family-based association study of 323 families from the University of Michigan PCGP(12).
Materials and Methods
Study Subjects
The PCGP is a large, ongoing family-based study designed to identify genes predisposing to inherited forms of prostate cancer. Enrollment into the PCGP is restricted to (1) families with two or more living members with prostate cancer in a first- or second-degree relationship or (2) men diagnosed with prostate cancer at ≤55 years of age without a family history of the disease. All participants are asked to provide a blood sample, extended family history information, and access to medical records. For the present investigation, we identified 338 families in which we had DNA from at least one pair of brothers discordant for prostate cancer. These discordant sibling pairs (DSPs) were selected from a single generation to mitigate potential cohort effects. We also preferentially enrolled the oldest available unaffected brother from each family to maximize the probability that unaffected men were truly unaffected and not simply unaffected by virtue of being younger than their affected brother(s). Additional male siblings as well as multiple sibships from the same family were included if DNA was available.
The majority of the PCGP families were recruited directly from the University of Michigan Comprehensive Cancer Center. Other sources included direct patient or physician referrals. Diagnosis of prostate cancer was confirmed by review of pathology reports or medical records, and age at diagnosis was calculated from the date of the first biopsy positive for prostate cancer. Cases were classified as clinically aggressive if they met at least one of the following criteria: (1) pathologic Gleason sum > 7, (2) pathologic stage T3b (pT3b) tumor (indicating seminal vesicle involvement) or pT4 or N1 (positive regional lymph nodes), (3) pathologic Gleason sum of 7 and a positive margin, or (4) pre-operative serum prostate-specific antigen (PSA) value >15 ng/ml, or a biopsy Gleason score > 7, or a serum PSA level > 10 ng/ml and a biopsy Gleason score > 6. Based on data from D’Amico et al.(13), these criteria were developed by the Southwest Oncology Group (protocol 9921) to identify men at intermediate to high risk of clinical recurrence after primary therapy. Disease status of the unaffected brothers was confirmed through serum PSA testing whenever possible.
The majority of the families were non-Hispanic white, although 13 African American and 2 Asian families were also recruited. All of the following results, however, were restricted to the sample of 323 non-Hispanic white families. This decision was supported by an analysis of the HapMap(14) samples, which revealed substantial allele frequency differences and dissimilar linkage disequilibrium (LD) patterns in the BRCA1 gene region between African, Asian, and European samples. The Institutional Review Board at the University of Michigan Medical School approved all aspects of the protocol, and all participants gave written informed consent, including permission to release their medical records.
Genotyping Assays
Genomic DNA was isolated from whole blood using the Puregene kit (Gentra Systems Inc., Minneapolis, MN). A validated SNP, rs7223952, located downstream of BRCA1, was selected and genotyped from the Assays-On-Demand catalog (Applied Biosystems, Foster City, CA). After this SNP displayed significant association with prostate cancer, we used the haplotype tagging strategy described below to select and genotype six additional SNPs: rs2271539 in intron 2 of the RPL27 gene; rs691144 in intron 1 of the IFI35 gene; and rs1799966, rs3737559, rs1799950, and rs799923 in exon 17, intron 13, exon 11, and intron 6, respectively, of the BRCA1 gene. We genotyped all 7 SNPs with the TaqMan allelic discrimination assay, and we used the ABI PRISM 7900HT Sequence Detection System (Applied Biosystems) to distinguish SNP alleles as previously described(12). Assay details are available from the authors on request. On average, we achieved a genotyping call rate of 98.94%, with call rates >97.46% for each SNP. We sequenced SNPs that were undetermined by the assay for a final genotyping call rate of 100%. A subset of samples were also duplicated and verified by either TaqMan SNP genotyping or direct sequencing. We observed 3 discrepancies among 363 duplicate genotype pairs by TaqMan SNP genotyping and 1 discrepancy among 166 duplicate genotype pairs by direct sequencing, yielding genotyping reproducibility rates of 99.2% and 99.4%, respectively.
Haplotype Tagging Strategy
Based on the HapMap(14) data (February 2005 release), a total of 71 SNPs mapped to the 200 kb interval around our originally associated SNP, rs7223952, and 41 of these SNPs were present in at least one of 60 unrelated individuals from a U.S. Utah population with northern and western European ancestry (abbreviated CEU). Using these data, we applied the dynamic programming algorithm proposed by Zhang et al.(15) and implemented in HapBlock(16) (version 3.0) to partition this 200 kb region into blocks and select a maximally informative set of SNPs. Specifically, we defined common haplotypes as those having frequency >3% (i.e., haplotypes inferred to be present at least 4 times among 120 chromosomes), and we defined a consecutive set of SNPs as a block if common haplotypes accounted for at least 80% of all predicted haplotypes. We then determined haplotype-tag SNPs (htSNPs) as the minimum set of SNPs that distinguished all common haplotypes inferred within each block. Based on these criteria, the 41 SNPs clustered into two, non-overlapping blocks of limited haplotype diversity, and six htSNPs (rs2271539 and rs691144 in the first block and rs1799966, rs3737559, rs1799950, and rs799923 in the second block) distinguished 94% and 96% of all haplotypes inferred within the first and second blocks, respectively (Figure 1). SNP rs7223952, which is not present in the HapMap database, is located in the second block.
Figure 1.

