

Summary
Background and objectives
Mycophenolate mofetil (MMF) is an immunosuppressive drug used in renal transplant patients. Upon oral administration it is hydrolyzed to the active agent mycophenolic acid (MPA). In renal transplant recipients, MMF therapy is optimal when the area under the curve of MPA is 30 to 60 mg·h/L. When MMF doses are adjusted, a linear relationship between dose and MPA exposure is assumed. In this study, the linearity of MMF pharmacokinetics was investigated.
Design, setting, participants, & measurements
MPA concentration-time profiles from renal transplant recipients cotreated with cyclosporine (n = 140) or tacrolimus (n = 101) were analyzed retrospectively using nonlinear mixed-effects modeling. The correlation between the MMF dose and the pharmacokinetics parameters was evaluated.
Results
In the developed population pharmacokinetics model MPA clearance and the central volume of distribution were correlated with cyclosporine coadministration and time posttransplantation. The pharmacokinetics of MPA were not linear. Bioavailability decreased with increasing MMF doses. Compared with an MMF dose of 1000 mg (=100%), relative bioavailability was 123%, 111%, 94%, and 90% in patients receiving MMF doses of 250, 500, 1500, and 2000 mg in combination with cyclosporine (P < 0.001); respective values in tacrolimus-cotreated patients were 176%, 133%, 85%, and 76% (P < 0.001). Because of the decreasing relative bioavailability, MPA exposure will increase less than proportionally with increasing MMF doses.
Conclusions
MMF exhibits nonlinear pharmacokinetics. This should be taken into account when performing therapeutic drug monitoring.
Introduction
Mycophenolate mofetil (MMF) is an immunosuppressive drug used in renal transplant patients. Upon oral administration it is hydrolyzed to the active agent mycophenolic acid (MPA). Although introduced as a fixed-dose drug, therapeutic drug monitoring (TDM) of the MPA area under the MPA concentration versus time curve (AUC) was found to improve clinical outcome. The large interpatient variability in MPA pharmacokinetics at fixed dose and the observation that the risk for acute rejection increases with lower MPA plasma concentrations suggest that a strategy of TDM would improve outcome (1–3). In 2007, Le Meur et al. published the results of the APOMYGRE study, which showed that TDM of MPA reduces the risk of treatment failure and acute rejection in renal allograft recipients without an increase in adverse events and without adding any extra costs (4,5). To provide for clinicians an objective and balanced clinical interpretation of the current scientific evidence on TDM of MPA, recently in this journal a consensus document was published (6).
When the MMF dose is adjusted, a linear relationship between dose and MPA exposure, that is, linear pharmacokinetics, is assumed. So far, this linearity has however not been evaluated. This may be caused by the fact that MPA exerts complex pharmacokinetics properties (7). Furthermore, factors complicating the assessment of linear pharmacokinetics are the changes in MPA clearance in the first 3 months posttransplantation, the influence of comedication and the within-patient variability (8,9).
In an early dose-ranging study in cyclosporine-treated patients Sollinger et al. compared a 1000-mg MMF two times daily dose with a 1500-mg MMF two times daily dose and found that with the 50% higher dose the AUC was also about 50% higher (12.3 ± 5.8 mg·h/L versus 19.5 ± 13.9 mg·h/L). (10) These results are in accordance with a linear relationship between MMF dose and MPA AUC. In the randomized concentration controlled trial (RCCT), a total of 154 cyclosporine-cotreated adult recipients of a deceased kidney graft were randomly allocated to receive MMF treatment targeted at three predefined MPA AUC values. At day 3 posttransplantation the median-assigned daily doses of MMF were 0.90, 1.90, and 3.40 g and corresponding MPA AUC values were 13.9, 24.6, and 39.1 mg·h/L. In this study, the increase in AUC is less than proportional, which supports nonlinear pharmacokinetics for MMF. With a convex relationship between dose and exposure, an increase of the MMF dose may produce a less than expected increase in MPA exposure. Alternatively, the decrease of MPA AUC may be overestimated when the MMF dose is reduced. Clearly, this may have significant implications for TDM of MPA.
In this study a population pharmacokinetics model was developed in which the effect of calcineurin inhibitor cotreatment on MPA disposition and the time dependency of the pharmacokinetics was quantified. The developed population model was used to evaluate the relationship between MMF dose and the pharmacokinetics parameters of MPA.
