

Abstract
The tyrosine kinase Wee1 is part of a key cellular sensing mechanism that signals completion of DNA replication, ensuring proper timing of entry into mitosis. Wee1 acts as an inhibitor of mitotic entry by phosphorylating cyclin-dependent kinase CDK1. Wee1 activity is mainly regulated at the protein level through its phosphorylation and subsequent degradation by the ubiquitin proteasome pathway. To facilitate identification of small molecules preventing Wee1 degradation, a homogeneous cell-based assay was developed using HeLa cells transiently transfected with a Wee1-Luciferase fusion protein. To insure uHTS compatibility, the assay was scaled to 1,536-well plate format and cells were transfected in bulk and cryopreserved. This miniaturized homogenous assay demonstrated robust performance, with a calculated Z′ factor of 0.65±0.05. The assay was screened against a publicly available library of ~218,000 compounds in order to identify Wee1 stabilizers. Nonselective, cytotoxic and promiscuous compounds were rapidly triaged through the use of a similarly formatted counterscreen that measured stabilization of a N-cyclin B-Luciferase fusion protein, as well as execution of viability assessment in the parental HeLa cell line. This screening campaign led to the discovery of four unrelated cell-permeable small molecules that showed selective Wee1-Luciferase stabilization with micromolar potency. One of these compounds, SID4243143, was shown to inhibit cell cycle progression, underscoring the importance of Wee1 degradation to the cell cycle. Our results suggest that this uHTS approach is suitable for identifying selective chemical probes that prevent Wee1 degradation, and generally applicable to discovering inhibitors of the ubiquitin proteasome pathway.
Keywords: Wee1, degradation, stabilizer, reporter assay, transient transfection, cryopreserved cells, ubiquitin, proteasome
INTRODUCTION
Wee1 is a highly conserved tyrosine kinase that inhibits mitotic entry by inactivating the mitosis-specific kinase Cdk1/Cyclin B complex during the S and G2 phases though Cdk1 phosphorylation at tyrosine 15.1 By contrast, the phosphatase Cdc25 abrogates Wee1-mediated effect by removing Cdk1 phosphorylation.2, 3 Therefore, there is a competition between Wee1 and Cdc25 in controlling Cdk1/Cyclin B complex activity, which ultimately determines mitotic entry or division arrest.4 Upon the onset of mitosis, Wee1 is inactivated both by protein phosphorylation on specific residues and subsequent degradation via the ubiquitin proteasome pathway.5–7 This mechanism tips the balance in favor of Cdc25, triggering a positive feedback loop driven by activated Cdc25 and Cdk1/Cyclin B, thus conferring unidirectionality to mitosis.8 Maintaining the right amount of Wee1 is essential for cell growth and proliferation and hence Wee1 is likely to participate in tumor progression. Lung cancer biopsies have low levels of Wee1 protein.9 By contrast, increasing Wee1 levels by reducing its degradation in a prostate cancer model was beneficial as it limited cell growth.10 Moreover, an anti-cancer compound that increases the steady-state levels of Wee1 by inhibiting Plk-1 dependent Wee1 turnover is now in Phase I clinical trials.11 In addition, many cancer cells are lacking Wee1 dependent checkpoint pathways needed to ensure proper correction of DNA defects prior to mitosis, causing the cells to divide with incompletely replicated DNA.12 Tight regulation of Wee1 activity in these cells may prevent the genomic instability caused by premature mitosis entry. Taken together, these studies suggest Wee1 is a promising target in cancer, and the regulation of its degradation a point of choice for chemotherapeutic intervention. In addition to providing potential novel drug leads, small-molecule inhibitors of Wee1 degradation could yield valuable probes to decipher pathways controlling Wee1 turnover and cell cycle transit. However, no effort to identify these small molecules has been reported thus far.
In this report, we describe a novel homogeneous 1536-well plate assay to monitor Wee1 degradation using cryopreserved transiently transected cells. We also demonstrate the excellent performance of this assay in the context of an uHTS campaign that led to the identification of potential selective cell-permeable Wee1-Luc stabilizers.
MATERIALS AND METHODS
Vector construction
The construct allowing expression of the K328M (kinase inactive) mutant of the Wee1-Luciferase fusion protein (K328M-Wee1-Luc) was created by standard cloning methods as previously described.7 The N-cyclin B-Luc expressing construct has also been previously reported.13, 14
Cell culture
HeLa cells (American Type Culture Collection, Manassas, VA) were routinely cultured in T-175 flasks (Corning Life Sciences, Acton, MA) in Dulbecco’s Modified Eagle’s Media (DMEM, Gibco, Carlsbad, CA) supplemented with 10% (v/v) fetal bovine serum (FBS, Hyclone, Logan, UT) and 1% penicillin/streptomycin/neomycin mix (PSN, Gibco) at 37°C, 5% CO2, 95% relative humidity (RH). For small scale experiments, HeLa cells were transiently transfected with the k328M-Wee1-Luc expression vector by batches of 6×106 cells prepared in T75 flasks (Corning) containing 24 mL of a 1:1 ratio of OptiMEM and DMEM supplemented with 10% FBS, 1% PSN, 29 μg of K328M-Wee1-Luc plasmid and 87 μL of TransIT-LT1 reagent, according to the manufacturer’s protocol (Mirus Bioproducts, Madison, WI). Cells were then incubated for two days at 37°C, 5% CO2, 95% RH. For large-scale experiments, cells were prepared in larger quantities (≈0.5–2×109) and transfected using the same transfection reagents and media quantities relative to the cell number. Two days post-transfection, cells were cryopreserved.15 Briefly, cells were trypsinized, counted and resuspended in freezing medium (DMEM supplemented with 10%DMSO, 10% FBS, and 1% PSN) at a concentration ranging from 1.5 to 2×107 cells per mL. Cells were then dispensed into 1.8 mL cryovials (Nalgene) and transferred into a −80°C freezing unit using a device allowing a cooling rate of roughly 1°C per minute (Mr. Frosty, Nalgene, Rochester, NY).
