

Abstract
In mammals, the second messenger cAMP is synthesized by a family of transmembrane isoforms (tmACs) and one known cytoplasmic enzyme, “soluble” adenylyl cyclase (sAC). Understanding the individual contributions of these families to cAMP signaling requires tools which can distinguish them. Here, we describe the structure-based development of isoform discriminating AC inhibitors. Docking calculations using a library of small molecules with the crystal structure of a sAC homologue complexed with the noncompetitive inhibitor catechol estrogen identified two novel inhibitors, 3,20-dioxopregn-4-en-21-yl 4-bromobenzenesulfonate (2) and 1,2,3,4,5,6,7,8,13,13,14,14-dodecachloro-1,4,4a,4b,5,8,8a,12b-octahydro-11-sulfo-1,4:5,8-dimethanotriphenylene-10-carboxylic acid (3). In vitro testing revealed that 3 defines a novel AC inhibitor scaffold with high affinity for human sAC and less inhibitory effect on mammalian tmACs. 2 also discriminates between sAC and tmACs, and it appears to simultaneously block the original binding pocket and a neighboring interaction site. Our results show that compounds exploiting the catechol estrogen binding site can produce potent, isoform discriminating AC inhibitors.
Introduction
The ubiquitous second messenger cAMP regulates a diverse set of essential biological processes in mammals,1 and its dysfunction contributes to a variety of human diseases. In mammals, it is generated by two families of enzymes from the class III adenylyl cyclase superfamily (AC; E.C. 4.6.1.1).2,3 A family of transmembrane ACsa is encoded by nine distinct genes (tmACs AC1 to AC9), and a second family of cytoplasmic enzymes, referred to as “soluble” ACs (sAC), is generated by alternative splicing of a single gene.2,4,5 The tmACs play key roles in cellular responses to extracellular signals:1 they are regulated through heterotrimeric G-proteins in response to the stimulation of G-protein coupled receptors (GPCRs). sAC enzymes, in contrast, are directly activated by calcium and by the cellular metabolites bicarbonate and ATP6,7 thus, sAC has been postulated to act as an intracellular metabolic sensor.8
All known mammalian class III ACs are comprised of two related catalytic domains, C1 and C2, and the crystal structure of a tmAC enzyme revealed that these domains are structurally very similar.9 The C1/C2 heterodimer therefore resembles a homodimer, and the shared active site at the dimer interface has a pseudosymmetric site that is catalytically inactive. Sequence conservations and the crystal structure of the cyano-bacterial sAC homologue CyaC showed that sAC enzymes, despite their unique regulation, have the same overall structure as tmACs and employ the same two-metal ion mechanism for catalysis.9–11 The active site at the dimer interface contains two magnesium ions in the so-called ion A and ion B sites. Ion A acidifies the ribose 3′ hydroxyl and stabilizes the transition state, while ion B serves as an anchoring point for the ATP β- and γ-phosphates.10,11
Because of the biological importance of cAMP signal transduction, it is essential to understand the processes that regulate the cellular levels of this second messenger. There has been significant progress toward development of pharmacological tools to distinguish among the various phosphodiesterases, the enyzme responsible for cAMP degradation.12 In contrast, there has been much less progress toward the development of specific tools to distinguish among the various means to synthesize cAMP. The limited recent progress toward development of specific AC inhibitors has not yet yielded compounds with both high potency and high selectivity.13,14 Also, pharmacological modulation of tmAC isoforms has been proposed for treatment of acute heart failure,14 whereas sAC is a target for contraception15 and possibly for the treatment of hypercalciuria.16 The most widely characterized class of AC inhibitors are the so-called P-site inhibitors, nucleotide analogues that occupy the binding site for the substrate ATP.17 P-sites show a moderate AC isoform specificity and bear the potential to bind to many cellular nucleotide binding proteins.18,19 A second inhibitor class contains an adenine linked to ion chelators. In general, this class suffers from the same predicament;20 however, a new compound series from this class can discriminate between some tmAC isoforms.21 Related AC inhibitors are derivatives of 9-(2-phosphonylmethoxyethyl)adenine,22 but these were initially identified as potent inhibitors of nucleotide polymerases.22 A distinct nucleotide modification, the methylan-thranyloyl (MANT) group in MANT-GTP, MANT-inosine-5′-(γ-thio)triphosphate and other nucleotides, showed promising discrimination between tmACs and sAC.19 The MANT group makes MANT-GTP a potent tmAC inhibitor, whereas sAC and, surprisingly, even heterotrimeric G-proteins show little sensitivity against this compound.19 The MANT nucleotides show little selectivity among tmAC isoforms,19 but they may be useful complements to the sAC-selective compound, KH7, which was identified in a chemical screen.23 The nucleotide part of MANT-GTP occupies the ATP binding site, but the MANT group binds to a less conserved pocket at the C1/C2 interface,24 suggesting that varying the group in this pocket may prove to be an attractive approach for achieving greater isoform specificity. The same holds true for the binding pocket for the noncompetitive inhibitors of the catechol estrogen (CE) family. These compounds bind close to the AC catalytic site and inhibit sAC and tmACs by chelating the catalytic ion A.25 The residues lining the binding pocket show little conservation between mammalian AC isoforms; therefore, we postulated that exploiting this binding site may yield more specific compounds.
Here, we describe the structure-based development of novel AC inhibitors that exploit the CE binding site. Using the crystal structure of an AC/CE complex, we searched a compound library through docking calculations. Potential inhibitors were characterized in vitro for their inhibition potency and specificity. Two novel compounds were identified that inhibit sAC but show little to no inhibitory effect on a variety of transmembrane ACs. These inhibitors reveal a novel AC inhibitor scaffold and a substituent that enables a steroid-based compound to exploit an additional binding site.