Materials and Methods
Patients
MPA plasma concentration-time profiles obtained from renal transplant recipients treated with MMF and cyclosporine (n = 140) or MMF and tacrolimus (n = 101) were combined and analyzed simultaneously. The data were obtained from the RCCT and the IMPDH-activity study, which were published earlier (1,11). In the RCCT study (1), de novo renal transplant recipients were divided into three MPA AUC target groups. All patients in this study received cyclosporine and corticosteroids as concomitant immunosuppressive therapy. In this study plasma MPA concentrations were measured at days 3, 7, 11, 21, 28, 56, 84, 112, and 140 posttransplantation. On days 3, 7, and 11 posttransplantation, sample times were before dose and 0.33, 0.66, 1.25, 2, 6, 8, and 12 hours after oral intake of MMF. On the remaining occasions, sample times were before dose and 0.33, 0.66, 1.25, and 2 hours after dose. The MMF dose was adjusted on the basis of the measured MPA concentrations. In the IMPDH-activity study (12), de novo renal transplant recipients started with 1000 mg of MMF two times daily, combined with tacrolimus and corticosteroids. The MMF dose was adjusted on the basis of clinical evaluations. In the IMPDH-activity study MPA plasma concentrations were measured at days 6, 21, 49, and 140 after transplantation. On day 6, samples were taken before dose and 0.5, 1, 2, 6, and 12 hours after dose. On the remaining occasions, sample times were before dose and 0.5 and 2 hours after oral intake of MMF.
Pharmacokinetics Analysis
Because of the heterogeneous nature of the data, the pharmacokinetics analysis was performed using nonlinear mixed-effects modeling (NONMEM, Verion VI, level 1.0; GloboMax LLC, Ellicott City, MD). By application of this technique, general pharmacokinetics parameters and their relationship with covariates can be estimated as well as their inter- and intrapatient variability. This technique allows the combined analysis of concentration-time data from patients at several times posttransplantation, with different covariates, and with sparse sampling schemes.
Basic Model.
Concentration data were logarithmically transformed, and the first-order estimation method was used throughout the entire model-building process. A two-compartment model with lag time (TLAG) and first-order absorption and elimination was fitted to the MPA concentrations, as reported earlier (9,13). Pharmacokinetics parameters were estimated in terms of volume of distribution of the central compartment (Vc), clearance (CL), volume of distribution of the peripheral compartment (Vp), and intercompartmental clearance (Q). Because no intravenous data were available, absolute bioavailability (F) cannot be estimated. As a result, Vc, CL, Vp, and Q correspond to the ratios Vc/F, CL/F, Vp/F, and Q/F, respectively. At some point in the analysis bioavailability was compared for the different doses (see below). In this case the relative bioavailability (Frel) of an MMF dose of 1000 mg was arbitrarily set at 100%. Interpatient variability (IPV) and interoccasion variability (IOV) of the pharmacokinetics parameters were modeled using an exponential error model. Residual variability between observed and predicted MPA plasma concentrations was described using an additional error model.
Covariate Model.
To explain variability, relationships were investigated between pharmacokinetics parameters and patient characteristics. The correlation between the use of calcineurin inhibitors and the pharmacokinetics parameters was tested using equation 1,
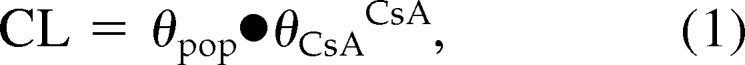 |
where θpop is the typical clearance in patients using tacrolimus (exponent CsA = 0) and θCsA is the fractional change in MPA clearance in patients cotreated with cyclosporine (exponent CsA = 1).
The time-dependent changes of MPA pharmacokinetics were modeled as described in equation 2,
where θΔ is the relative change of MPA CL at day 3 compared with its stabilized value at 6 months posttransplantation and θrate is a first-order rate constant describing the decrease of CL posttransplantation from day 0 to day 180.
The relationship between the MMF dose and the pharmacokinetics parameters of MPA was tested as follows (equation 3),
in which θpop is the Frel in individuals who received 1000 mg of MMF two times daily, which was arbitrarily set at the value of 1, and θdose is an exponent determining the shape of the relationship. The final model was developed by forward inclusion and backward elimination using the log likelihood test (14). Covariates were introduced one by one. When inclusion of a covariate caused a decrease in OFV >3.84 (P < 0.05), the covariate was considered to be added to the model. Subsequently, all covariates selected were included in an intermediate model. From this model covariates were deleted one by one. If the elimination of a covariate caused an increase in OFV >10.8 (P < 0.001), then the covariate remained in the model and was considered to be significant. The goodness of the fit was graphically evaluated using goodness-of-fit plots. (15).