Haplotype tag SNPs (htSNPs) and pair-wise linkage disequilibrium (LD) as measured by R2 in a 200 kb-region surrounding (and including) the BRCA1 gene. Blocks represent consecutive sets of SNPs for which common haplotypes (with a frequency of at least 3%) accounted for at least 80% of all predicted haplotypes. Two SNPs in the first block, rs2271539 and rs691144, and four SNPs in the second block, rs1799966, rs3737559, rs1799950, and rs799923, distinguished 94% and 96%, respectively, of haplotypes inferred within the first and second blocks. SNP rs7223952, which is not present in the HapMap database, is not an htSNP but is located in the second block and was in high LD with htSNP rs1799966 (R2=0.94). Data are based on 323 unrelated, unaffected non-Hispanic white men from our family-based association study.
Data Analysis Methods
The observed genotype distributions were tested for departures from Hardy-Weinberg equilibrium in a subset of unrelated men by selecting the oldest unaffected man from each family. Two-SNP haplotype frequencies were estimated using the expectation-maximization algorithm and were used to calculate the LD measure R2 between each pair of SNPs. For comparison, we also estimated minor allele frequencies, two-SNP haplotypes, and LD in unrelated individuals from the HapMap CEU sample (n=60).
We used conditional logistic regression with family as the stratification variable and a robust variance estimate that incorporates familial correlations due to potential linkage(17) to estimate odds ratios (OR’s) and 95% confidence intervals (CI’s) for the association between genotypes and prostate cancer. In parallel, we used the Family-Based Association Test (FBAT) program (version 1.5.5) to test for association between genotypes and prostate cancer. FBATs are a class of generalized score statistics that utilize within- and between-family marker-inheritance patterns to test for association (18;19). Given our prior evidence of prostate cancer linkage to chromosome 17q21, we employed the empirical variance function in FBAT, which is a valid test of the null hypothesis of no association in the presence of linkage. To maximize power, we analyzed the combined sample of affected and unaffected men using the offset option. We also carried out affecteds-only analyses to allow for the possibility of misclassification of unaffected men (e.g., via reduced penetrance). Both conditional logistic regression analyses and FBATs were carried out assuming additive, dominant, and recessive genetic models. For conditional logistic regression and affecteds-only FBATs, we also examined a general (2 degrees of freedom) genotype model. Predetermined stratified analyses were also performed to explore the relationship between genotypes and prostate cancer, stratifying on clinically aggressive prostate cancer, age at diagnosis (<50 years), or number of confirmed cases of prostate cancer within a family (≥3).
To account for the number of correlated tests performed (i.e., 3 genetic models for each of 7 SNPs), we performed permutation tests to assess the overall significance of our primary FBAT results in the combined sample of affected and unaffected men. Specifically, we created 1,000 permuted samples by randomly permuting the affection status labels of genotyped men within each sibship (i.e., leaving intact the vector of correlated SNPs). For each permuted sample, we performed the FBATs described above assuming additive, dominant, and recessive genetic models. We then compared the number of significant associations in the permuted samples with the number of significant associations originally found. The overall p-value was computed as the proportion of permuted samples having at least as many tests with p-values less than or equal to the least significant p-value from the analyses of the original data.
To assess the association of haplotypes with prostate cancer, we divided each of the htSNPs by block and examined two- and four-SNP haplotypes corresponding to the first and second blocks, respectively. We also examined six-SNP haplotypes by combining SNPs from both blocks. Because our originally associated SNP, rs7223952, was in strong LD (R2=0.94) with one of the htSNPs, rs1799966, we excluded it from our haplotype-based analyses. All haplotypes were analyzed using the haplotype FBAT (HBAT) method(20). We jointly tested all n-SNP haplotypes (where n=2, 4, or 6) for association with prostate cancer (i.e., a global test). We also tested each individual haplotype for association with prostate cancer, assuming additive, dominant, and recessive genetic models. As described above for FBAT, we used the empirical variance option to account for prostate cancer linkage to this region and the offset option to weight the contribution of unaffected and affected subjects.
To determine whether our most strongly associated SNP, rs1799950, explained our prior evidence of prostate cancer linkage to chromosome 17q21, we genotyped it in 154 of the original 157 GWS families of non-Hispanic white descent(6). These 154 families included 411 affected and 72 unaffected men for whom we had sufficient DNA. We then used the Genotype-IBD Sharing Test (GIST) proposed by Li et al.(21) and implemented in version 0.3 of their program to determine if the linkage signal was explained by rs1799950. The GIST tests for a positive correlation between family-specific weights (based on genotypes of affected family members, e.g., at rs1799950, and the model of interest, e.g., dominant) and family-based identity-by-descent (IBD) sharing as represented by the non-parametric linkage (NPL) score. Using the 15 microsatellite markers on chromosome 17 from our original GWS(6), we re-calculated non-parametric multipoint LOD scores using Merlin(22) (version 1.0.0) with the ‘pairs’ scoring statistic, the exponential model, and equal weights for each of the 154 families. To investigate the evidence for linkage in the families without (with) the risk allele, we re-computed non-parametric multipoint LOD scores in the subset of families in which no affected men (at least one affected man) carried the risk allele.
All statistical tests were two-sided, and p-values < 0.05 were considered statistically significant. Conditional logistic regression was conducted using version 8.2 of the SAS programming language (SAS Institute, Cary, NC). All remaining analyses (except where noted above) were conducted using the R language (version 2.1.1).