Results
Identification of Novel AC Inhibitors through Docking Calculations
For the identification of novel inhibitors of class III AC enzymes, we started with the crystal structure of the catalytic core of CyaC, a cyanobacterial homologue of human sAC, bound to the CE compound 2-hydroxy-17β-estradiol (1; Figure 1a), a nonselective inhibitor (PDB entry 2BW7).25 In this structure of CyaC with the ATP analogue α, β-Me-ATP (α, β-methylene-ATP) and the noncompetitive inhibitor 1, the catalytic magnesium ion A is moved out of its proper location due to a chelating interaction with the inhibitor hydroxyl groups (Figure 1b). This structure further displays a partial active site closure upon inhibitor binding. To identify other compounds that can exploit the binding site of 1, we performed our docking study with this partially closed protein structure; the inhibitor 1 was removed, but the substrate analogue and the divalent active site ions (ion A: Mg2+; ion B: Ca2+) were included in the target structure. The ligand target site was defined as a cube covering the binding site of 1 as well as the complete ATP binding pocket. We tested the system by redocking 1 into the protein. This control resulted in a complex close to the experimental structure (rmsd 0.3–0.5 Å for 8 of the 9 top solutions).
Figure 1.
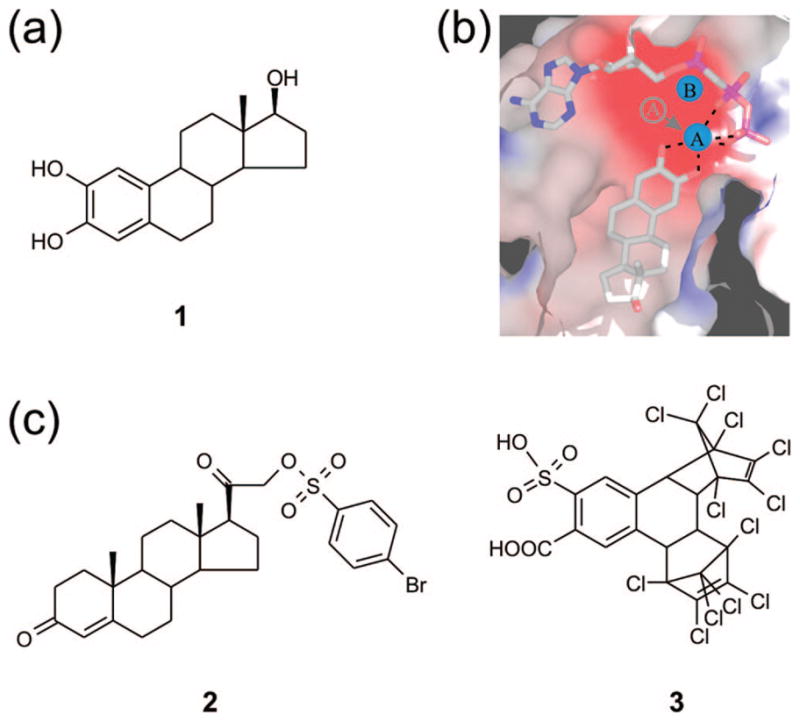
Novel AC inhibitors developed based on the CE binding site. (a) Chemical structure of the noncompetitive AC inhibitor 1. (b) Crystal structure of the active site of CyaC in complex with a substrate analogue and the inhibitor 1. The catechol moiety of the inhibitor chelates the active site magnesium ion A and removes it from its normal position. This figure was reproduced from ref 25. (c) Chemical structures of the novel AC inhibitors 2 and 3 identified through docking calculations with the 1 binding pocket of CyaC, followed by in vitro activity assays.
For identifying potential novel inhibitor leads, we screened the NCI diversity set (http://www.dtp.nci.nih.gov) of 1990 structurally diverse organic compounds. The compound structures were docked into the CyaC target site by using the genetic algorithm of AutoDock 3.0.26 The 17 hits with highest predicted affinity (Table 1), except for hit number 10 (compound 11), which is an alkylating agent, were then tested in an in vitro activity assay for inhibition of CyaC. Six compounds showed significant inhibition at or below 250 μM, corresponding to a high hit rate of 38%, comparable to hit rates in other high-throughput docking studies.27,28 Five of these six hits were among the nine top-ranked compounds, indicating that the virtual screen indeed yielded a significant enrichment of inhibitors at the top of the hit list. Three compounds inhibited CyaC with an IC50 ≤ 70 μM (Table 1), and the two compounds with highest affinities (docking hits 9 and 4; Figure 1c), 3,20-dioxopregn-4-en-21-yl 4-bromobenzenesulfonate (2) and 1,2,3,4, 5,6,7,8,13,13,14,14-dodecachloro-1,4,4a,4b,5,8,8a,12b-octahy-dro-11-sulfo-1,4:5,8-dimethanotriphenylene-10-carboxylic acid (3), were characterized further.
Table 1.