Model Validation
As an internal validation method, a bootstrap resampling method (16) was applied, using the Wings for NONMEM software (Dr. N. Holford, version 612, March 2007, Auckland, New Zealand). Two hundred bootstrap data sets were generated by sampling randomly from the original data set with replacement. The median and 95th percentile range from the bootstrap analysis were compared with the final model.
As an external validation data set, a subset of data from a fixed-dose concentration-controlled (FDCC) trial were available. (17) This database contained 1443 concentration-time curves, obtained from 289 patients. The MMF dose of these patients ranged from 250 to 2500 mg. In 460 of the 1443 occasions the patients were cotreated with cyclosporine. Concentration-time profiles were obtained at day 3, day 10, month 3, month 6, and month 12 posttransplantation. Concentrations of these pharmacokinetics profiles were simulated using the different models derived in this study.
Results
Patients
The population pharmacokinetics model was developed using 7739 MPA samples originating from 1402 concentration-time profiles obtained from 241 renal transplant recipients. The pharmacokinetics profiles of patients cotreated with cyclosporine (n = 140) and tacrolimus (n = 101) are presented in Figure 1, a and b. Sampling occasions varied from day 3 to day 168 after renal transplantation. MMF doses ranged from 250 to 2200 mg two times daily. Patient characteristics are presented in Table 1.
Figure 1.

Concentration-time profiles of patients cotreated with cyclosporine (A) and tacrolimus (B).
Table 1.
Patient characteristics
| Cotreatment | Cyclosporine | Tacrolimus |
|---|---|---|
| Gender (men/women) | 88/52 | 73/28 |
| Age (years) | 50 (19 to 70) | 53 (19 to 76) |
| Length (cm) | 170 (150 to 190) | 175 (150 to 195) |
| Body weight (kg) | 69 (37 to 104) | 80 (44 to 145) |
| Time posttransplantation (days) | 26 (1 to 152) | 23 (3 to 168) |
| Creatinine (μmol/L) | 141 (53 to 1238) | 145 (61 to 1190) |
| Albumin (g/L) | 36 (20 to 53) | 39 (10 to 50) |
| ASAT (U/L) | 14 (2 to 289) | 21 (8 to 236) |
| ALAT (U/L) | 14 (1 to 653) | 24 (1 to 534) |
| PK-day | 26 (4 to 152) | 23 (3 to 168) |
| MMF dose two times daily (mg) | 1150 (250 to 2200) | 750 (250 to 1500) |
| CNI daily dose (mg) | 450 (125 to 2100) | 8 (1 to 20) |
Parameters are presented as median (range), separated for the cotreatment of the calcineurin inhibitors, cyclosporine, and tacrolimus. ASAT, aspartate aminotransferase; ALAT, alanine transaminase; CNI, calcineurin inhibitors; PK-day, number of days between the transplantation and the pharmacokinetic assessment.
Pharmacokinetics Analysis
A two-compartment model with first-order elimination adequately described the data. In the basic model, Frel was fixed on 100%. Introduction of IPV for TLAG, ka, Vc, CL, and Frel significantly improved the fit of the model for each parameter (P < 0.001). IOV could be estimated for ka, Vc, CL, and Frel; each improved the model even further (P < 0.001).
Equation 1 was used to evaluate possible differences in the pharmacokinetics parameters of MPA between patients receiving either cyclosporine or tacrolimus. Significant differences were observed for CL and Vc. CL was significantly higher in patients receiving cyclosporine, whereas Vc was significantly lower (P < 0.001). Introduction of the differences in CL and Vc between patients cotreated with cyclosporine or tacrolimus improved the fit of the model to the data (P < 0.001). IPV decreased from 44% to 38% for MPA CL and from 124% to 105% for Vc. The respective reductions for IOV were from 9.3% to 7.3% and from 57% to 52%. As a result, part of the IPV and IOV in Vc and CL could be explained by the type of comedication used.
A significant decrease in CL and Vc was seen in time during the first 6 months after transplantation (P < 0.001). MPA CL and Vc decreased from 35.3 to 18.6 L/h and from 104 to 49 L in patients cotreated with cyclosporine and from 26.9 to 13.6 L/h and from 297 to 54 L in patients cotreated with tacrolimus (Figure 2, Table 2). The time-dependent phenomena explained some IPV; IPV decreased from 38% to 31% for MPA CL and from 105% to 71% for Vc.
Figure 2.

Post hoc changes in time posttransplantation of MPA (A) clearance (CL) and (B) volume of distribution of the central compartment (VC). The median and 90th percentile range are presented for patients cotreated with cyclosporine (black) and tacrolimus (gray).