Results
Characteristics of the Families and Men
For this investigation, we identified 323 non-Hispanic white families with at least one discordant sibling pair (DSP), resulting in a total of 516 DSPs. Of the 323 families, 316 included only the index case and one or more of his brothers. The remaining 7 families included additional DSPs unrelated to the index case as a brother (e.g., a pair of DSPs related as first cousins). Approximately 32%, 38%, and 30% of families included one, two, and three or more men with prostate cancer, respectively. The total sample consisted of 817 men (434 affected and 383 unaffected men). The clinical characteristics of the men with prostate cancer are shown in Table 1. The median age at diagnosis was 55 years (inter-quartile range=50–63 years). The median age of unaffected men at their time of consent was 56 years (inter-quartile range=50–63 years). Approximately 76% of unaffected men reported their most recent PSA testing results and/or had their PSA values confirmed by medical record review, and ~95% of them reported and/or had a PSA level <4.0 mg/dL or normal. At the time of consent, unaffected men were significantly older than their affected brothers were at their time of diagnosis (p<0.0001 for paired t-test of within family means), with a mean age difference of ~3 years.
Table 1.
Characteristics of men with prostate cancer (n=434)
| Trait | No.* (%) |
|---|---|
| Age at diagnosis† | 55 [50–63] |
| Pre-diagnosis PSA† | 5.7 [4.3–9.6] |
| Surgery‡ (% yes) | 333 (77%) |
| Stage: | |
| Localized | 323 (77%) |
| Locally advanced | 78 (19%) |
| Metastasized | 16 (4%) |
| Gleason: | |
| ≤6 | 209 (49%) |
| 7 | 171 (40%) |
| >7 | 45 (11%) |
| Clinically aggressive CaP (%) | 153 (35%) |
Column subtotals do not sum to 434 due to missing data.
Median and [interquartile range] are reported.
Number and (percentage) of men with prostate cancer who underwent a radical prostatectomy.
Allele Frequencies and Disequilibrium Analyses
Initially, we genotyped SNP rs7223952, which is located ~1.6 kb downstream of the poly-adenylation site of the BRCA1 gene on chromsome 17q21. Based on the haplotype tagging strategy described above (see Materials and Methods), we then genotyped six additional SNPs spanning a 200 kb region around rs7223952. Positions and minor allele frequencies for all seven SNPs are given in Table 2 for affected and unaffected men. The observed genotype data were consistent with Hardy-Weinberg equilibrium. The pattern of LD in the 200-kb region surrounding rs7223952 is shown in Figure 1 for the sample of unrelated, unaffected men. Consistent with our haplotype tagging strategy, 16 of all 21 SNP pairs exhibited weak LD (R2<0.2). SNP rs7223952, which was not present in the HapMap database and therefore did not inform our htSNP selection strategy, was in strong LD with htSNP rs1799966 (R2=0.94). In general, the LD patterns in Figure 1 were consistent with those predicted by the HapMap CEU sample (data not shown).
Table 2.
Allele frequencies for SNPs in or near the BRCA1 gene by prostate cancer affection status
| Minor allele frequency in | ||||||
|---|---|---|---|---|---|---|
| Gene | SNP name | Location | Position* | Major>minor allele | Affected men (n=434) | Unaffected men (n=383) |
| RPL27 | rs2271539 | Intron 2 | −43.936 | T>C | 0.28 | 0.30 |
| IFI35 | rs691144 | Intron 1 | −31.236 | G>A | 0.19 | 0.19 |
| Intergenic | rs7223952 | - | 0 | A>G | 0.29 | 0.31 |
| BRCA1 | rs1799966 | Exon 17 | 28.209 | T>C | 0.28 | 0.30 |
| BRCA1 | rs3737559 | Intron 13 | 39.419 | G>A | 0.06 | 0.08 |
| BRCA1 | rs1799950 | Exon 11 | 51.596 | A>G | 0.08 | 0.06 |
| BRCA1 | rs799923 | Intron 6 | 57.046 | G>A | 0.28 | 0.26 |
Kilobase position relative to SNP rs7223952 according to the reference sequence (UCSC Genome Browser, NCBI Build 36.1, March 2006).
Single SNP and Haplotype-Based Association Analyses
Table 3 summarizes results for all seven SNPs using both conditional logistic regression and family-based association tests. Conditional logistic regression analyses were based on a single sib-ship per family (799 affected and unaffected men or 506 DSPs). Initial FBAT analyses included only affected men (n=434), while the primary, combined analyses (reported below) included all affected and unaffected men (n=817). We initially identified a weak association between prostate cancer and SNP rs7223952 downstream of the BRCA1 gene. However, after genotyping an additional 6 htSNPs, our strongest evidence for prostate cancer association was for SNP rs1799950, which results in a glutamine-to-arginine substitution at codon 356 (Gln356Arg) in exon 11 of the BRCA1 gene. The minor allele of rs1799950 was preferentially transmitted to affected men (z = 2.79; p=0.005 for a dominant model), with an odds ratio of 2.25 (95% CI = 1.21–4.20; p-value=0.011). There was an insufficient number of Arg/Arg genotypes to evaluate other genetic models for rs1799950. Another SNP in the BRCA1 gene, rs3737559 in intron 13, also revealed significant evidence of prostate cancer association (z=−2.37; p=0.018 for a dominant model). This SNP was less strongly but still significantly associated with prostate cancer under an additive model (data not shown). In contrast, under an additive (but not dominant) genetic model, the minor allele at SNP rs799923 in intron 6 of the BRCA1 gene was preferentially transmitted to affected men (z=2.07; p=0.039), with an odds ratio of 1.39 (95% CI = 0.99,1.95; p=0.056). Each of these associated SNPs (rs1799950, rs3737559, and rs799923) were in weak LD (R2<0.2) with each other and with rs7223952 in our sample of unrelated, unaffected men (Figure 1). We found no evidence of an association between SNPs rs2271539, rs691144, or rs1799966 and prostate cancer. Thus, a total of 3 SNPs exhibited significant prostate cancer association in 5 FBATs for the combined sample of affected and unaffected men. Among 1,000 permuted samples, only 24 samples had 5 or more FBAT results with a p-value ≤ 0.039 (our least significant FBAT p-value), indicating that these association results are unlikely to be due to chance alone (p=0.024).