Highest Ranked Docking Hits
| Docking rank | NSC number (Compound no.) | Chemical structure | Compound name | IC50 against CyaC in vitro |
|---|---|---|---|---|
| 1 | 28081 (4) | 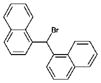 |
1-(bromo(1-naphthyl)methyl)naphthalene | ~200 μMa |
| 2 | 150289 (5) |  |
2,3-dibromo-1-(4-(hydroxy(oxido)amino)phenyl)-3-(4-quinolinyl)-1-propanone | ~ 100 μMa |
| 3 | 371884 (6) | 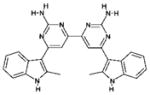 |
6-(2-methyl-2H-indol-3-yl)-6′-(2-methyl-3aH-indol-3-yl)-4,4′ -bipyrimidine-2,2′-diamine |
No significant inhibition up to 250 μM |
| 4 | 270718 (3) | 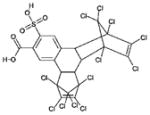 |
1,2,3,4,5,6,7,8,13,13,14,14-dodecachloro-1,4,4a,4b,5,8,8a, 12b-octahydro-11-sulfo-1,4:5,8 -dimethanotriphenylene-10-carboxylic acid |
5 μM |
| 5 | 123994 (7) | 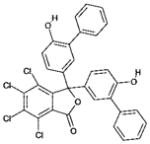 |
4,5,6,7-tetrachloro-3,3-bis(6-hydroxy[1,1′-biphenyl]-3-yl)-2- benzofuran-1 (3H)-one |
No significant inhibition up to 250 μM |
| 6 | 88135 (8) |  |
2,3,6,23-tetrahydroxyurs-12-en-28-oic acid | ~100 μM a |
| 7 | 7524 (9) | 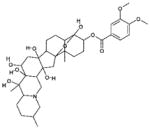 |
4, 9-Epoxycevane-3,4,12,14,16,17,20-heptol 3-(3,4-dimethoxybenzoate) | No significant inhibition up to 250 μM |
| 8 | 371878 (10) | 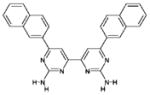 |
6,6′-di(naphthalen-2-yl)-4,4′-bipyrimidine-2,2′-diamine | No significant inhibition up to 250 μM |
| 9 | 88915 (2) | 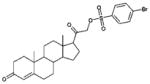 |
3,20-dioxopregn-4-en-21-yl 4-bromobenzenesulfonate | 15 μM |
| 10 | 46529 (11) | 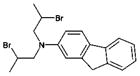 |
N,N-bis(2-bromopropyl)-9H-fluoren-2-amine | Not tested (non-specific alkylating agent) |
| 11 | 50352 (12) | 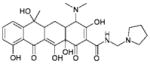 |
4-(dimethylamino)-3,6,10,12,12a-pentahydroxy-6-methyl-1,11-dioxo-N-(1-pyrrolidinylmethyl) -1,4,4a,5,5a,6,11,12a-octahydro-2-naphthacenecarboxamide |
No significant inhibition up to 250 μM |
| 12 | 159628 (13) | 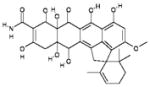 |
{Spiro[2-cyclohexene-1,} {2′(1′H)- cyclopenta[de]naphthacene]-9′-carboxamide,} 7′,7′a,8′,11′, 11′a, 12′-hexahydro-5′,6′,7′a,10′,11′a, 12′-hexahydroxy-3′-methoxy-2,6,6-trimethyl-7′,8′-dioxo-, (2′α,7′aβ,11′aβ,12′β)-(−)- |
No significant inhibition up to 250 μM |
| 13 | 371876 (14) | 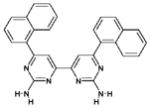 |
6,6′-di(naphthalen-1-yl)-4,4′-bipyrimidine-2,2′-diamine | No significant inhibition up to 250 μM |
| 14 | 44480 (15) |  |
2-bromo-1-(9-bromo-3-phenanthryl)-1-propanone | No significant inhibition up to 250 μM |
| 15 | 259968 (16) |  |
17,24-dihydroxy-10-(4-methoxybenzyl)-4,7,9,13,15,29-hexamethyl-22-oxa-O3,6,9,12,15,29- hexaazatetracyclo[14.12.2.2~18,21~.1~2 3,27~]tritriaconta-18,20,23(31),24,26,32-hexaene-2,5,8,11,14,30-hexone |
No significant inhibition up to 250 μM |
| 16 | 168656 (17) |  |
2,3-dibromo-4-(4-methoxyphenyl)-4-oxobutanoic acid | No significant inhibition up to 250 μM |
| 17 | 95090 (18) |  |
5′-benzyl-12′-hydroxy-2′-methyl-3′,6′, 18-trioxoergotaman |
~70 μMa |
IC50 estimated from experiments without inhibitor and with two different inhibitor concentrations, respectively.
2 Shows a High Affinity to Mammalian sAC and Exploits a Second Binding Site
2 resembles the AC inhibitor 1, having a steroid scaffold, but it lacks the catechol arrangement in ring A thought to be essential for AC inhibition (see below). Of the 16 compounds tested in vitro, however, the compound showed the second highest potency at inhibiting CyaC, with an IC50 of 15 μM (Figure 2a). CyaC is a cyanobacterial AC that we have used for modeling mammalian sAC; the two enzymes are closely related at both the amino acid sequence and regulatory levels.10 Therefore, we tested whether 2 also inhibits mammalian sAC. On purified, recombinant mammalian sAC, 2 revealed an even greater affinity, with an IC50 of 1.1 μM (Figure 2b) compared to the reported IC50 of ~3 μM observed with 1.25,29 Thus, this compound is among the highest affinity inhibitors known for human sAC with an IC50 comparable to the 0.7 μM reported for the nucleotide 2′,5′-didesoxy-3′-ATP.19
Figure 2.

Inhibition of CyaC and mammalian sAC and tmACs by compound 2. (a) Dose–response curve for 2 inhibition of CyaC activity which shows an IC50 of 15 μM. (b) The dose–response curve for the effect of 2 on mammalian sAC activity, which shows that this compound inhibits with high potency (IC50 of 1.1 μM). (c) Forskolin-stimulated AC activity of HEK293T cell lysates in presence of 2, 3, or DMSO (control). (d) Forskolin-stimulated (AC1 and AC5) or basal (AC2 and AC9) AC activity of cell lysates from HEK293T cells transfected with the indicated tmAC isoform in absence (control) and presence of 100 μM compund 2.