Table 2.
Pharmacokinetics parameter estimations
| Parameter | Without Effect Dose | Final Model (CV in %) | Bootstrap (95th Percentile) |
|---|---|---|---|
| TLAG (h) | 0.21 (2) | 0.21 (2) | 0.21 (0.15 to 0.26) |
| ka (hours−1) | 3.9 (10) | 3.9 (10) | 3.8 (2.5 to 5.0) |
| Vc (L) | 54 (11) | 68 (14) | 70 (52 to 98) |
| CL (L/h) | 13.6 (6) | 17.0 (9) | 16.8 (13.9 to 20.2) |
| Vp (L) | 222 (9) | 229 (9) | 244 (202 to 326) |
| Q (L/h) | 37 (7) | 38 (7) | 38 (32 to 53) |
| Residual error | 0.45 (2) | 0.45 (2) | 0.44 (0.42 to 0.47) |
| Calcineurin inhibitor effect | |||
| θCsA of CL | 1.37 (9) | 1.13 (11) | 1.16 (0.93 to 1.40) |
| θCsA of Vc | 0.91 (15) | 0.72 (18) | 0.73 (0.50 to 0.97) |
| Time dependency | |||
| CsA: θΔ of CL | 0.90 (11) | 0.87 (11) | 0.90 (0.74 to 1.14) |
| CsA: θrate of CL (day−1) | 0.018 (19) | 0.019 (19) | 0.020 (0.013 to 0.031) |
| TCL: θΔ of CL | 0.98 (16) | 0.50 (36) | 0.55 (0.26 to 1.00) |
| TCL: θrate of CL (day−1) | 0.046 (22) | 0.040 (35) | 0.038 (0.003 to 0.085) |
| CsA: θΔ of Vc | 1.11 (28) | 1.13 (27) | 1.25 (0.58 to 2.15) |
| θrate of Vc (day−1) | 0.050 (19) | 0.045 (22) | 0.047 (0.026 to 0.072) |
| TCL: θΔ of Vc | 4.5 (22) | 3.2 (27) | 3.2 (1.8 to 5.4) |
| Relationship of MMF dose Frel | |||
| CsA: θdose of F | −0.15 (30) | −0.15 (−0.24 to −0.06) | |
| TCL: θdose of F | −0.41 (25) | −0.41 (−0.62 to −0.19) | |
| Interpatient variability | |||
| TLAG (%) | 7.3 (189) | 7.4 (207) | 17 (4 to 155) |
| ka (%) | 124 (18) | 125 (18) | 140 (102 to 199) |
| Vc (%) | 71 (29) | 71 (29) | 70 (41 to 108) |
| CL (%) | 31 (24) | 31 (24) | 34 (23 to 49) |
| Frel (%) | 43 (17) | 41 (17) | 44 (34 to 59) |
| Interoccasion variability | |||
| ka (%) | 117 (10) | 117 (10) | 120 (106 to 134) |
| Vc (%) | 50 (20) | 50 (21) | 48 (19 to 70) |
| CL (%) | 3.3 (452) | 3.8 (297) | 5.3 (0.1 to 12) |
| Frel (%) | 25 (12) | 24 (12) | 24 (20 to 26) |
Values of estimated parameters with coefficient of variation (CV). IPV, interpatient variability; IOV, interoccasion variability; OFV, minimum value of objective function; TLAG, lag time; ka, absorption rate constant; Vc, volume of distribution of the central compartment; CL, clearance; Vp, volume of distribution of the peripheral compartment; Q, intercompartmental clearance; CsA, cyclosporine; TCL, tacrolimus; Frel, relative bioavailability; θCsA, fractional change in patients cotreated with CsA; θΔ, relative change in time posttransplantation; θrate, first-order rate constant describing the relative change of a parameter posttransplantation; θdose, exponent determining the shape of the relationship between a parameter and the MMF dose. For example, MPA clearance in cyclosporine-cotreated patients: CL = θpop × θCsACsA = 17.0 × 1.13 = 19.2 L/h; MPA clearance at 30 days posttransplantation in tacrolimus-cotreated patients: CL = θpop(1 + θΔ × e−θrate×time) = 17.0 × (1 + 0.50 × e−0.040×30) = 19.6 L/h; relative bioavailability of 1.5 g of MMF in patients cotreated with tacrolimus: Frel = θpop × (dose/1000)θdose = 1 × (1500/1000)−0.41 = 0.85.