Table 3.
Association results from conditional logistic regression and family-based association tests (FBATs)
| Discordant sib pairs (DSPs) | Affected men | Affected and unaffected men | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| SNP name | Genotype* | Odds ratio | 95% CI | P-value | Families† | Z-score | P-value | Families† | Z-score | P-value |
| rs2271539 | CC vs CT or TT | 0.69 | (0.47,1.01) | 0.06 | 117 | −1.73 | 0.08 | 119 | −1.78 | 0.08 |
| rs691144 | GG vs GA or AA | 0.82 | (0.54,1.24) | 0.34 | 97 | −0.91 | 0.36 | 100 | −0.93 | 0.35 |
| rs7223952 | AA vs AG or GG | 0.70 | (0.47,1.04) | 0.08 | 112 | −1.89 | 0.06 | 113 | −1.84 | 0.07 |
| rs1799966 | TT vs TC or CC | 0.70 | (0.47,1.05) | 0.08 | 114 | −1.83 | 0.07 | 115 | −1.81 | 0.07 |
| rs3737559 | GG vs GA or AA | 0.43 | (0.21,0.89) | 0.02 | 45 | −2.20 | 0.03 | 47 | −2.37 | 0.02 |
| rs1799950 | AA vs AG or GG | 2.25 | (1.21,4.20) | 0.01 | 49 | 2.66 | <0.01 | 49 | 2.79 | <0.01 |
| rs799923 | GG vs GA or AA | 1.46 | (0.98,2.17) | 0.06 | 106 | 1.90 | 0.06 | 106 | 1.89 | 0.06 |
NOTE: Results are based on 323 families (506 and 516 DSPs for conditional logistic regression and FBAT analyses, respectively).
First genotype is the referent group.
Number of informative families.
As described above, analyses were repeated after stratifying on clinically aggressive prostate cancer, age at diagnosis (<50 years), and number of confirmed cases of prostate cancer within a family (≥3). After stratification, the minor allele at SNP rs1799950 was preferentially transmitted to affected men in the subset of families in which affected men were diagnosed with prostate cancer at <50 years of age (z= 2.89; p=0.004 for a dominant model), with an odds ratio of 7.51 (95% CI = 0.74–76.23; p=0.082). In the subset of families with 3 or more confirmed affected men, the minor allele at SNP rs3737559 was preferentially transmitted to unaffected men (z=−2.49; p=0.013 for a dominant model), with an odds ratio of 0.23 (95% CI = 0.03,2.14; p=0.200). These stratified results are consistent with the FBAT results based on all families and are statistically significant despite a substantial loss of informative families, over half in both cases (12 versus 49 families for rs1799950 and 20 versus 47 families for rs3737559). Although SNP rs2271539 was not significantly associated with prostate cancer in the un-stratified analyses, the major allele was preferentially transmitted to affected men in the subset of families with 3 or more confirmed affected men (z=−2.53; p=0.012 for a dominant model), with an odds ratio of 0.40 (95% CI = 0.17–0.95; p=0.038). Results for SNPs rs3737559 and rs2271539 were also statistically significant under an additive model. We found no significant evidence of an association between SNPs rs691144, rs7223952, rs1799966, or rs799923 and prostate cancer in any of the stratified analyses.
Haplotype analysis did not reveal a risk haplotype or set of haplotypes that explained our prostate cancer associations substantially more than individual SNPs (data not shown). Further, the only significantly associated haplotype contained the most significantly associated SNP, rs1799950. For example, the 4-SNP haplotype uniquely defined by the minor allele at rs1799950 was over-transmitted to affected men (z=2.812; p=0.005 for a dominant model), consistent with single-SNP results for rs1799950. Similarly, the 6-SNP haplotype uniquely defined by the minor allele of rs1799950 was also significantly over-transmitted to affected men under a dominant model (z=2.317; p=0.020 for a dominant model).
Linkage Analyses
We followed up the most significant SNP, rs1799950, by genotyping it in our 154 non-Hispanic white GWS families and using the GIST to test whether it explained our original linkage signal on chromosome 17q21. It should be noted that 74 of these 154 GWS families overlapped with the 323 families from the current family-based association study (Table 4), in which we have already established evidence of prostate cancer linkage to rs1799950 (via FBAT). At least one affected man carrying at least one copy of the minor allele at rs1799950 was present in 25 of the 154 GWS families. Using the GIST, we found evidence for an association between the presence of the risk (or minor) allele in affected men and increased IBD allele sharing among brothers affected with prostate cancer (p=0.022 for a dominant model), suggesting that SNP rs1799950 contributes to our originally reported linkage signal on chromosome 17q21(6). Notably, evidence of suggestive prostate cancer linkage to chromosome 17q21 remained after we removed families in which affected men carried one or more copies of the minor allele at rs1799950 (MLS of 1.45 in 129 non-carrier families versus 1.93 in all 154 families) (Figure 2). By comparison, we observed a LOD score of 0.85 at rs1799950 in the 25 carrier families.