Wee1 degradation uHTS assay
When using cryopreserved cells, frozen stocks of transiently transfected cells were rapidly thawed prior to the assay by transferring them in a centrifuge tube containing pre-heated media composed of phenol-red free DMEM supplemented with 10% FBS and 1% PSN. Transiently transfected cells -either from frozen or regular stocks- were then centrifuged, counted and resuspended at a concentration of 800,000 cells per mL. A stepwise assay protocol is presented in Table 1. Briefly, the Wee1 degradation assay was performed by dispensing 5 μL of cell-suspension into each well of 1536-well plates (i.e. 4,000 cells per well). After a 4 hour incubation period at 37°C, 5% CO2 and 95% RH, plates received 25 nL per well of either test compounds from the MLSMR library (final nominal concentration of 5μM), positive control MG132 at a final concentration of 30μM (serves as a 100% activation control), or DMSO alone (0.5% final DMSO concentration, 0% activation control) using a Pin Tool transfer unit (GNF/Kalypsys). Plates were then incubated at 37°C, 5% CO2 and 95% RH for twenty hours. At the end of the incubation time, plates were equilibrated at room temperature for 15 minutes before receiving 5 μL/well of SteadyLite HTS reagent (PerkinElmer Life and Analytical Sciences, Waltham, MA) into each well. Fifteen minutes later, light emitted by the conversion of luciferase’s substrate D-luciferin was measured on the ViewLux reader for 60 seconds (PerkinElmer Life and Analytical Sciences).
Table 1.
Wee1 degradation assay protocol in 1,536 well plate format
| Order | Step | Condition | Comments |
|---|---|---|---|
| 1 | Cell dispensing | 5 μL/well | 4,000 cells/well |
| 2 | Primary incubation time | 4 hours | 37°C, 95% R.H., 5% CO2 |
| 3 | Compound addition | 25 nL/well | Test concentration: 5 μM Final DMSO concentration: 0.5% |
| 4 | Secondary incubation time | 20 hours | 37°C, 95% R.H., 5% CO2 |
| 5 | Luciferase detection reagent addition | 5 μL/well | |
| 6 | Tertiary incubation | 15 min | Room temperature |
| 7 | Luminescence acquisition | 60 s/plate |
uHTS screen using the Wee1 assay
Details relative to each step of the uHTS campaign are shown Table 2. During the primary screen, test compounds from the library were screened as singlicate in the miniaturized K328M-Wee1-Luc assay at a final nominal test concentration of 5μM (final DMSO concentration of 0.5%) using the automated GNF/Kalypsys robotic platform at the Scripps Research Institute Molecular Screening Center (The Scripps Research Institute, Jupiter, FL). The percent activation of each test compound was calculated on a per-plate basis as further described in the Data Normalization section. The hit-cutoff used to qualify active compounds was calculated as the average percentage activation of all compounds tested plus three times their standard deviation16. The confirmation screen was run in the exact same conditions as the primary uHTS, except that plates were assessed in triplicate and that results for each compound were reported as the average percentage activation of the three measurements, plus or minus the associated standard deviation. For titration experiments, assay protocols were identical to those described above, with the following exception that compounds were prepared in 10 point, 1:3 serial dilutions starting at a nominal test concentration of 50 μM, and assessed in triplicate.17
Table 2.
uHTS campaign summary and results
| Step | Screen type | Target | Number of compound tested | Selection criteria | Number of selected compounds | PubChem AIDc | Assay statistics |
|
|---|---|---|---|---|---|---|---|---|
| Z′ | S/B | |||||||
| 1 | Primary screen | Wee1 | 218,117 | %inh > 8.82%a | 2,610 | 1321 | 0.65±0.05 | 17.76±2.68 |
| 2 | Confirmation | Wee1 | 1,090 | %inh > 8.82%b | 39 | 1410 | 0.62±0.02 | 8.42±0.39 |
| 3a | Titration | Wee1 | 1412 | 0.62±0.03 | 8.23±1.01 | |||
| 3b | Selectivity | N-Cyclin B | 38 | %inh>50% in Wee1 and <50% in N-cyclin B | 4 | 1414 | 0.52±0.03 | 6.61±0.25 |
| 3c | Cytotoxicity | N.A. | 1413 | 0.78±0.02 | 38.23±2.26 | |||
The primary screen hit-cutoff was calculated at the average percent activation of all test compounds plus three times the standard deviation.
The hit-cutoff calculated for the primary run was also applied to the confirmation run.
PubChem AIDs are accessible on-line at http://www.ncbi.nlm.nih.gov/sites/entrez?db=pcassay&term=xxxx, where xxxx represents the PubChem AID number listed in the table.