Finally, the correlation between MMF dose and the different pharmacokinetics parameters was evaluated. Significant relations with dose were detected for CL, Vp, and Frel (P < 0.05). After combining and backward elimination, only the relationship between MMF dose and Frel remained significant (Figure 3, Table 2). Compared with 1000 mg of MMF two times daily, median Frel (and 95% confidence interval) was 123% (109% to 139%), 111% (104% to 118%), 94% (91% to 98%), and 90% (85% to 96%) in patients receiving MMF doses of 250, 500, 1500, and 2000 mg two times daily, respectively, in combination with cyclosporine (P < 0.001); corresponding values for patients receiving tacrolimus were 176% (134% to 233%), 133% (116% to 153%), 85% (78% to 92%), and 76% (65% to 87%) (P < 0.001). IPV and IOV of relative bioavailability were 41% and 24%, respectively. The goodness-of-fit plots of the final model (Figure 4) showed no structural bias.
Figure 3.

Correlation between MMF dose and relative bioavailability (Frel). Patients cotreated with cyclosporine are represented in black and patients cotreated with tacrolimus are in gray.
Figure 4.

Goodness-of-fit plots of the final model. Model-predicted MPA concentration versus observed MPA concentration (A), individual-predicted MPA concentration versus observed concentration (B), and weighted residuals versus time (C). The solid line in (A) and (B) is the line of identity. The solid line in (C) is the line for x = 0. Patients cotreated with cyclosporine are represented in black and patients cotreated with tacrolimus are in gray.
Model Validation
The median estimates and 95th percentile range from the bootstrap procedure were similar to the population estimates of the final model, demonstrating the accuracy of the model (Table 2).
In the external validation, data were simulated using the final model, taking into account the relationship between dose and Frel, and the model without this correlation (Table 2). The differences between the predicted and observed concentrations for both models are graphically presented in Figure 5. For the final model, no trend in the deviation of the concentration is seen over the whole MMF dose range. In contrast, an underestimation of the concentration after a low MMF dose and an overestimation after a high MMF dose are seen when the relationship between dose and Frel is not taken into account.
Figure 5.

External validation. Median and interquartile range of the difference between the estimated and observed MPA concentrations in the external validation data set. The differences are represented for several MMF doses, after estimations with the final model (right) and the model in which the correlation between MMF dose and bioavailability is not included (left).
Post hoc Analysis
The exponents in equation 3 were −0.15 for cyclosporine and −0.41 for tacrolimus. This indicates that bioavailability of MMF decreases when doses increase. The correlation between MMF dose (“administered MMF dose”) and MMF dose multiplied by Frel (“relative MMF dose”) is shown in Figure 6. For instance, a patient receiving 500 mg of MMF cotreated with tacrolimus exhibits a MPA exposure corresponding to 665 mg of “relative MMF dose”. When exposure has to be doubled to 1330 mg, a MMF dose of 1620 mg has to be administered (Figure 6). In this case, to double the MPA exposure, the administered dose needs to be increased to 3.2 times its original value.
Figure 6.

Conversion from administered MMF dose to the relative MMF dose, corrected for relative bioavailability (Frel). The median and 90th percentile range are presented for patients cotreated with cyclosporine (black) and tacrolimus (gray).
Discussion
A population pharmacokinetics analysis of MPA was performed in renal transplant recipients, who were followed for 6 months posttransplantation. Differences were seen in MPA CL and Vc between patients cotreated with either cyclosporine or tacrolimus (P < 0.001). The pharmacokinetics of MPA were time-dependent with decreasing CL and Vc posttransplantation. Finally, in the developed model a relationship between MMF dose and relative bioavailability, Frel, was observed. Because of the decrease of Frel with rising MMF doses, an increase of the MMF dose will produce an increase in MPA AUC that is less than proportional.
The basic model and estimated pharmacokinetics parameters of the present model were comparable with those of previously published models for MPA in renal transplant recipients (18–20). The pharmacokinetics of MPA are dependent on the calcineurin inhibitor that is coadministered. MPA CL was increased in patients cotreated with cyclosporine (19.2 L/h) compared with tacrolimus-cotreated patients (17.0 L/h). The enterohepatic recirculation is decreased by inhibition of the multidrug resistance–associated protein 2 (MRP2) transporter by cyclosporine. MRP2 is responsible for the excretion of MPAG in bile, which can be reabsorbed in the gut as MPA. (21) The intact enterohepatic recirculation contributes to MPA exposure, and explains the reduced clearance, in tacrolimus-cotreated patients.