To examine effects of 2 on tmACs, we measured its ability to inhibit the forskolin-stimulated activities of the endogenous tmACs expressed in HEK293T cells; forskolin exclusively stimulates tmACs.4 2 showed no significant inhibition of forskolin-stimulated AC activity in whole cell extracts of HEK293T cells at a concentration of 100 μM (Figure 2c). To examine its potency on tmACs further, we used whole cell extracts of HEK293T cells transfected with a representative of each of the tmAC subfamilies30 in the presence of forskolin. At 100 μM, 2 did not significantly inhibit the activities of AC1 (representing tmACs 1, 3, and 8), AC2 (representing tmACs 2, 4, and 7), or AC5 (representing tmACs 5 and 6) (Figure 2d). To examine whether 2 inhibited AC9, which is the lone tmAC insensitive to forskolin stimulation, we tested its effect on basal activity, and once again, 2 did not appreciably affect AC9 activity at a concentration of 100 μM (Figure 2d). Thus, 1 shows high potency versus sAC and discriminates between sAC and tmACs, which makes it an attractive lead for the further development of potent, isoform-specific AC inhibitors.
The crystal structure of 1 inhibiting CyaC revealed that the two ortho-hydroxyl groups chelate the catalytic magnesium, ion A and remove it from its correct position.25 Other compounds with a catechol group, including catechol steroids and norad-renalin derivatives, were also able to inhibit soluble adenylyl cyclase activity.31 Therefore, it was thought that an undisturbed catechol moiety would be essential for sAC inhibition. But 2, which has a steroid scaffold but no catechol in ring A, inhibits sAC with even higher potency than 1. Analysis of the docked CyaC/2 complexes hints at an explanation for this finding (Figure 3a,b,c). The most favorable predicted binding orientation (representing cluster comprising 54% of all poses, top binding energy −13.4 kcal/mol) is rotated ~180° compared to 1, bringing the D ring of the steroid scaffold with its hydrophilic groups, the keto function and the 4-bromobenzenesulfonate moiety, into position for interactions with the active site magnesium (Figure 3a); slightly moving the ion down would then again result in a tight chelator interaction with the inhibitor. This binding mode predicts, however, a localization for the 4-bromobenzene group of 2 in a more polar, possibly unfavorable environment next to the ATP phosphates and ribose and the loop between β2 and β3, as more favorable binding pockets appear to be blocked by the substrate. Redocking of 2 into CyaC without bound substrate analogue indicates that 2, in addition to exploiting the CE binding site and inhibition mechanism, might also block the ATP binding site with its 4-bromobenze-nesulfonate group (Figure 3b,c). The inhibitor could either shift toward the ATP site, placing the 4-bromobenzene group into the ribose and part of the adenine binding pocket (Figure 3b), or the inhibitor might occupy the second 1 binding site of the homodimer and the complete adenine binding cleft (Figure 3c). Both binding orientations would predict 2 should compete with ATP for binding. Activity assays with various substrate concentrations in the presence of different fixed amounts of inhibitor had previously revealed that 1 is a noncompetitive inhibitor with respect to ATP,25 and by doing these experiment with 2, we found that this compound also acts as noncompetitive inhibitor (Figure 3d). Consistently, most docking orientations of 2 (80% of 30 poses) in the absence of a ligand for the nucleotide binding site did not occupy this site, and solutions exploiting the site were predicted to have relatively low binding energies (top binding energy −9.7 kcal/mol). Instead, the top cluster of poses (27% of poses; top binding energy −11.2 kcal/mol) positions the 4-bromobenzene group again close to the phosphate/sugar binding site. Thus, 2 indicates that steroids can take advantage of an alternative binding orientation at the 1 binding site, and it suggests that steroid derivatization to simultaneously use the 1 site and a neighboring pocket may facilitate development of potent, isoform discriminating inhibitors. The docking results suggest that this additional site might be formed by side-chains around the loop between β2 and β3, but we will have to await future structural studies for the definite identification of this additional interaction site.
Figure 3.

Mode of inhibition of CyaC by compound 2. (a) Model of the inhibitor 2 docked into the CyaC/α, β-Me-ATP complex. (b) Crystal structure of CyaC in complex with the substrate analogue α, β-Me-ATP, overlaid with 2 docked into CyaC without ligand, showing a potential steric clash predicted between ATP and 2. (c) Alternative conformation for 2 docked into unliganded CyaC, with the 4-bromobenzene in the adenine binding cleft. (d) Saturation curves for CyaC activity at various substrate concentrations in the presence of different amounts of 2 (●: 0 μM; Δ: 1 μM; ■: 2.5 μM; □: 15 μM). The depicted nonlinear regression using the Michaelis–Menten equation resulted in Km values fluctuating between 0.5 and 1 mM, whereas the Vmax values decreased with increasing inhibitor concentration (0.51, 0.38, 0.29, and 0.08 nmol/(mg · min)), indicating noncompetitive inhibition by 2.
3 Reveals an Alternative to the Estrogen Scaffold
Of the 16 compounds tested in vitro, 3 was the most potent inhibitor of CyaC, with an IC50 of 5 μM (Figure 4a). The steep slope of the dose–response curve hints at cooperativity, which might be expected due to the homodimeric architecture of the bacterial CyaC (see Discussion). The modeled complex of CyaC with 3 predicts an inhibition mode comparable to 1 (Figure 4b,c,d) despite its novel inhibitor scaffold. The more hydrophobic ring systems of 3 occupy the pocket that also binds the steroid ring systems B to D of 1; this pocket is formed by atoms from the central helix α4 (Asn1146, Ala1149, Arg1150, Gln1152, Glu1153), the active site strands β2 and β3 (Val1059, Ala1062, Val1059*; * indicates the partner monomer of the dimeric catalytic core), and strand β1 (Phe1015, Asp1017). The polar carboxyl group and sulfonic acid moiety point toward the active site region harboring the two divalent ions A and B. All top clusters of docked conformations showed an interaction between 3 and ion A, although various arrangements were observed for this interaction (Figure 4b,c,d). Both the carboxyl and the sulfonic acid groups may interact with ion A in a chelating interaction (Figure 4b), as previously observed for 1, or either group may interact with ion A by itself (Figure 4c,d).