N.A., not applicable
N-cyclin B counterscreen assay
The N-cyclin B assay has previously been reported elsewhere.13, 14 To be used as a counterscreen for potential Wee1 degradation inhibitors, the original protocol was modified to match the one followed for the Wee1 assay as described above.
Cell viability assay
Five microliters of non-transfected HeLa cell suspension (100,000 cells per mL) in DMEM supplemented with 10% FBS and 1% PSN was dispensed into each well of a 1,536-well plate. Four hours after dispensing, 25 nL of test compounds prepared as 10-point, 1:3 serial dilutions were added to the cells using the Pin Tool. Plates were incubated for 20 hours at 37°C, 5% CO2, 95% RH and 5 μL of CellTiter-Glo (Promega, Madison, WI) was then added to each well. After 15-minute incubation at room temperature, luminescence was measured for 30 s using the ViewLux reader (PerkinElmer Life and Analytical Sciences). Cell survival was expressed as a percentage relative to wells containing media only (no cells, 0% survival) and wells containing cells treated with DMSO at 0.5% final only (100% survival).
Screening data acquisition, normalization, representation and analysis
Raw fluorescent data from the ViewLux plate reader were uploaded into our institutional HTS database (MDL Information Systems, San Ramon, CA). Activity of each well was normalized on a per-plate basis using the following equation:
where High Control represents wells from the same plate containing 30 μM MG132 (100% activation control, n = 24) and Low Control represents wells from the same plate treated with DMSO only (0% activation, n = 24).
A Z′ value greater than 0.5 was required for plate validation during the quality control process.18 For dose-response experiments, triplicate percentage activations were plotted against compound concentration. A four-parameter equation describing a sigmoidal dose-response curve was then fitted with adjustable baseline using Assay Explorer software (MDL Information Systems). All data showed here was represented using Prism version 4.03 (GraphPad Software, San Diego, CA). Fitting of dose-response curves and EC50 determination were performed using the variable slope sigmoidal curve analysis tool of Prism. Results of screening assays presented in this text can be found the NIH’s PubChem website (http://pubchem.ncbi.nlm.nih.gov/).
Chemicals
The Molecular Libraries Small Molecule Repository (MLSMR) library was provided by BioFocus DPI (South San Francisco, CA) through the NIH’s Roadmap: Molecular Libraries Initiative. Details regarding compound selection for this library can be found online at http://mli.nih.gov/mli/compound-repository/mlsmr-compounds/. Briefly, the MLSMR library is a highly diversified collection of small molecules (more that 50% of compounds exhibit molecular weights between 350 and 410 g/mol) comprising both synthetic and natural products, from either commercial or academic sources, that can be grouped into the three following categories: (1) specialty sets of know bioactive compounds such as drugs and toxins, (2) focused libraries aimed at specific target classes and (3) diversity sets covering a large area of the chemical space. Reference control MG132 (Z-Leu-Leu-Leu-CHO) was purchased from the American Peptide Company (Sunnyvale, CA). Synthesis of SID4243143, SID3713089, SID4256064 are detailed in the supplemental section of this manuscript.
FACS analysis
Compound treated Hela cells were harvested by collecting culture media, washing once in PBS, and trypsinizing. Cells were centrifuged at 400 g for five minutes at 4°C. Cells were then washed with cold PBS, centrifuged at 400 g for 5 minutes at 4°C, and rapidly resuspended with 7.5 mL 70% Ethanol and incubated at −20°C overnight. Cells were then centrifuged at 400 g for 10 minutes at 4°C and washed with 10 mL cold PBS. The supernatant was removed and the cell pellet was resuspended in 38 mM sodium citrate containing 69 mM of propidium iodide (Invitrogen) and 19mg/mL of Rnase A. FACS analysis was performed on a BD Bioscience LSR II system and analyzed using Flowjo 8.7.3 software.
Kinase profiling
The kinase profiling assay was performed by Reaction Biology Corporation (Malvern, PA) using a radiometric-based filtration binding assay as previously described.19 Briefly, compound SID4243143 was tested in duplicate at 10 μM on a panel of purified kinases in presence of 32P-γ-ATP. Incorporation of 32P-γ-ATP into peptide substrates was measured and compared to DMSO controls. The kinase pan-inhibitor staurosporine was used as a positive control for these experiments.
RESULTS
Development, validation and miniaturization of a cell-based HTS assay to monitor Wee1 stabilization
The primary screening assay was designed to measure the effect of test compounds on the stabilization of intracellular Wee1. To facilitate Wee1 protein level monitoring in cells, the firefly luciferase protein was fused in frame with the C-terminal end of Wee1. We used a kinase inactive mutant of Wee1, Wee1K328M, to prevent known toxicity issues associated with Wee1 overexpression.6 Prior studies demonstrated that this fusion protein, K328M-Wee1-Luc, had similar degradation characteristics as wild-type endogenous Wee1.7 HeLa cells transiently transfected with the K328M-Wee1-Luc encoding plasmid were incubated with test compounds for 20 hours and lysed to measure their luciferase content via the use of a light-emitting D-luciferin substrate (Fig. 1A). K328M-Wee1-Luc expressing cells treated with 30 μM MG132 (Fig. 1B), a widely used proteasome inhibitor20, showed a significant increase (>9-fold) in luminescent levels consistent with a blockade of K328M-Wee1-Luc degradation through the ubiquitin-proteasome pathway (Fig. 1C). In absence of a previously described reference compound targeting Wee1 degradation, we used MG132 as a positive control for all the experiments described in this study.