Time-dependent changes in MPA pharmacokinetics cause at least a 30% to 50% increase in MPA AUC during the first weeks posttransplantation (22). The decrease in Vc might be explained by the increase in renal function and albumin levels in time posttransplantation. (9,18) MPA CL decreased from 35.9 to 19.2 L/h for cyclosporine and from 25.5 to 17.0 L/h for tacrolimus-cotreated patients. This is caused by a combination of improving renal function, increasing albumin levels, increasing hemoglobin, and decreasing cyclosporine before dose target concentrations during the first 6 months posttransplantation (8).
Because of inclusion of cyclosporine cotreatment and time posttransplantation as covariates, the pharmacokinetics model can be used in the whole population. There is no need to develop separate models for the different periods after transplantation. Compared with separated models for each covariate, this integrated model includes as much information as possible, allowing multivariate analysis and increasing the statistical power. Because of the known decrease in MPA clearance over time, the needed MMF dose to reach a certain AUC will decrease. However, in this study the range of the MMF dose varies from at least 300 to 2200 mg two times daily for every period. Although the developed model takes these changes in time into account, a significant correlation between MMF dose and bioavailability was still detected for the whole period, also when the different periods posttransplantation were analyzed separately.
The population pharmacokinetics analysis demonstrated that bioavailability was not constant over the studied dose range of MMF. Bioavailability decreased significantly with rising MMF doses. As a result, MMF exhibits nonlinear pharmacokinetics. The correlation between MMF dose and bioavailability was confirmed in the external validation, where concentrations were predicted in an independent data set. The decrease in bioavailability with higher doses may be caused by a saturable absorption process of MPA from the gut. Hereby, a limited amount of MPA can be absorbed when high doses are administered. Another possible explanation might be saturation of the enterohepatic circulation, which is responsible for the reabsorption of MPAG in the gut as MPA. At higher doses, less MPAG is recirculated and more will be excreted by the kidney, producing less exposure to MPA. Consequently, the effect of this mechanism will be less in patients treated with cyclosporine, which may explain the slightly different relationship between MMF dose and Frel in cyclosporine-cotreated patients compared with tacrolimus-cotreated patients. More support for our findings comes from a recent trial that investigated whether an intensified dosing regimen of enteric-coated mycophenolate sodium (EC-MPS) resulted in a higher MPA AUC compared with standard dosing. (23) In this cyclosporine-based study, treatment with 2724 mg of EC-MPS (the equivalent of 2000 mg of MMF) resulted in a MPA-AUC that was only 37% higher compared with treatment with 1440 mg of EC-MPS (42.8 versus 31.3 mg·h/L).
Figure 6 may be used to calculate the MMF dose needed for a certain change in MPA AUC in an individual patient. With a dose of 1000 mg of MMF two times daily and a MPA AUC of 20 mg·h/L, the patient is underexposed. Doubling the dose to 2000 mg of MMF (to obtain an AUC value of 40 mg·h/L) will result in a “relative MMF dose” of 1520 mg in tacrolimus- and 1800 mg in cyclosporine-cotreated patients. This increase in MMF dose will on average result in an AUC of 30 mg·h/L for the tacrolimus-cotreated patient and 36 mg·h/L for the cyclosporine-cotreated patient. Clearly, decreasing bioavailability may produce underexposure to MPA when the MMF dose is increased and this may have consequences for the efficacy of MMF therapy.
The bioavailability of MPA has been improved through ester derivatization to MMF (24). MMF absorption has been reported to be almost complete. In healthy patients, bioavailability after single-dose oral administration of 1.5 g of MMF was 85.7% for the MPA AUC0–24 and 93.3% for the MPA AUC0–∞ in comparison with the intravenous formulation (25). However, MMF bioavailability seems to be decreased to 48.5% in liver transplant patients treated with 1 g of oral MMF two times daily (26). Also, in hematopoietic stem cell transplant recipients treated with 1 g of MMF in combination with cyclosporine, a decreased and highly variable bioavailability was seen (F = 72.3%, range 20.5% to 172%) (27). In combination with our findings, it would seem that changes in bioavailability might contribute to the variability seen in patients treated with MMF.
In conclusion, the population pharmacokinetics analysis of MPA demonstrated that bioavailability of MMF is not constant. Increasing the MMF dose will produce a less than proportional increase of the MPA exposure. This should be taken into account when performing TDM.
Disclosures
None.
Footnotes
Published online ahead of print. Publication date available at www.cjasn.org.