Figure 4.

Compound 3 reveals a novel scaffold and chelating group for AC inhibition. (a) Dose–response curve for the inhibition of CyaC AC activity by 3, yielding an IC50 of 5 μM. (b) Representative of a cluster of docked conformations of 3, which leads to a chelating interaction of the 3 carboxyl group and sulfonyl moiety with ion A. (c) Representative of a cluster of docked conformations of 3, which leads to an interaction of ion A with the 3 carboxyl group. (d) Representative of a cluster of docked 3 conformations, which leads to an interaction of the inhibitor sulfonyl group with ion A. (e) A dose–response curve for the inhibition of mammalian sAC activity by 3, yielding an IC50 of 11 μM. (f) Forskolin-stimulated (AC1 and AC5) or basal (AC2 and AC9) AC activities of cell lysates from HEK293T cells transfected with the indicated tmAC isoform in absence (control) and presence of 100 μM 3.
3 strongly inhibits the cyanobacterial sAC enzyme CyaC, and we next tested whether it also inhibits mammalian sAC. The compound inhibited purified, recombinant mammalian sAC in a concentration-dependent manner with an IC50 of 11 μM (Figure 4e). We then analyzed its potency on mammalian tmACs, first by testing forskolin-stimulated endogenous tmAC activity in HEK293T whole cell extracts. 3 inhibited this activity only slightly at a concentration of 100 μM (Figure 2c). We then tested 3 on lysates of HEK293T cells expressing representatives of the mammalian tmAC subfamilies.30 3 at a concentration of 100 μM showed only moderate inhibition of basal AC2 and AC9 activity and of forskolin-stimulated AC1 and AC5 activity (Figure 4f). Interestingly, 3 displayed a different effect on AC2 (stimulation) in the presence of forskolin (data not shown), which will be the focus of a separate study. We can conclude, however, that 3 shows a high selectivity for inhibition of mammalian sAC versus tmAC isoforms.
Discussion
The 10 mammalian AC subclasses control diverse sets of crucial physiological processes, rendering them interesting targets for therapeutic intervention.1,14 Successful AC-targeted drug development has been limited so far, as most known AC inhibitors bind to the enzyme’s ATP binding site and thus tend to bind to a wide range of nucleotide binding proteins.1,14,18 In contrast, inhibitors binding outside the active site, such as the non-nucleoside inhibitor nevirapine for reverse transcriptase,32 exploit more specific features of a target protein. CEs such as 1 are AC inhibitors binding to a conserved pocket different from the substrate binding site, which displays features varying between individual AC enzymes.25 1 shows little isoform specificity,25 however, and thus does not exploit the full potential of this binding site. Therefore, we explored the possibility of targeting the CE binding site with novel compounds in order to achieve greater specificity and higher potency.
Because of its steroid scaffold, the novel inhibitor 2 is structurally related to CEs such as 1. However, 2 displays some significant differences compared to CEs. It has no catechol moiety in the A ring and therefore appears to bind in a 180° rotated orientation, with its D-ring pointing toward the active site. This variation would be consistent with previous studies that indicated that the hydrophobic part of the ligand attached to the chelating group can vary widely. Compounds such as tyrphostatins33 and epinephrine derivatives,31,34 which contain a catechol completely different from steroids, inhibit sAC activity and are likely to exploit the CE binding site and inhibition mechanism. Our docking system reflected the imperfect steric fit of the steroid scaffold to the CE site indicated by these two possible binding orientations. Although nine of the ten top solutions for redocking 1 into the CyaC part of the CyaC/1 structure reproduced the experimentally determined orientation, one docking orientation corresponded to the orientation predicted for 2. It appears that the steroid scaffold offers a hydrophobic, nonspecific binding partner for the CE interaction site, but as seen for 2, variations and substitutions on this scaffold can increase the specificity of these compounds.
A second interesting feature discriminating 2 from 1 is 2’s ability to simultaneously exploit a neighboring binding sites in addition to the CE site. While the steroid part of 2 and the chelating groups at the D-ring appear to mimic CEs, the bulky extension of 2 likely adds interactions to a neighboring pocket of the enzyme. This extension of the binding area provides an opportunity to find compounds with both increased affinity and specificity. Consistently, 2 indeed shows a higher specificity and a slightly increased potency against sAC as compared to CEs.29 A similar principle is seen for the inhibition of ACs by MANT-GTP, where the nucleotide binds to the ATP binding site and the MANT group occupies a hydrophobic patch at the C1/C2 dimer interface.24 The interactions of this GTP substituent have not yet been refined, and MANT-GTP, in fact, is well-known as a ligand for heterotrimeric G-proteins35 and many other GTP-binding proteins (see, e.g., refs 36, 37). Thus, MANT-GTP is likely to inhibit many nucleotide binding proteins. However, although they are not yet fully developed, 2 and MANT-GTP indicate a huge potential for potent and specific AC inhibition by compounds exploiting the substrate or CE binding site and an additional, apparently AC isoform specific pocket.
The steroid scaffold of CEs and 2 renders these compounds potential ligands for the cellular steroid binding proteins. 3 reveals an alternative to the steroid scaffold for binding to the CE site. The geometry of 3 indeed would be a good starting point for further improvements due to the large space that can be probed by modifications at various positions, although its chemical nature makes it hard to introduce modifications at specific positions. In particular, the potential cooperativity observed for 3 on CyaC hints at an attractive idea for inhibitor improvement. The Hill coefficient of 3.2 indicates several binding sites for 3, most likely the two symmetry-related CE sites of the homodimer and possibly even the substrate binding site. The Hill coefficient of 2.0 for sAC inhibition also hints at a second binding site for 3; the effect of 3 on AC2 in presence of forskolin indicates that the pseudosymmetric substrate site which can accommodate forskolin and related diterpenes9,38 is still available for forskolin. Assuming that AC2, like sAC, has two sites for 3, we speculate that this compound can additionally occupy the second, pseudosymmetric CE site in C2 of the mammalian C1C2 heterodimer, although definitive answers will have to await further studies. In any case, in the homodimeric CyaC/1 crystal structure,25 the flat steroid binds with one side to the protein and faces with its opposite side the second 1 molecule, which is bound to the CE site of the partner monomer. Thus, bulkier compounds should be able to exploit the CE binding site and the second site—the symmetry-related CE site in homodimers and the pseudosymmetric site in heterodimers—simultaneously for higher affinity and specificity.