FIG. 1.

(A) HeLa cells were transfected with a plasmid allowing the expression of a chimeric Wee1 protein fused to the firefly luciferase at its C-terminal end. Upon ubiquitinylation, the Wee1-Luc fusion protein is subjected to degradation through the ubiquitin-proteasome pathway. Wee1-Luc protein levels were determined by measuring the conversion of the luciferase substrate D-luciferin into light-emitting oxiluciferin after cell lysis. A compound able to prevent Wee1 degradation is detected by an increase in measured luminescence. Ub: ubiquitin; Luc: luciferase; Wee1-Luc: K328M-Wee1-Luc. (B) Structure of the reference compound MG132. (C) Raw luminescence counts measured on the ViewLux (PerkinElmer) after HeLa cells have been transfected with the K328M-Wee1-Luc expressing plasmid and treated with DMSO or 20 μM MG132. Bars represent the average ± SD (n=4). *** indicates a P value greater than 0.0001 by a paired t test. The actual P value was 0.0007 and the calculated Z′ was 0.56.
In order to reduce cost per well and allow higher throughput, we miniaturized the assay to a 1536-well plate format in a final volume of 10 μL/well. Critical variables of the assay, such as transfection conditions, cell seeding density, DMSO tolerance, primary and secondary incubation times were systematically assessed and optimized during the miniaturization phase in order to provide the best compromise between assay performance, reagent consumption and protocol compliance with the robotic platform. Figure 2A exemplifies such systematic assay variable optimization through the determination of the most advantageous cell seeding density. In this case, the luminescent signal was found to be linear up to 5,000 cells per well upon treatment with 30 μM of the reference activator MG132. Assay performance, as determined by Z′ factor18, was optimal at 4,000 cells per well, the cell density giving the highest S/B ratio. The stepwise protocol resulting from the optimization of each assay variable is detailed Table 1. Under these conditions, MG132 (Fig. 2b) yielded reproducible EC50 values exhibiting a coefficient of variation <5% (4,686 ± 184 nM, n=5).
FIG. 2.

Wee1 degradation assay optimization and validation. (A) Cell seeding density optimization. After transfection with the K328M-Wee1-Luc contruct, HeLa cells were seeded at different densities ranging from 1,000 to 5,000 cells per well. Cells were either treated with DMSO alone (black bars) or with 30 μM of reference compounds MG132 (grey bars) for 20 hours. Z′ (○) and signal-to-background ratio (□) calculated based on relative luminescence unit (RLU) values between DMSO and MG132-treated cells are shown for each tested cell density. The star indicates the selected optimal cell density. Error bars represent the S.D. of each test condition (n=4). (B) Concentration-dependent activation of the K328M-Wee1-Luc reporter upon MG132 treatment in HeLa cells. Results in 1536-well plate format protocol using non-frozen cells (○) or frozen cells (●). Twenty-five nanoliters of a 10-point, 3:2 serial dilution starting at 30 μM were dispensed into an assay plate using a PinTool. Each data point is mean ± S.D. (n=16). The calculated EC50 was 4.54 ± 0.44 μM. (C) Scatter diagram of the controls of a representative 1536-well screening plate. Each plate contains three different controls: wells treated with DMSO only, noted as Low Controls (▼), wells receiving 30 μM of the reference compound MG132 (▲) and wells receiving a concentration of MG132 equivalent to its EC50, i.e. 5 μM (●). The calculated signal-to-background ratio was 15.53 and the Z′ factor 0.71. Solid lines represent the average luminescence value for each control type and the dashed lines the associated 95% prediction interval. Note the absence of overlap between the prediction intervals for all three control types.
To uncouple the transfection step from the rest of the assay, large amounts (>0.5×109 cells) of K328M-Wee1-Luc-expressing cells were produced and cryopreserved. Cells were transfected and cultured as described above, the only difference being that they were frozen down 48 hours post-transfection. Upon thawing, plating and treatment with a serial dilution of MG132, the thawed cells displayed pharmacological properties comparable to those of non-cryopreserved suspensions (4,540 ± 440 nM vs. 5,330 ± 360 nM, Fig. 2C). Moreover, Wee1 assays that used different thawed vials from a single batch yielded excellent reproducibility with consistent Z′ (0.65 ± 0.18), S/B (14.74 ± 1.03) and EC50 (4,699 ± 266 nM) values (n=4).