References
- 1. van Gelder T, Hilbrands LB, Vanrenterghem Y, Weimar W, de Fijter JW, Squifflet JP, Hene RJ, Verpooten GA, Navarro MT, Hale MD, Nicholls AJ: A randomized double-blind, multicenter plasma concentration controlled study of the safety and efficacy of oral mycophenolate mofetil for the prevention of acute rejection after kidney transplantation. Transplantation 68: 261–266, 1999 [DOI] [PubMed] [Google Scholar]
- 2. van Gelder T, Shaw LM: The rationale for and limitations of therapeutic drug monitoring for mycophenolate mofetil in transplantation. Transplantation 80[2 Suppl]: S244–S253, 2005 [DOI] [PubMed] [Google Scholar]
- 3. de Winter BC, Mathot RA, van Hest RM, van Gelder T: Therapeutic drug monitoring of mycophenolic acid: does it improve patient outcome? Expert Opin Drug Metab Toxicol 3: 251–261, 2007 [DOI] [PubMed] [Google Scholar]
- 4. Le Meur Y, Buchler M, Thierry A, Caillard S, Villemain F, Lavaud S, Etienne I, Westeel PF, de Ligny BH, Rostaing L, Thervet E, Szelag JC, Rerolle JP, Rousseau A, Touchard G, Marquet P: Individualized mycophenolate mofetil dosing based on drug exposure significantly improves patient outcomes after renal transplantation. Am J Transplant 7: 2496–2503, 2007 [DOI] [PubMed] [Google Scholar]
- 5. Rousseau A, Laroche ML, Venisse N, Loichot-Roselmac C, Turcant A, Hoizey G, Compagnon P, Hary L, Debruyne D, Saivin S, Jacqz-Aigrain E, Buchler M, Villeneuve C, Vergnenegre A, Le Meur Y, Marquet P: Cost-effectiveness analysis of individualized mycophenolate mofetil dosing in kidney transplant patients in the APOMYGRE trial. Transplantation 89: 1255–1262, 2010 [DOI] [PubMed] [Google Scholar]
- 6. Kuypers DR, Le Meur Y, Cantarovich M, Tredger MJ, Tett SE, Cattaneo D, Tonshoff B, Holt DW, Chapman J, Gelder T: Consensus report on therapeutic drug monitoring of mycophenolic acid in solid organ transplantation. Clin J Am Soc Nephrol 5: 341–358, 2010 [DOI] [PubMed] [Google Scholar]
- 7. West-Thielke P, Kaplan B: Therapeutic monitoring of mycophenolic acid: Is there clinical utility? Am J Transplant 7: 2441–2442, 2007 [DOI] [PubMed] [Google Scholar]
- 8. van Hest R, van Gelder T, Bouw R, Goggin T, Gordon R, Mamelok R, Mathot R: Time-dependent clearance of mycophenolic acid in renal transplant recipients. Br J Clin Pharmacol 63: 741–752, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. van Hest RM, Mathot RA, Pescovitz MD, Gordon R, Mamelok RD, van Gelder T: Explaining variability in mycophenolic acid exposure to optimize mycophenolate mofetil dosing: A population pharmacokinetic meta-analysis of mycophenolic acid in renal transplant recipients. J Am Soc Nephrol 17: 871–880, 2006 [DOI] [PubMed] [Google Scholar]
- 10. Sollinger HW, Deierhoi MH, Belzer FO, Diethelm AG, Kauffman RS: RS-61443–a phase I clinical trial and pilot rescue study. Transplantation 53: 428–432, 1992 [DOI] [PubMed] [Google Scholar]
- 11. Sombogaard F, Peeters AMA, Baan CC, Mathot RA, Quaedackers M, Vulto AG, Weimar W, Van Gelder T: Inosine monophosphate dehydrogenase messenger RNA expression is correlated to clinical outcomes in mycophenolate mofetil-treated kidney transplant patients, whereas inosine monophosphate dehydrogenase activity is not. Ther Drug Monit 31: 549–556, 2009 [DOI] [PubMed] [Google Scholar]
- 12. Sombogaard F, van Schaik RH, Mathot RA, Budde K, van der Werf M, Vulto AG, Weimar W, Glander P, Essioux L, van Gelder T: Interpatient variability in IMPDH activity in MMF-treated renal transplant patients is correlated with IMPDH type II 3757T>C polymorphism. Pharmacogenet Genomics 19: 626–634, 2009 [DOI] [PubMed] [Google Scholar]
- 13. de Winter BC, Neumann I, van Hest RM, van Gelder T, Mathot RA: Limited sampling strategies for therapeutic drug monitoring of mycophenolate mofetil therapy in patients with autoimmune disease. Ther Drug Monit 31: 382–390, 2009 [DOI] [PubMed] [Google Scholar]