Table 4.
Number of families and men with and without prostate cancer from Genome-Wide Scan (GWS) and Discordant Sibling Pair (DSP) studies
| GWS study | DSP study | Overlap | |
|---|---|---|---|
| All: | |||
| Families | 154 | 323 | 74 |
| Affected men | 411 | 434 | 119 |
| Unaffected men | 72 | 383 | 57 |
|
| |||
| Arg356 carrier*: | |||
| Families | 25 | 61 | 13 |
| Affected men | 44 | 71 | 15 |
| Unaffected men | 8 | 47 | 7 |
NOTE: All families were enrolled in the University of Michigan Prostate Cancer Genetics Project (PCGP). GWS families were recruited for the purpose of conducting linkage analyses and were previously genotyped for 15 microsatellite markers on chromosome 17(6). DSP families were recruited for the purpose of conducting family-based association tests as described elsewhere(12). Numbers correspond to non-Hispanic white families and men, which represented >90% of all families and men from each study.
Carriers of the Arg356 allele of BRCA1 Gln356Arg (or equivalently, the minor allele at SNP rs1799950); excludes families with no affected male carriers.
Figure 2.
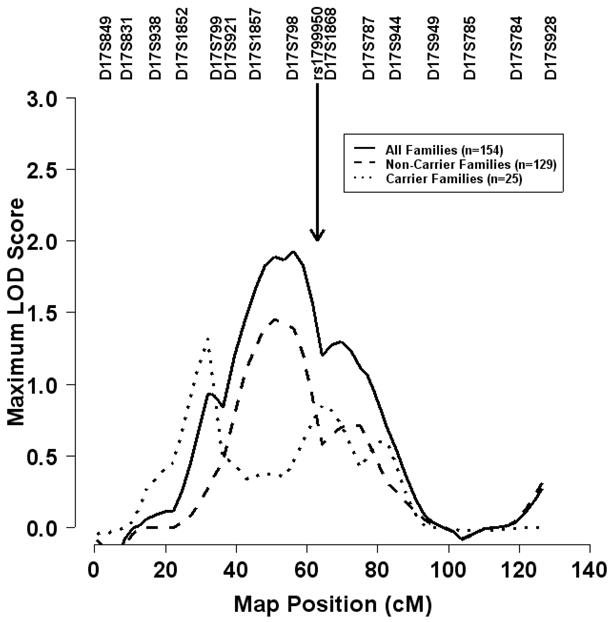
Non-parametric multipoint linkage analysis for prostate cancer on chromosome 17. Results are based on the exponential model, the ‘pairs’ scoring statistic, and equal weights for each of the families. The solid curve represents the maximum LOD (logarithm of the odds) score for 154 of the 157 non-Hispanic white families previously published by Lange et al(6). The dashed (dotted) curve represents the maximum LOD score for the 129 (25) families in which no affected men (at least one affected man) carried one or more copies of the Arg356 allele at BRCA Gln356Arg (or equivalently, SNP rs1799950).
Discussion
In summary, we found three common SNPs (i.e., minor allele frequency greater than 5%) in the BRCA1 gene, rs1799950, rs3737559, and rs799923, which are associated with prostate cancer and are in weak LD with each other. Our strongest evidence for prostate cancer association was for SNP rs1799950, or equivalently Gln356Arg. We estimate that men carrying at least one Arg356 allele are approximately twice as likely to develop prostate cancer as non-carriers. If BRCA1 Gln356Arg is causally related to prostate cancer, we estimate a population attributable fraction (PAF) of ~13%. This PAF is based on the frequency of the Arg356 allele in our sample of unaffected men and applies to the population of early onset, familial prostate cancer. Remarkably, this SNP also contributes, in part, to our prior evidence of prostate cancer linkage to chromosome 17q21(6). Thus, we now have evidence of both linkage and association between prostate cancer and common variation in the BRCA1 gene. Not unexpectedly, our results also suggest that BRCA1 Gln356Arg accounts only partially for prostate cancer linkage to chromosome 17q21, indicating that there are likely multiple functional variants in this region that influence prostate cancer susceptibility.
Notably, we found significant evidence of association in several different strata, including the subset of families in which men were diagnosed with prostate cancer at < 50 years of age (for SNP rs1799950) and the subset of families with 3 or more affected men (for SNPs rs2271539 and rs3737559). The former is consistent with the findings of Thompson et al.(5) that suggested an increased risk of prostate cancer in younger men carrying known BRCA1 truncating mutations (relative to the general population), and the latter is consistent with our previous report of a stronger linkage signal on chromosome 17q21 in families enriched for prostate cancer(6). Together, these findings suggest that particular subsets of families, possibly those with more heritable forms of prostate cancer, contributed disproportionately to our results. Of note, these strata defined only partially overlapping sets of families (i.e., less than 50% of informative families in one stratum were included in another stratum), suggesting the presence of genetic heterogeneity.