K328M-Wee1-Luc uHTS assay performance
Three types of controls (n=24 each) were placed on every assay plate: a positive control (30 μM MG132, serving as a 100% activation reference), a negative control (DMSO only, 0% activation reference), as well as a 50% activation control (using a MG132 concentration equivalent to its EC50 in the assay, i.e. 5 μM). These controls were used to (1) ensure cell responsiveness upon MG132 treatment, (2) monitor quality control through Z′ and S/B calculation, (3) verify proper compound dispensing and (4) normalize data on a per-plate basis. A scattergram of the raw luminescent counts for the different controls from a typical assay plate are shown Figure 2C, illustrating good segregation between the three control types and the absence of positional effects that are commonly associated with evaporation issues, liquid dispensing errors, or plate-reader bias. The primary screen was conducted on three separate runs employing a single batch of frozen, transiently transfected cells over two consecutive days at an average speed of ~15,000 test compounds per hour. Raw RLU values of the different controls and the associated Z, Z′ and S/B of each of the 190 plates from the primary screen are reported Figure 3 (panel A and B, respectively). Although a 20–35% decline in raw RLU values was observed over the course of each individual run, it did not translate in a degradation of assay performance as shown by the steady Z′ and S/B values measured across the entire campaign. Despite a slight slope at the beginning of each run, initial raw RLU values showed <25% variation between runs, as well as Z′ and S/B values kept within a 5% variation window, demonstrating a very limited run-to-run and day-to-day variability. As expected when screening a non-biased compound collection that encompasses a large chemical space, the percent activation of all test compounds displayed a distribution approximating a normal distribution, with an average of 0.32 ± 2.83 and a majority of compounds showing little or no activity (Fig. 3C). Overall, the screen yielded satisfactory assay statistics, with an average Z′ value of 0.65 ± 0.05 and S/B ratio of 17.76 ± 2.68. Remarkably, all assay plates exhibited Z′ values higher than 0.5, a cut-off widely accepted to validate assay plates and no plate rescheduling was required.
FIG. 3.

Wee1-Luc primary uHTS campaign performance.
(A) Scatter plot of the raw RLU values of intra-plate controls during the K328M-Wee1-Luc primary screen. One hundred and ninety 1536-well plates were run on the automated robotic platform of the Scripps Research Institute Molecular Screening Center. Plates were run in three separate batches noted Run #1, 2 and 3. Designated control wells on each plate received either 30 μM MG132 (High Control, △), 3 μM MG132 (Medium Control, □) or DMSO only (Low Control, ▽). (B) Scatter plot of the calculated Z, Z′ and S/B values of the K328M-Wee1-Luc primary screen. Low and High Control wells raw values were used to calculate the signal-to-background ratio (◆, right Y-axis) and the Z′ value (○, left Y-axis). Z values (●, left Y-axis) were calculated in reference to High Control wells and wells within the sample field of the assay plate. The low Z values observed at the beginning and at the end of each run are typical of the use of a concentration-response control plate. Indeed, each run was starting and finishing with a series of three control plates, respectively a first plate containing DMSO only in the sample field, a second one containing a 10-point, 3:2 serial dilution of MG132 starting at 30 μM a third plate identical to the first one to check for carry-over. (C) Scatter plot of the normalized percent activation of test compounds (n=218,180, 5 μM test concentration). The dashed grey line represents the hit-cutoff, which was calculated at 8.82% activation. Selected hits correspond to test compounds located above the dashed line.
K328M-Wee1-Luc screening campaign results
A stepwise description of the uHTS funnel employed to identify Wee1 degradation inhibitors is presented in Table 2. The K328M-Wee1-Luc assay was used as a first pass to screen a total of 218,117 distinct chemical entities from the National Institutes of Health’s Molecular Libraries Small Molecules Repository21 at a final nominal concentration of 5 μM (Fig. 4). A set of 2,610 compounds (i.e. 1.20% of the full library) exhibiting percentage activation greater than that calculated by a nominal cutoff algorithm (8.82%, see Material and Methods for details) were designated as primary hits (Fig. 4). Of these 2,610 primary hits, 1,280 were selected for retesting (the MLSMR limits the number of compounds that can be requested to 1280) and 1,090 of these compounds were available from the MLSMR.
FIG. 4.

The N-cyclin B-Luc counterscreen. (A) Titration results of reference control MG132 on HeLa cells transiently transfected with either the K328M-Wee1-Luc (●) or the N-cyclin B-Luc (○) expressing construct. MG132 was tested as a 10-point, 3:2 serial dilution starting at 30 μM. Data points show mean ± SD (n=3). (B) Scatter plot of the percent activation of the selected 1,090 primary hits measured in the Wee1 confirmation assay (X-axis) vs. the N-cyclin B-Luc counterscreen (Y-axis). Compounds were tested in triplicate at a nominal screening concentration of 5 μM. (C) Example of titration results of a false-positive, non-selective compound. Compound SID14743125 was titrated in a 10-point, 3:1 serial dilution starting at 50 μM in the Wee1 (○), Cyclin B (□) and viability (◇) assays. Error bars represent the standard deviation (n=3).
In order to eliminate compounds affecting Wee1 turnover in a non-selective manner, all 1,090 available compounds were retested in triplicate at 5 μM in both the original K328M-Wee1-Luc assay and in a similarly formatted counterscreen assay employing a N-cyclin B-luciferase fusion protein.14 Consistent with its ability to block the ubiquitin-proteasome pathway, the reference control MG132 exhibited comparable dose-response curves in both the K328M-Wee1-Luc and the N-cyclin B-Luc assays (4.7μM and 5.0μM EC50, respectively), hence facilitating the comparison of normalized results between the two assays (Fig. 4A).
From triplicate screening results, 39 compounds confirmed activity (i.e. yielded in average percentage activation greater than the primary hit-cutoff of 8.82%) and exhibited selective activity in the K328M-Wee1-Luc assay (Fig. 4B). These 39 compounds were selected for titration and testing in the K328M-Wee1-Luc and the N-cyclin B-Luc assays. Inspection of the individual concentration-response curves offered a better insight of the activation profiles in the different assays, facilitating the triage of non-selective compounds such as SID14743125. This compound’s promiscuity would not have been detected in a single-dose experiment because of its abrupt activation profile (Fig. 4C).