Taken together, our docking and inhibition studies have identified two novel AC inhibitors, 2 and 3. Both compounds inhibit mammalian sAC potently. In contrast, they show weak or no inhibition of mammalian tmACs. 2 shows the potential of exploiting the CE binding site and an additional pocket simultaneously, whereas 3 indicates that bulkier compound scaffolds also can exploit the CE binding site with an increased specificity compared to 1.
Materials and Methods
Target Preparation and Virtual Ligand Screening
The crystal structure of CyaC in complex with α, β-Me-ATP and 1 (PDB entry 2BW7)25 was used for this docking study. All water molecules as well as the two molecules of 1 were removed from the catalytic dimer. Polar hydrogen atoms were added and Kollman charges assigned to all atoms with AutoDockTools (http://autodock.scripps.edu/resources/adt). The divalent active site ions A and B were treated as found in the target structure, i.e., as Mg2+ and Ca2+, respectively. One of the two active sites of the homodimer was chosen as docking site, and 60 Å × 50 Å × 66 Å affinity grids centered on this active site plus 1 binding pocket were calculated with 0.375 Å spacing by using Autogrid326 for each of the following atom types: C, A (aromatic C), N, O, S, H, F, Cl, Br, I, P, and e (electrostatic). The NCI diversity set of 1990 compounds with unique scaffolds selected from the NCI-3D structural database (http://www.dtp.nci.nih.gov) was then docked into the target site. The modified set with all hydrogens added and assignments of Gasteiger charges and rotate able bonds was obtained from http://autodock.scripps.edu/resources/databases. The docking calculations were done by using the Lamarckian genetic algorithm (LGA) for ligand conformational searching of AutoDock version 3.0.26 For each compound, the docking parameters were as follows: trials of 100 dockings, population size of 150, starting position, orientation, and conformation randomized using the default AutoDock randomization, translation step ranges of 1.5 Å, rotation step ranges of 35°, elitism of 1, mutation rate of 0.02, crossover rate of 0.8, local search rate of 0.06, and 10 million energy evaluations. The calculations were done on an Opteron Linux workstation. Final docked conformations were clustered by use of a tolerance of 2.5 Å root-mean-square deviation (rmsd). The top 17 compounds with the best simulated binding energies were selected for the in vitro inhibition assay. Figures of structural models were made with PyMol.39
Protein Purification and Activity Assay
The compounds corresponding to the top docking hits were obtained from Division of Cancer Treatment and Diagnosis at the National Cancer Institute. The immunogenic cAMP assay kit used for cAMP measurements was from Assay Designs (Ann Arbor, MI), and all other chemicals from Sigma (Saint Louis, MO). Forskolin and inhibitors were dissolved in DMSO (10 mM stock solutions), yielding a maximum DMSO concentration of 2% v/v for the tmAC experiments and 0.5% v/v for CyaC experiments; control reactions were done with the same solvent without compound. A GST fusion of human sAC7 and his-tagged rat sAC6 were expressed and purified as described previously. Dose–response curves were measured with both sAC enzymes, and the IC50 values obtained did not differ significantly between both enzymes. Data shown are representative results obtained with either human or rat sAC. The catalytic domain of CyaC from S. platensis was expressed and purified with an N-terminal his-tag as described10 and stored at −80 °C or supplemented with 50% (v/v) glycerol and stored at −20 °C for activity assays.
Mammalian sAC assays were done in 50 mM Tris-HCl (pH 7.5) with 2.5 mM ATP as substrate, 5 mM MgCl2, 2.5 mM CaCl2, and 40 mM bicarbonate. Assays were started by addition of purified mammalian sAC, incubated 30 min at room temperature, and stopped through 400-fold dilution into 0.1 M HCl. The cAMP produced was quantitated by cAMP ELISA. Activity assays with CyaC were done in 50 mM Tris-HCl (pH 7.5), 5 mM ATP, 10 mM MgCl2, and 5 mM CaCl2. Reactions were incubated 30 min at 37°C, diluted 500-fold into 0.1 M HCl, and tested with the cAMP ELISA. Activity assays with the various tmACs were performed on 50 μg of protein of whole cell lysates of HEK293T cells transfected with the indicated mammalian tmAC. Assays were performed in 50 mM Tris-HCl (pH 7.5), 1 mM ATP, 5 mM MgCl2, 80 μM CaCl2, and creatine kinase ATP regenerating system in the presence of 100 μM forskolin. Reactions were incubated 30 min at 37 °C, diluted 20-fold into 0.1 M HCl, and cAMP was measured with the cAMP ELISA.
Acknowledgments
Supply of chemicals from the Drug Synthesis and Chemistry Branch, Developmental Therapeutics Program, Division of Cancer Treatment and Diagnosis at the National Cancer Institute, is greatly acknowledged. This work was supported by funds from National Institutes of Health (L.R.L. and J.B.), Hirschl Weill-Caulier Trust (L.R.L.), the American Diabetes Association (L.R.L.), and grant STE1701/1 of Deutsche Forschungsgemeinschaft (C.S.).
Footnotes
Abbreviations: α,β-Me-ATP, α, β-methylene-ATP; AC, adenylyl cyclase; C, catalytic domain; CE, catechol estrogen; GST, glutathione S-alkyl transferase; MANT, methylanthranyloyl; sAC, soluble adenylyl cyclase; tmAC, transmembrane adenylyl cyclase.