Several groups have previously examined the role of BRCA1 Gln356Arg in cancer susceptibility. For example, some(23;24) but not all groups(25–27) have reported an association between Gln356Arg and breast cancer in case-control studies, with the Arg356 allele conferring a reduction in breast cancer risk. At least three other groups(28–30) have also examined the role of BRCA1 Gln356Arg in case-control studies of ovarian cancer but found no evidence of an association. To our knowledge, no case-control study has been conducted to evaluate the role of BRCA1 Gln356Arg in prostate cancer, although Sinclair et al.(11) and we(8) previously identified BRCA1 Gln356Arg in two small-scale studies of high-risk prostate cancer families. Specifically, Sinclair et al.(11) found the Arg356 allele in 1 of 22 families after screening 43 individuals from families with at least three cases of prostate cancer and at least two cases of breast and/or ovarian cancer. Similarly, after screening for rare BRCA1 mutations in 93 unrelated men with prostate cancer from families with a strong history of the disease and evidence of linkage to chromosome 17q21, we initially reported only 1 family carrying the Arg356 allele(8) but subsequently confirmed that Arg356 was also present in 13 other families. Notably, we did not identify any known deleterious mutations in these 14 families, making it unlikely that they carry an obvious loss-of-function mutation (e.g., a protein truncating mutation) in BRCA1.
Analysis of the HapMap samples suggests that the Arg356 allele is absent in samples of African and Asian ancestry. Still, we observed 2 heterozygous men with prostate cancer in 1 of our 13 African American families (data not shown). In an independent case-control sample of unrelated African American men with (n=128) and without (n=342) prostate cancer from the Flint Men’s Health Study (FMHS)(31), we also identified 7 case (5.5%) and 5 control (1.5%) carriers. These data are consistent with European admixture in the African American population. They also suggest that African American men carrying the Arg356 allele are more likely to develop prostate cancer than non-carriers, similar to their European counterparts (based on the FMHS sample, age-adjusted OR = 4.17; 95% CI = 1.27–13.72; p=0.02). Of note, a previous analysis of population substructure in the FMHS sample did not reveal any significant differences in European ancestry between cases and controls(32), suggesting that genetic admixture is unlikely to generate false positive evidence for association.
Data from the FMHS add to the initial evidence from this study that genetic variation in the BRCA1 gene is associated with prostate cancer. Still, we acknowledge several study limitations. First, because our study included only a small number of families of African and Asian ancestry, we were unable to evaluate the role of BRCA1 Gln356Arg in prostate cancer susceptibility in these populations in the context of a family-based association design. Second, the absence of the Arg356 allele in HapMap samples of non-European descent suggests that the practical implications of our findings may be limited to specific populations. Third, our findings may not generalize to sporadic and/or late-onset prostate cancer since men in our study were ascertained from families with early-onset or familial prostate cancer. In fact, preliminary data (October 10, 2006 release) from the Cancer Genetic Markers of Susceptibility (CGEMS) project (a whole genome association study of prostate cancer in men from the Prostate, Lung, Colorectal, and Ovary study) suggests that the BRCA1 intronic SNPs that we tested, rs3737559 and rs799923, are not associated with sporadic prostate cancer. It is worth noting, however, that SNP rs1799950 (BRCA1 Gln356Arg) has not been examined in the CGEM project.
BRCA1 Gln356Arg is located in a region of exon 11 that binds Rad50 (which is part of the DNA damage repair complex) and the transcriptional repressor ZBRK1. Some(33;34) but not all(35) computational tools predict that the Gln356Arg substitution adversely affects BRCA1 protein function. These tools compare orthologous sequences to establish whether mutations occur in regions that are evolutionarily conserved and/or are evolving under selective pressure. Alignment of exon 11 BRCA1 sequences for 57 placental mammals indicates ~81% amino acid identity for the Gln allele at Gln356Arg (37). At present, however, there are insufficient experimental data available to evaluate the functional consequence, if any, of BRCA1 Gln356Arg.
It is important to note that we chose BRCA1 Gln356Arg not because of any prior knowledge of potential function but because it tags a common haplotype. Thus, it is possible that the associations observed in our study are due to LD with adjacent loci. In fact, it has been previously documented that the BRCA1 gene falls within a 200–400 kb region of chromosome 17q21 with suppressed recombination and strong LD(36;37). However, based on the HapMap CEU sample (January 2006 release), Gln356Arg does not show evidence of strong LD (R2≥ 0.5) with any known SNP on chromosome 17. Additionally, sequence analysis of the 93 unrelated men with prostate cancer from our previous screening study(8) indicates that Gln356Arg is not in LD (R2<0.07) with any coding SNP in the BRCA1 gene (data not shown). Our other two associated SNPs, rs3737559 and rs799923, are located in non-coding sequences with no obvious functional effect, and based on the HapMap CEU sample, have a pair-wise R2 ≥ 0.5 with only 3 and 11 other known SNPs, respectively, on chromosome 17, none of which are located in coding or known regulatory regions.
In summary, our findings support the hypothesis that common variation in the BRCA1 gene plays a role in prostate cancer susceptibility. Several lines of evidence suggest that BRCA1 Gln356Arg, in particular, may influence prostate cancer-susceptibility. These include: prostate cancer linkage to the BRCA1 region on chromosome 17q21 in two samples with limited overlap; significant prostate cancer association with the Gln356Arg substitution; evidence that Gln356Arg partially accounts for prostate cancer linkage to chromosome 17q21; and the absence of strong LD between Gln356Arg and any known SNP. Our results suggest that future studies of familial cancer risk should include careful consideration of common polymorphisms in genes implicated in hereditary cancer syndromes.