Wee1 selective compounds were chosen by applying the following set of criteria: (1) a maximal percent activity greater than 50% and an EC50 below 10 μM in the K328M-Wee1-Luc assay; and (2) a maximal percent activity lower than 50% in the N-cyclin B-Luc assay. Four structurally unrelated compounds passed these criteria with EC50 values ranging from ~2 to 8 μM, namely SID26664802, SID4256064, SID3713089, and SID4243143 (Table 3). The three last compounds were synthesized in our lab for confirmation purposes (please see supplemental materials for detailed syntheses). SID26664802 synthesis was not pursued, since this compound contains structural features that suggest it could be reactive towards many biological targets (e.g., as a Michael acceptor). Upon retesting, all newly synthesized compounds confirmed their selective activity in the K328M-Wee1-Luc assay. In contrast to the three other compounds, SID4243143 did not significantly affect cell viability at any of the different test concentrations as determined by an ATP-content measurement method (Fig. 5A) and was hence considered for further characterization.
Table 3.
Activity profile of selective K328M-Wee1-Luc degradation inhibitors
| Compound | Structure | K328M-Wee1-Luc assay |
N-cyclin B-Luc assay |
Cytotox |
|||
|---|---|---|---|---|---|---|---|
| EC50 | Max act. | EC50 | Max act. | EC50 | Max cytotox. | ||
| μM | % | μM | % | μM | % | ||
| SID4243143 | 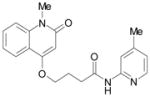 |
7.9 | 113 ± 3 | >50 | 5 ± 2 | >50 | 12 ± 4 |
| SID3713089 | 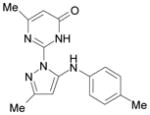 |
2.3 | 166 ± 19 | >50 | 34 ± 7 | >50 | 35 ± 2 |
| SID4256064 | 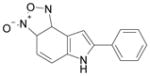 |
5.9 | 157 ± 17 | >50 | 39 ± 8 | >50 | 10 ± 2 |
| SID26664802 | 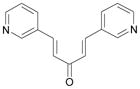 |
>1,800 | 83 ± 18 | >50 | 36 ± 14 | >50 | 25 ± 4 |
FIG. 5. Characterization of SID4243143.

(A) Titration results of SID4243143 in the Wee1 (○), N-cyclin B (□) and viability (◇) assays. Note the limited cytotoxicity and activity in the N-cyclin-B assay at higher test concentrations. Data points represent mean ± SD (n=3). (B) FACS analysis on HeLa cells treated for 20 hours with various concentrations of SID4243143 or DMSO alone. Note the dose-dependent increase in the G2/M population. (C) Profiling of SID4243143 against a panel of mitotic kinases. Black bars indicate activities lower than the average of activities measured for all kinases minus three times their standard deviation. Error bars represent the standard deviation (n=2).
Profiling SID4243143 activity
Our prediction was that a small molecule inhibitor of Wee1 degradation would inhibit cell cycle progression, since overexpression of non-degradable Wee1 induces a G2-M arrest6. FACS analysis was performed in order to test the effect of SID4243143 on the cell cycle (see Materials and Methods for details). Identical to the protocol that was used during the course of the screen, HeLa cells were treated for 20 hours with different concentrations of SID4243143. As shown Figure 5B, SID4243143 dose-dependently increased the G2-M cell population. At 5 μM, the highest concentration tested, 30.5% of cells were in the G2-M phase compared with 16.7% of the same cell population when treated with DMSO only. Furthermore, we did not observe an increase in the sub-G1 population upon treatment, confirming that SID4243143 was not toxic at the tested concentration.
Wee1 degradation has been shown to be regulated by complex pathways involving kinases and phosphatases22. In an attempt to identify potential new proteins involved in Wee1 phosphorylation, we profiled SID4243143 against a panel of kinases involved in cell cycle progression, including Wee1 itself. As shown Figure 5C, the hit compound we identified significantly inhibited CDK9, either coupled to cyclin K or cyclin T1.
In addition to characterization of the biological properties of SID4243143, a chemical optimization effort was initiated. Preliminary results of a structure-activity relationship (SAR) study revealed an unusually stringent profile of compound SID4243143 (see Supplemental Section). Indeed, initial SAR efforts focused on moving the methyl to the 2- position of the pyridine ring resulted in complete loss of activity. Furthermore, replacement of the pyridine ring with various phenyl derivatives resulted in loss of activity in both the Wee1 assay. Finally, amides based on 2-amino-6-methylpyridine and 2-amino-5-methylpyridine with only one methylene between the phenolic oxygen and carbonyl group were also found to be inactive.
DISCUSSION
We report here the development of a simple homogeneous screening protocol for identifying small-molecule inhibitors of Wee1 turnover using a luciferase reporter system. With a Z′ value greater than > 0.6 and a throughput of ~15,000 compounds per hour, the K328M-Wee1-Luc 1536-well plate assay proved to adapt very well in the context of automated uHTS and demonstrated excellent assay performance over the course of screening a small molecule collection encompassing ~218,000 distinct chemical entities.
