

Abstract
Blockade of growth hormone (GH), decreased insulin-like growth factor-1 (IGF1) action and increased insulin sensitivity are associated with life extension and an apparent slowing of the aging process. We examined expression of genes involved in insulin action, IR, IRS1, IRS2, IGF1, IGF1R, GLUT4, PPARs and RXRs in the hearts of normal and GHR−/− (KO) mice fed ad libitum or subjected to 30% caloric restriction (CR). CR increased the cardiac expression of IR, IRS1, IGF1, IGF1R and GLUT4 in normal mice and IRS1, GLUT4, PPARα and PPARβ/δ in GHR-KO animals. Expression of IR, IRS1, IRS2, IGF1, GLUT4, PPARγ and PPARα did not differ between GHR-KO and normal mice. These unexpected results suggest that CR may lead to major modifications of insulin action in the heart, but high insulin sensitivity of GHR-KO mice is not associated with alterations in the levels of most of the examined molecules related to intracellular insulin signaling.
Keywords: Caloric restriction, aging, GHR-KO, insulin, fatty acid
INTRODUCTION
Growth hormone (GH) resistance or insensitivity with low circulating levels of insulin-like growth factor-1 (IGF1), insulin and glucose, all of which imply increased insulin sensitivity, are among the key characteristics of long-lived GH receptor/binding protein knockout (GHR-KO) mice (1,2). These knockout mice live approximately 40% longer than their normal siblings (3). Caloric restriction (CR) is known to increase lifespan and delay aging (4). The effects of CR are very similar to key phenotypic characteristics of GHR-KO mice. The similarities include reductions in body weight, plasma levels of insulin, IGF1, glucose and thyroid hormone. Results obtained in animals subjected to CR and in various long-lived mutants suggest that perturbations in the insulin signaling pathway may play a key role in extended longevity (5). We have recently described the effects of long-term 30% CR on the expression of genes related to insulin and IGF1 signaling in the liver and skeletal muscle of these animals (6-8). Effects of insulin on protein metabolism in skeletal muscle and the liver are believed to be important in regulating the substrate availability for the heart (9). Insulin resistance is associated with increased risk of chronic heart failure (10). Insulin action through AKT signaling is also responsible for physiological heart growth during postnatal development (11). The study in rat hearts showed that CR restores the cardio-protective effects of preconditioning by increasing tolerance to ischemia (12). In view of the strong dependence of heart functions on insulin actions, it was of interest to include this organ in our ongoing studies of the effects of CR in normal and in long-lived mutant mice.
In the present study, in order to analyze relationships between GH resistance, CR and insulin signaling in the heart, we have examined the effects of GHR-KO and CR on the expression of insulin receptor (IR), insulin receptor substrate 1(IRS1), insulin receptor substrate 2 (IRS2), IGF1, glucose transporter 4 (GLUT4), peroxisome proliferator-activated receptor γ (PPARγ), PPARα, PPARδ/β (also known as PPARδ or PPARβ), retinoid X receptor γ (RXRγ), RXRα and RXRβ/δ in cardiac muscle.
Insulin action depends on the presence of the IR on the cell surface. Insulin induced intracellular signaling starts by insulin binding to IR and results in autophosphorylation of IR. This action leads to phosphorylation of IRS1 and IRS2, which can bind p85 regulatory subunit of PI 3-kinase and cause activation of this enzyme (13,14). GLUT4 is a regulator of glucose uptake in muscle and has a role in the maintenance of whole body glucose homeostasis (15, 16). As a result of activation of the insulin signaling pathway GLUT4 is translocated to the cell surface.
Genes related to IGF1 signaling were also of interest because prolonged longevity of different types of mutant mice, strongly suggests that IGF1 may play a key role in regulation and longevity (17-21).