References
- 1.Hanoune J, Defer N. Regulation and role of adenylyl cyclase isoforms. Annu Rev Pharmacol Toxicol. 2001;41:145–174. doi: 10.1146/annurev.pharmtox.41.1.145. [DOI] [PubMed] [Google Scholar]
- 2.Kamenetsky M, Middelhaufe S, Bank EM, Levin LR, Buck J, et al. Molecular details of cAMP generation in mammalian cells: a tale of two systems. J Mol Biol. 2006;362:623–639. doi: 10.1016/j.jmb.2006.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Linder JU, Schultz JE. The class III adenylyl cyclases: multipurpose signalling modules. Cell Signalling. 2003;15:1081–1089. doi: 10.1016/s0898-6568(03)00130-x. [DOI] [PubMed] [Google Scholar]
- 4.Buck J, Sinclair ML, Schapal L, Cann MJ, Levin LR. Cytosolic adenylyl cyclase defines a unique signaling molecule in mammals. Proc Natl Acad Sci US A. 1999;96:79–84. doi: 10.1073/pnas.96.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jaiswal BS, Conti M. Identification and functional analysis of splice variants of the germ cell soluble adenylyl cyclase. J Biol Chem. 2001;276:31698–31708. doi: 10.1074/jbc.M011698200. [DOI] [PubMed] [Google Scholar]
- 6.Chen Y, Cann MJ, Litvin TN, Iourgenko V, Sinclair ML, et al. Soluble adenylyl cyclase as an evolutionarily conserved bicarbonate sensor. Science. 2000;289:625–628. doi: 10.1126/science.289.5479.625. [DOI] [PubMed] [Google Scholar]
- 7.Litvin TN, Kamenetsky M, Zarifyan A, Buck J, Levin LR. Kinetic properties of “soluble” adenylyl cyclase. Synergism between calcium and bicarbonate. J Biol Chem. 2003;278:15922–15926. doi: 10.1074/jbc.M212475200. [DOI] [PubMed] [Google Scholar]
- 8.Zippin JH, Levin LR, Buck J. CO(2)/HCO(3)(−)-responsive soluble adenylyl cyclase as a putative metabolic sensor. Trends Endocrinol Metab. 2001;12:366–370. doi: 10.1016/s1043-2760(01)00454-4. [DOI] [PubMed] [Google Scholar]
- 9.Tesmer JJ, Sunahara RK, Gilman AG, Sprang SR. Crystal structure of the catalytic domains of adenylyl cyclase in a complex with Gsalpha. GTPgammaS Science. 1997;278:1907–1916. doi: 10.1126/science.278.5345.1907. [DOI] [PubMed] [Google Scholar]
- 10.Steegborn C, Litvin TN, Levin LR, Buck J, Wu H. Bicarbonate activation of adenylyl cyclase via promotion of catalytic active site closure and metal recruitment. Nat Struct Mol Biol. 2005;12:32–37. doi: 10.1038/nsmb880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tesmer JJ, Sunahara RK, Johnson RA, Gosselin G, Gilman AG, et al. Two-metal ion catalysis in adenylyl cyclase. Science. 1999;285:756–760. doi: 10.1126/science.285.5428.756. [DOI] [PubMed] [Google Scholar]
- 12.Menniti FS, Faraci WS, Schmidt CJ. Phosphodiesterases in the CNS: targets for drug development. Nat Rev Drug Discovery. 2006;5:660–670. doi: 10.1038/nrd2058. [DOI] [PubMed] [Google Scholar]
- 13.Ishikawa Y. Isoform-targeted regulation of cardiac adenylyl cyclase. J Cardiovasc Pharmacol. 2003;41(Suppl 1):S1–4. [PubMed] [Google Scholar]
- 14.Iwatsubo K, Okumura S, Ishikawa Y. Drug therapy aimed at adenylyl cyclase to regulate cyclic nucleotide signaling. Endocr Metab Immune Disord Drug Targets. 2006;6:239–247. doi: 10.2174/187153006778249994. [DOI] [PubMed] [Google Scholar]
- 15.Sinclair ML, Wang XY, Mattia M, Conti M, Buck J, et al. Specific expression of soluble adenylyl cyclase in male germ cells. Mol Reprod Dev. 2000;56:6–11. doi: 10.1002/(SICI)1098-2795(200005)56:1<6::AID-MRD2>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 16.Geng W, Wang Z, Zhang J, Reed BY, Pak CY, et al. Cloning and characterization of the human soluble adenylyl cyclase. Am J Physiol Cell Physiol. 2005;288:C1305–1316. doi: 10.1152/ajpcell.00584.2004. [DOI] [PubMed] [Google Scholar]
- 17.Tesmer JJ, Dessauer CW, Sunahara RK, Murray LD, Johnson RA, et al. Molecular basis for P-site inhibition of adenylyl cyclase. Biochemistry. 2000;39:14464–14471. doi: 10.1021/bi0015562. [DOI] [PubMed] [Google Scholar]
- 18.Dessauer CW, Tesmer JJ, Sprang SR, Gilman AG. The interactions of adenylate cyclases with P-site inhibitors. Trends Pharmacol Sci. 1999;20:205–210. doi: 10.1016/s0165-6147(99)01310-3. [DOI] [PubMed] [Google Scholar]
- 19.Gille A, Lushington GH, Mou TC, Doughty MB, Johnson RA, et al. Differential inhibition of adenylyl cyclase isoforms and soluble guanylyl cyclase by purine and pyrimidine nucleotides. J Biol Chem. 2004;279:19955–19969. doi: 10.1074/jbc.M312560200. [DOI] [PubMed] [Google Scholar]