In optimizing the assay, we found that the use of cryopreserved transiently transfected cells was beneficial to the screening process, as previously reported by others15, 23, 24. In the case of the research presented here, it uncoupled the labor and time-consuming transfection step from the rest of the assay, greatly facilitating the organization of the screen and allowing measurement of results as fast as 20 hours after initiating the assay instead of 72 hours (i.e. if the transfection step was kept part of the process). In addition, the use of cryopreserved cells, as long as they are prepared in sufficient quantities, samples a single batch of transiently transfected cells for all the different steps involved in the lead discovery and optimization effort, hence increasing assay data consistency along the entire process.
As judged by the amount of compounds that reproduced activity in the secondary assay (Table 2), the K328M-Wee1-Luc assay yielded an unusually low confirmation rate. A retrospective analysis of the data indicated that 40 out of the 4,560 negative controls (0.87%) showed percent activation above the hit-cutoff. This suggests that ~1,900 hits out of the 2,610 found, i.e. 73%, were potentially false-positives. A primary explanation for this fact is that the calculated hit-cutoff of 8.82% is unusually low for a cell-based assay, causing compounds with marginal activity to be selected as hits.
Another explanation for the low confirmation rate lies in the fact that activation profiles observed for all active compounds, including the reference control MG132, displayed very steep dose-response curves with hill slopes >2.5. In some instances, compounds displayed a bell-shaped dose-response curve, as exemplified by compound SID14743125 (Fig. 4C). The cell viability data for this compound indicated that this bell-shaped curve profile is likely associated with cytotoxicity at higher concentration. Taken together, this suggests that even small deviations from a compound’s expected test concentration negatively impact the confirmation rate. Nevertheless, the fact that the newly synthesized compounds confirmed their activities in our assay indicated that active compounds can be reliably identified by the K328M-Wee1-Luc assay.
The use of the N-cyclin B-Luc assay as a counterscreen proved to be an effective way of rapidly eliminating non-selective and promiscuous compounds early in the probe discovery process, allowing us to focus on a limited set of compounds that reduced Wee1 degradation. Among them, SID4243143 appeared to be the most attractive. Although this compound has also been reported as active in a non-trivial number of other cell-based assays available on PubChem (13 assays out of 359, i.e. 3.6%), our results clearly show a selective effect on the K328M-Wee1-Luc fusion protein over the N-cyclin B-Luc construct, ruling out the possibility that SID4243143 acts broadly as a proteasome inhibitor or any other mechanism that is not specific to the Wee1 degradation pathway. Moreover, SID4243143 did not affect HeLa cell viability at concentrations up to 50 μM. Results of the FACS analysis indicated that SID4243143 inhibits cell cycle progression in HeLa cells (Fig. 5A). This preliminary insight about SID4243143’s mode of action may explain its pleiotropic effects in different cell-based assays without dismissing a selective effect on Wee1 stabilization, as indeed high levels of Wee1 prevent mitotic entry.
The confirmed robustness of the screening platform and its ability to rapidly identify and triage promising selective, non-toxic and cell-permeable compounds encourages us to further screen other chemical libraries. In addition to the possibility of identifying other potent, selective scaffolds that will be useful in cell cycle research or as possible starting points for chemotherapeutic drug development, our long-term goal is to uncover potential phospho-transferases involved in Wee1 degradation through the screen of focused-library designed at targeting kinases. Taken together, the assays and automated protocol described herein proved to be an excellent platform for interrogating large compounds libraries for inhibitors of Wee1 degradation.
Supplementary Material
Acknowledgments
We thank Pierre Baillargeon, Lina DeLuca and Louis Scampavia (Lead Identification Division, Scripps Florida) for compound management and quality control of compounds by LC-MS. The work of N.G.A., P.H. and S.S. was supported by the National Institutes of Health grant #NS05699. The National Institutes of Health Molecular Library Screening Center Network (5U54MH074404) supported the research efforts of P.C., P.H., F.M., J.K.M. and W.R.R.
References
- 1.Reed SI. Ratchets and clocks: the cell cycle, ubiquitylation and protein turnover. Nat Rev Mol Cell Biol. 2003;4(11):855–864. doi: 10.1038/nrm1246. [DOI] [PubMed] [Google Scholar]
- 2.Gautier J, Solomon MJ, Booher RN, Bazan JF, Kirschner MW. cdc25 is a specific tyrosine phosphatase that directly activates p34cdc2. Cell. 1991;67(1):197–211. doi: 10.1016/0092-8674(91)90583-k. [DOI] [PubMed] [Google Scholar]
- 3.Perry JA, Kornbluth S. Cdc25 and Wee1: analogous opposites? Cell Div. 2007;2:12. doi: 10.1186/1747-1028-2-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kraft C. Mitotic entry: tipping the balance. Curr Biol. 2003;13(11):R445–446. doi: 10.1016/s0960-9822(03)00367-1. [DOI] [PubMed] [Google Scholar]