Acknowledgments
The University of Michigan Prostate Cancer Genetics Project (PCGP) is made possible by funds from the National Cancer Institute SPORE in Prostate Cancer (P50 CA69568) and R01 CA79596 (to KAC). We gratefully acknowledge Joseph Washburn for technical advice and the University of Michigan Comprehensive Cancer Center (UMCCC) DNA Microarray Facility, which is funded (in part) by a National Institute of Health support grant to the UMCCC (P30 CA46592). We thank all men and their families who generously volunteered their time to participate in our study. We also thank Drs. Eric Fearon, Stephen Gruber, and Patricia Peyser for helpful comments on an earlier draft of this manuscript.
References
- 1.Risch HA, McLaughlin JR, Cole DE, et al. Prevalence and penetrance of germline BRCA1 and BRCA2 mutations in a population series of 649 women with ovarian cancer. Am J Hum Genet. 2001;68(3):700–710. doi: 10.1086/318787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brose MS, Rebbeck TR, Calzone KA, Stopfer JE, Nathanson KL, Weber BL. Cancer risk estimates for BRCA1 mutation carriers identified in a risk evaluation program. J Natl Cancer Inst. 2002;94(18):1365–1372. doi: 10.1093/jnci/94.18.1365. [DOI] [PubMed] [Google Scholar]
- 3.Ford D, Easton DF, Bishop DT, Narod SA, Goldgar DE. Risks of cancer in BRCA1-mutation carriers. Breast Cancer Linkage Consortium. Lancet. 1994;343(8899):692–695. doi: 10.1016/s0140-6736(94)91578-4. [DOI] [PubMed] [Google Scholar]
- 4.Struewing JP, Hartge P, Wacholder S, et al. The risk of cancer associated with specific mutations of BRCA1 and BRCA2 among Ashkenazi Jews. N Engl J Med. 1997;336(20):1401–1408. doi: 10.1056/NEJM199705153362001. [DOI] [PubMed] [Google Scholar]
- 5.Thompson D, Easton DF. Cancer Incidence in BRCA1 mutation carriers. J Natl Cancer Inst. 2002;94(18):1358–1365. doi: 10.1093/jnci/94.18.1358. [DOI] [PubMed] [Google Scholar]
- 6.Lange EM, Gillanders EM, Davis CC, et al. Genome-wide scan for prostate cancer susceptibility genes using families from the University of Michigan prostate cancer genetics project finds evidence for linkage on chromosome 17 near BRCA1. Prostate. 2003;57(4):326–334. doi: 10.1002/pros.10307. [DOI] [PubMed] [Google Scholar]
- 7.Xu J, Dimitrov L, Chang BL, et al. A combined genomewide linkage scan of 1,233 families for prostate cancer-susceptibility genes conducted by the international consortium for prostate cancer genetics. Am J Hum Genet. 2005;77(2):219–229. doi: 10.1086/432377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zuhlke KA, Madeoy JJ, Beebe-Dimmer J, et al. Truncating BRCA1 mutations are uncommon in a cohort of hereditary prostate cancer families with evidence of linkage to 17q markers. Clin Cancer Res. 2004;10(18 Pt 1):5975–5980. doi: 10.1158/1078-0432.CCR-04-0554. [DOI] [PubMed] [Google Scholar]
- 9.Wilkens EP, Freije D, Xu J, et al. No evidence for a role of BRCA1 or BRCA2 mutations in Ashkenazi Jewish families with hereditary prostate cancer. Prostate. 1999;39(4):280–284. doi: 10.1002/(sici)1097-0045(19990601)39:4<280::aid-pros8>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 10.Gayther SA, de Foy KA, Harrington P, et al. The frequency of germ-line mutations in the breast cancer predisposition genes BRCA1 and BRCA2 in familial prostate cancer. The Cancer Research Campaign/British Prostate Group United Kingdom Familial Prostate Cancer Study Collaborators. Cancer Res. 2000;60(16):4513–4518. [PubMed] [Google Scholar]
- 11.Sinclair CS, Berry R, Schaid D, Thibodeau SN, Couch FJ. BRCA1 and BRCA2 have a limited role in familial prostate cancer. Cancer Res. 2000;60(5):1371–1375. [PubMed] [Google Scholar]
- 12.Douglas JA, Zuhlke KA, Beebe-Dimmer J, et al. Identifying susceptibility genes for prostate cancer--a family-based association study of polymorphisms in CYP17, CYP19, CYP11A1, and LH-beta. Cancer Epidemiol Biomarkers Prev. 2005;14(8):2035–2039. doi: 10.1158/1055-9965.EPI-05-0170. [DOI] [PubMed] [Google Scholar]
- 13.D’Amico AV, Schultz D, Loffredo M, et al. Biochemical outcome following external beam radiation therapy with or without androgen suppression therapy for clinically localized prostate cancer. JAMA. 2000;284(10):1280–1283. doi: 10.1001/jama.284.10.1280. [DOI] [PubMed] [Google Scholar]
- 14.A haplotype map of the human genome. Nature. 2005;437(7063):1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang K, Deng M, Chen T, Waterman MS, Sun F. A dynamic programming algorithm for haplotype block partitioning. Proc Natl Acad Sci U S A. 2002;99(11):7335–7339. doi: 10.1073/pnas.102186799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang K, Qin Z, Chen T, Liu JS, Waterman MS, Sun F. HapBlock: haplotype block partitioning and tag SNP selection software using a set of dynamic programming algorithms. Bioinformatics. 2005;21(1):131–134. doi: 10.1093/bioinformatics/bth482. [DOI] [PubMed] [Google Scholar]