- 20.Levy DE, Bao M, Cherbavaz DB, Tomlinson JE, Sedlock DM, et al. Metal coordination-based inhibitors of adenylyl cyclase: novel potent P-site antagonists. J Med Chem. 2003;46:2177–2186. doi: 10.1021/jm0205604. [DOI] [PubMed] [Google Scholar]
- 21.Iwatsubo K, Minamisawa S, Tsunematsu T, Nakagome M, Toya Y, et al. Direct inhibition of type 5 adenylyl cyclase prevents myocardial apoptosis without functional deterioration. J Biol Chem. 2004;279:40938–40945. doi: 10.1074/jbc.M314238200. [DOI] [PubMed] [Google Scholar]
- 22.Shoshani I, Laux WH, Perigaud C, Gosselin G, Johnson RA. Inhibition of adenylyl cyclase by acyclic nucleoside phosphonate antiviral agents. J Biol Chem. 1999;274:34742–34744. doi: 10.1074/jbc.274.49.34742. [DOI] [PubMed] [Google Scholar]
- 23.Hess KC, Jones BH, Marquez B, Chen Y, Ord TS, et al. The “soluble” adenylyl cyclase in sperm mediates multiple signaling events required for fertilization. Dev Cell. 2005;9:249–259. doi: 10.1016/j.devcel.2005.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mou TC, Gille A, Fancy DA, Seifert R, Sprang SR. Structural basis for the inhibition of mammalian membrane adenylyl cyclase by 2′(3′)-O-(N-Methylanthraniloyl)-guanosine 5′-triphosphate. J Biol Chem. 2005;280:7253–7261. doi: 10.1074/jbc.M409076200. [DOI] [PubMed] [Google Scholar]
- 25.Steegborn C, Litvin TN, Hess KC, Capper AB, Taussig R, et al. A novel mechanism for adenylyl cyclase inhibition from the crystal structure of its complex with catechol estrogen. J Biol Chem. 2005;280:31754–31759. doi: 10.1074/jbc.M507144200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, et al. Automated Docking Using a Lamarckian Genetic Algorithm and and Empirical Binding Free Energy Function. J Comput Chem. 1998;19:1639–1662. [Google Scholar]
- 27.Li C, Xu L, Wolan DW, Wilson IA, Olson AJ. Virtual screening of human 5-aminoimidazole-4-carboxamide ribonucleotide transformylase against the NCI diversity set by use of AutoDock to identify novel nonfolate inhibitors. J Med Chem. 2004;47:6681–6690. doi: 10.1021/jm049504o. [DOI] [PubMed] [Google Scholar]
- 28.McInnes C. Virtual screening strategies in drug discovery. Curr Opin Chem Biol. 2007;11:494–502. doi: 10.1016/j.cbpa.2007.08.033. [DOI] [PubMed] [Google Scholar]
- 29.Pastor-Soler N, Beaulieu V, Litvin TN, Da Silva N, Chen Y, et al. Bicarbonate-regulated adenylyl cyclase (sAC) is a sensor that regulates pH-dependent V-ATPase recycling. J Biol Chem. 2003;278:49523–49529. doi: 10.1074/jbc.M309543200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sunahara RK, Taussig R. Isoforms of mammalian adenylyl cyclase: multiplicities of signaling. Mol Interv. 2002;2:168–184. doi: 10.1124/mi.2.3.168. [DOI] [PubMed] [Google Scholar]
- 31.Braun T. Inhibition of the soluble form of testis adenylate cyclase by catechol estrogens and other catechols. Proc Soc Exp Biol Med. 1990;194:58–63. doi: 10.3181/00379727-194-43055. [DOI] [PubMed] [Google Scholar]
- 32.Esnouf R, Ren J, Ross C, Jones Y, Stammers D, et al. Mechanism of inhibition of HIV-1 reverse transcriptase by non-nucleoside inhibitors. Nat Struct Biol. 1995;2:303–308. doi: 10.1038/nsb0495-303. [DOI] [PubMed] [Google Scholar]
- 33.Jaleel M, Shenoy AR, Visweswariah SS. Tyrphostins are inhibitors of guanylyl and adenylyl cyclases. Biochemistry. 2004;43:8247–8255. doi: 10.1021/bi036234n. [DOI] [PubMed] [Google Scholar]
- 34.Ebina T, Toya Y, Oka N, Schwencke C, Kawabe J, et al. Isoform-specific regulation of adenylyl cyclase by oxidized catecholamines. J Mol Cell Cardiol. 1997;29:1247–1254. doi: 10.1006/jmcc.1996.0362. [DOI] [PubMed] [Google Scholar]
- 35.Remmers AE, Posner R, Neubig RR. Fluorescent guanine nucleotide analogs and G protein activation. J Biol Chem. 1994;269:13771–13778. [PubMed] [Google Scholar]
- 36.Hutchinson JP, Eccleston JF. Mechanism of nucleotide release from Rho by the GDP dissociation stimulator protein. Biochemistry. 2000;39:11348–11359. doi: 10.1021/bi0007573. [DOI] [PubMed] [Google Scholar]
- 37.Pisareva VP, Pisarev AV, Hellen CU, Rodnina MV, Pestova TV. Kinetic analysis of interaction of eukaryotic release factor 3 with guanine nucleotides. J Biol Chem. 2006;281:40224–40235. doi: 10.1074/jbc.M607461200. [DOI] [PubMed] [Google Scholar]
- 38.Pinto C, Papa D, Hubner M, Mou TC, Lushington GH, et al. Activation and inhibition of adenylyl cyclase isoforms by forskolin analogs. J Pharmacol Exp Ther. 2008;325:27–36. doi: 10.1124/jpet.107.131904. [DOI] [PubMed] [Google Scholar]
- 39.DeLano W. The PyMol User’s Manual. DeLano Scientific; San Carlos, CA: 2002. p. JM800481Q. [Google Scholar]