- 5.Watanabe N, Arai H, Iwasaki J, Shiina M, Ogata K, Hunter T, et al. Cyclin-dependent kinase (CDK) phosphorylation destabilizes somatic Wee1 via multiple pathways. Proc Natl Acad Sci U S A. 2005;102(33):11663–11668. doi: 10.1073/pnas.0500410102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Watanabe N, Arai H, Nishihara Y, Taniguchi M, Watanabe N, Hunter T, et al. M-phase kinases induce phospho-dependent ubiquitination of somatic Wee1 by SCFbeta-TrCP. Proc Natl Acad Sci U S A. 2004;101(13):4419–4424. doi: 10.1073/pnas.0307700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Owens L, Simanski S, Squire C, Smith A, Cartzendafner J, Cavett V, et al. Activation Domain-dependent Degradation of Somatic Wee1 Kinase. J Biol Chem. 2010;285(9):6761–6769. doi: 10.1074/jbc.M109.093237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pomerening JR, Kim SY, Ferrell JE., Jr Systems-level dissection of the cell-cycle oscillator: bypassing positive feedback produces damped oscillations. Cell. 2005;122(4):565–578. doi: 10.1016/j.cell.2005.06.016. [DOI] [PubMed] [Google Scholar]
- 9.Yoshida T, Tanaka S, Mogi A, Shitara Y, Kuwano H. The clinical significance of Cyclin B1 and Wee1 expression in non-small-cell lung cancer. Ann Oncol. 2004;15(2):252–256. doi: 10.1093/annonc/mdh073. [DOI] [PubMed] [Google Scholar]
- 10.Kiviharju-af Hallstrom TM, Jaamaa S, Monkkonen M, Peltonen K, Andersson LC, Medema RH, et al. Human prostate epithelium lacks Wee1A-mediated DNA damage-induced checkpoint enforcement. Proc Natl Acad Sci U S A. 2007;104(17):7211–7216. doi: 10.1073/pnas.0609299104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lenart P, Petronczki M, Steegmaier M, Di Fiore B, Lipp JJ, Hoffmann M, et al. The small-molecule inhibitor BI 2536 reveals novel insights into mitotic roles of polo-like kinase 1. Curr Biol. 2007;17(4):304–315. doi: 10.1016/j.cub.2006.12.046. [DOI] [PubMed] [Google Scholar]
- 12.Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434(7035):907–913. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- 13.Verma R, Peters NR, D’Onofrio M, Tochtrop GP, Sakamoto KM, Varadan R, et al. Ubistatins inhibit proteasome-dependent degradation by binding the ubiquitin chain. Science. 2004;306(5693):117–120. doi: 10.1126/science.1100946. [DOI] [PubMed] [Google Scholar]
- 14.Harmey D, Smith A, Simanski S, Moussa CZ, Ayad NG. The anaphase promoting complex induces substrate degradation during neuronal differentiation. J Biol Chem. 2009;284(7):4317–4323. doi: 10.1074/jbc.M804944200. [DOI] [PubMed] [Google Scholar]
- 15.Chen J, Lake MR, Sabet RS, Niforatos W, Pratt SD, Cassar SC, et al. Utility of large-scale transiently transfected cells for cell-based high-throughput screens to identify transient receptor potential channel A1 (TRPA1) antagonists. J Biomol Screen. 2007;12(1):61–69. doi: 10.1177/1087057106295220. [DOI] [PubMed] [Google Scholar]
- 16.Hodder P, Cassaday J, Peltier R, Berry K, Inglese J, Feuston B, et al. Identification of metabotropic glutamate receptor antagonists using an automated high-throughput screening system. Anal Biochem. 2003;313(2):246–254. doi: 10.1016/s0003-2697(02)00608-5. [DOI] [PubMed] [Google Scholar]
- 17.Madoux F, Li X, Chase P, Zastrow G, Cameron MD, Conkright JJ, et al. Potent, selective and cell penetrant inhibitors of SF-1 by functional ultra-high-throughput screening. Mol Pharmacol. 2008;73(6):1776–1784. doi: 10.1124/mol.108.045963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang JH, Chung TD, Oldenburg KR. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen. 1999;4(2):67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 19.Ma H, Horiuchi KY, Wang Y, Kucharewicz SA, Diamond SL. Nanoliter homogenous ultra-high throughput screening microarray for lead discoveries and IC50 profiling. Assay Drug Dev Technol. 2005;3(2):177–187. doi: 10.1089/adt.2005.3.177. [DOI] [PubMed] [Google Scholar]
- 20.Lee DH, Goldberg AL. Proteasome inhibitors: valuable new tools for cell biologists. Trends Cell Biol. 1998;8(10):397–403. doi: 10.1016/s0962-8924(98)01346-4. [DOI] [PubMed] [Google Scholar]
- 21.Inglese J, Johnson RL, Simeonov A, Xia M, Zheng W, Austin CP, et al. High-throughput screening assays for the identification of chemical probes. Nat Chem Biol. 2007;3(8):466–479. doi: 10.1038/nchembio.2007.17. [DOI] [PubMed] [Google Scholar]
- 22.Watanabe N, Broome M, Hunter T. Regulation of the human WEE1Hu CDK tyrosine 15-kinase during the cell cycle. EMBO J. 1995;14(9):1878–1891. doi: 10.1002/j.1460-2075.1995.tb07180.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu J, Chen T, Norris T, Knappenberger K, Huston J, Wood M, et al. A high-throughput functional assay for characterization of gamma-aminobutyric acid(A) channel modulators using cryopreserved transiently transfected cells. Assay Drug Dev Technol. 2008;6(6):781–786. doi: 10.1089/adt.2008.161. [DOI] [PubMed] [Google Scholar]
- 24.Kunapuli P, Zheng W, Weber M, Solly K, Mull R, Platchek M, et al. Application of division arrest technology to cell-based HTS: comparison with frozen and fresh cells. Assay Drug Dev Technol. 2005;3(1):17–26. doi: 10.1089/adt.2005.3.17. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.