- 17.Siegmund KD, Langholz B, Kraft P, Thomas DC. Testing linkage disequilibrium in sibships. Am J Hum Genet. 2000;67(1):244–248. doi: 10.1086/302973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Laird NM, Horvath S, Xu X. Implementing a unified approach to family-based tests of association. Genet Epidemiol. 2000;19 (Suppl 1):S36–S42. doi: 10.1002/1098-2272(2000)19:1+<::AID-GEPI6>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 19.Rabinowitz D, Laird N. A unified approach to adjusting association tests for population admixture with arbitrary pedigree structure and arbitrary missing marker information. Hum Hered. 2000;50(4):211–223. doi: 10.1159/000022918. [DOI] [PubMed] [Google Scholar]
- 20.Horvath S, Xu X, Lake SL, Silverman EK, Weiss ST, Laird NM. Family-based tests for associating haplotypes with general phenotype data: application to asthma genetics. Genet Epidemiol. 2004;26(1):61–69. doi: 10.1002/gepi.10295. [DOI] [PubMed] [Google Scholar]
- 21.Li C, Scott LJ, Boehnke M. Assessing whether an allele can account in part for a linkage signal: the Genotype-IBD Sharing Test (GIST) Am J Hum Genet. 2004;74(3):418–431. doi: 10.1086/381712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin--rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002;30(1):97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 23.Dunning AM, Chiano M, Smith NR, et al. Common BRCA1 variants and susceptibility to breast and ovarian cancer in the general population. Hum Mol Genet. 1997;6(2):285–289. doi: 10.1093/hmg/6.2.285. [DOI] [PubMed] [Google Scholar]
- 24.Soucek P, Borovanova T, Pohlreich P, Kleibl Z, Novotny J. Role of single nucleotide polymorphisms and haplotypes in BRCA1 in breast cancer: Czech case-control study. Breast Cancer Res Treat. 2006 doi: 10.1007/s10549-006-9367-9. [DOI] [PubMed] [Google Scholar]
- 25.Menzel HJ, Sarmanova J, Soucek P, et al. Association of NQO1 polymorphism with spontaneous breast cancer in two independent populations. Br J Cancer. 2004;90(10):1989–1994. doi: 10.1038/sj.bjc.6601779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cox DG, Kraft P, Hankinson SE, Hunter DJ. Haplotype analysis of common variants in the BRCA1 gene and risk of sporadic breast cancer. Breast Cancer Res. 2005;7(2):R171–R175. doi: 10.1186/bcr973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Freedman ML, Penney KL, Stram DO, et al. A haplotype-based case-control study of BRCA1 and sporadic breast cancer risk. Cancer Res. 2005;65(16):7516–7522. doi: 10.1158/0008-5472.CAN-05-0132. [DOI] [PubMed] [Google Scholar]
- 28.Janezic SA, Ziogas A, Krumroy LM, et al. Germline BRCA1 alterations in a population-based series of ovarian cancer cases. Hum Mol Genet. 1999;8(5):889–897. doi: 10.1093/hmg/8.5.889. [DOI] [PubMed] [Google Scholar]
- 29.Wenham RM, Schildkraut JM, McLean K, et al. Polymorphisms in BRCA1 and BRCA2 and risk of epithelial ovarian cancer. Clin Cancer Res. 2003;9(12):4396–4403. [PubMed] [Google Scholar]
- 30.Auranen A, Song H, Waterfall C, et al. Polymorphisms in DNA repair genes and epithelial ovarian cancer risk. Int J Cancer. 2005;117(4):611–618. doi: 10.1002/ijc.21047. [DOI] [PubMed] [Google Scholar]
- 31.Cooney KA, Strawderman MS, Wojno KJ, et al. Age-specific distribution of serum prostate-specific antigen in a community-based study of African-American men. Urology. 2001;57(1):91–96. doi: 10.1016/s0090-4295(00)00873-6. [DOI] [PubMed] [Google Scholar]
- 32.Amundadottir LT, Sulem P, Gudmundsson J, et al. A common variant associated with prostate cancer in European and African populations. Nat Genet. 2006;38(6):652–658. doi: 10.1038/ng1808. [DOI] [PubMed] [Google Scholar]
- 33.Ng PC, Henikoff S. Accounting for human polymorphisms predicted to affect protein function. Genome Res. 2002;12(3):436–446. doi: 10.1101/gr.212802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Burk-Herrick A, Scally M, mrine-Madsen H, Stanhope MJ, Springer MS. Natural selection and mammalian BRCA1 sequences: elucidating functionally important sites relevant to breast cancer susceptibility in humans. Mamm Genome. 2006;17(3):257–270. doi: 10.1007/s00335-005-0067-2. [DOI] [PubMed] [Google Scholar]
- 35.Fleming MA, Potter JD, Ramirez CJ, Ostrander GK, Ostrander EA. Understanding missense mutations in the BRCA1 gene: an evolutionary approach. Proc Natl Acad Sci U S A. 2003;100(3):1151–1156. doi: 10.1073/pnas.0237285100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu X, Barker DF. Evidence for effective suppression of recombination in the chromosome 17q21 segment spanning RNU2-BRCA1. Am J Hum Genet. 1999;64(5):1427–1439. doi: 10.1086/302358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Durocher F, Shattuck-Eidens D, McClure M, et al. Comparison of BRCA1 polymorphisms, rare sequence variants and/or missense mutations in unaffected and breast/ovarian cancer populations. Hum Mol Genet. 1996;5(6):835–842. doi: 10.1093/hmg/5.6.835. [DOI] [PubMed] [Google Scholar]