- 14. Wahlby U, Jonsson EN, Karlsson MO: Comparison of stepwise covariate model building strategies in population pharmacokinetic-pharmacodynamic analysis. AAPS PharmSci 4: E27, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jonsson EN, Karlsson MO: Xpose: An S-PLUS based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Comput Methods Programs Biomed 58: 51–64, 1999 [DOI] [PubMed] [Google Scholar]
- 16. Ette EI, Williams PJ, Kim YH, Lane JR, Liu MJ, Capparelli EV: Model appropriateness and population pharmacokinetic modeling. J Clin Pharmacol 43: 610–623, 2003 [PubMed] [Google Scholar]
- 17. van Gelder T, Silva HT, de Fijter JW, Budde K, Kuypers D, Tyden G, Lohmus A, Sommerer C, Hartmann A, Le Meur Y, Oellerich M, Holt DW, Tonshoff B, Keown P, Campbell S, Mamelok RD: Comparing mycophenolate mofetil regimens for de novo renal transplant recipients: The fixed-dose concentration-controlled trial. Transplantation 86: 1043–1051, 2008 [DOI] [PubMed] [Google Scholar]
- 18. van Hest RM, van Gelder T, Vulto AG, Mathot RA: Population pharmacokinetics of mycophenolic acid in renal transplant recipients. Clin Pharmacokinet 44: 1083–1096, 2005 [DOI] [PubMed] [Google Scholar]
- 19. de Winter BC, van Gelder T, Glander P, Cattaneo D, Tedesco-Silva H, Neumann I, Hilbrands L, van Hest RM, Pescovitz MD, Budde K, Mathot RA: Population pharmacokinetics of mycophenolic acid: A comparison between enteric-coated mycophenolate sodium and mycophenolate mofetil in renal transplant recipients. Clin Pharmacokinet 47: 827–838, 2008 [DOI] [PubMed] [Google Scholar]
- 20. Shum B, Duffull SB, Taylor PJ, Tett SE: Population pharmacokinetic analysis of mycophenolic acid in renal transplant recipients following oral administration of mycophenolate mofetil. Br J Clin Pharmacol 56: 188–197, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hesselink DA, van Hest RM, Mathot RA, Bonthuis F, Weimar W, de Bruin RW, van Gelder T: Cyclosporine interacts with mycophenolic acid by inhibiting the multidrug resistance-associated protein 2. Am J Transplant 5: 987–994, 2005 [DOI] [PubMed] [Google Scholar]
- 22. Shaw LM, Korecka M, Venkataramanan R, Goldberg L, Bloom R, Brayman KL: Mycophenolic acid pharmacodynamics and pharmacokinetics provide a basis for rational monitoring strategies. Am J Transplant 3: 534–542, 2003 [DOI] [PubMed] [Google Scholar]
- 23. Glander P, Sommerer C, Arns W, Ariatabar T, Kramer S, Vogel EM, Shipkova M, Fischer W, Zeier M, Budde K: Pharmacokinetics and pharmacodynamics of intensified versus standard dosing of mycophenolate sodium in renal transplant patients. Clin J Am Soc Nephrol 5: 503–511, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lee WA, Gu L, Miksztal AR, Chu N, Leung K, Nelson PH: Bioavailability improvement of mycophenolic acid through amino ester derivatization. Pharm Res 7: 161–166, 1990 [DOI] [PubMed] [Google Scholar]
- 25. Bullingham R, Monroe S, Nicholls A, Hale M: Pharmacokinetics and bioavailability of mycophenolate mofetil in healthy subjects after single-dose oral and intravenous administration. J Clin Pharmacol 36: 315–324, 1996 [DOI] [PubMed] [Google Scholar]
- 26. Jain A, Venkataramanan R, Kwong T, Mohanka R, Orloff M, Abt P, Kashyap R, Tsoulfas G, Mack C, Williamson M, Batzold P, Bozorgzadeh A: Pharmacokinetics of mycophenolic acid in liver transplant patients after intravenous and oral administration of mycophenolate mofetil. Liver Transpl 13: 791–796, 2007 [DOI] [PubMed] [Google Scholar]
- 27. Jacobson P, Green K, Rogosheske J, Brunstein C, Ebeling B, DeFor T, McGlave P, Weisdorf D: Highly variable mycophenolate mofetil bioavailability following nonmyeloablative hematopoietic cell transplantation. J Clin Pharmacol 47: 6–12, 2007 [DOI] [PubMed] [Google Scholar]