In the present study we have also analyzed the expression of the PPARs family and the RXRs genes. PPARs are members of the nuclear receptor superfamily that are ligand-dependent transcription factors. These factors are known to regulate the expression of target genes by binding to specific peroxisome proliferator response elements (PPREs) in enhancer sites of regulated genes. PPARs require RXRs to form heterodimers, which allow binding to their specific PPREs (22). There are three known isoforms in the PPAR superfamily, each encoded by a separate genes: PPARγ, PPARα and PPARδ/β. The most explored gene of this superfamily is PPARγ. PPARγ is the target receptor for thiazolidinediones (TZDs), drugs used in type 2 diabetic patients as insulin sensitizers (23-26). PPARα is highly expressed in the liver, skeletal muscle, heart and kidney, where it regulates a wide variety of target genes involved in cellular lipid catabolism and fatty acids β and ω-oxidation and containing PPREs in their promoter regions (27). In the heart, PPARα activation results in the accumulation of myocardial lipids that leads to other features of diabetic cardiomyopathy (28). However, PPARα deficient mice have increased levels of total and HDL cholesterol (29).
The function of PPARβ/δ is still unclear, but it is known to play an important role in epidermal maturation and skin wound healing (30, 31) and has a possible function in lipid metabolism (32). Retinoid X receptors have a potential to heterodimerize with many nuclear receptors. PPARs act by forming heterodimers with RXRs, which allows binding to their PPREs. RXR isoforms are encoded by three separate genes: RXRα, RXRβ and RXRγ (33).
In this study, expression of genes involved in the initial steps of insulin pathway, PPARs and RXR families were analyzed in the heart muscle of normal and GHR-KO mice fed ad libitum (AL) or subjected to long-term CR. The data were also analyzed based on a comparison of these findings to the expression levels of the same genes in the hearts of young, 3-month old GHR-KO mice, and in GH over-expressing transgenic mice. The results indicate that CR influences genes from the insulin pathway in the heart and potentially retards cardiac aging in these animals. The data also suggest that the high insulin sensitivity of GHR-KO mice is not related to alterations of this molecular pathway in the heart muscle.
MATERIALS AND METHODS
Animals
21-month-old calorically restricted mice
Normal and GHR-KO male mice, eight in each group, were subjected to 30% CR or fed AL starting at 8 weeks of age. The detailed protocol of the experiment was described previously (34, 35). Because differences between genotypes and/or after CR treatment tend to be more pronounced in older animals, we continued caloric restriction for the period of 19 months till the animals reached the age of 21 months. The animals were anesthetized using isoflurane; blood was taken by cardiac puncture and the mice were killed by decapitation. Hearts were collected and rapidly frozen on dry ice.
Young GHR-KO mice
The hearts from additional eight GHR-KO and eight normal male mice aged 3 months were collected to examine age-dependent changes.
hGH transgenic mice
The hearts from seven male Mt-hGH transgenic (GH-Tg) mice and eight of their normal male siblings were collected to examine the effects of over-expressed GH on analyzed genes. These animals were produced in our breeding colony derived from transgenic breeders kindly provided by Dr. Thomas Wagner and Jeung Yun (36-38).
Extraction of mRNA and cDNA synthesis
After grinding the heart in liquid nitrogen, 1xPBS was added and total RNA was extracted from a 250 μl aliquot using phenol/chloroform procedure of Chomczynski and Sacchi (39). Using electrophoresis, the quantity and quality of total RNA was analyzed on agarose gel. Afterwards, cDNA was synthesized from 2 μg of total RNA using iScript™ cDNA Synthesis Kit (BIO-RAD) according to the manufacturer's protocol.
Real-Time PCR
The mRNA relative expression was analyzed by real-time PCR (RT-PCR) using the “Smart Cycler” instrument (Cepheid, Sunnyvale, CA) with iQ™ SYBR Green Supermix (BIO-RAD) (6, 7). The three steps of the RT-PCR included denaturation at 94°C for 2 min, annealing at 62°C for 30 sec with fluorescence reading, and extension at 72°C for 30 sec. All primers used in the study are listed in table 1. To evaluate the potential of nonspecific products, a melting curve was done for each reaction. All samples were analyzed in a single reaction for each gene to avoid false differences caused by different efficiency in separate PCR reactions.
Table 1.
Sequence of the primers used for real-time PCR. There is a gene bank sequence number by each forward primer.
| IR- forward (NM_010568) | 5′ - ATGAGGCCAACCTTCCTGGAA |
| IR- backward | 5′ - ACGGGACATTCTCCATGTCT |
| IRS1- forward (L24563) | 5′ - AGCCCAAAAGCCCAGGAGAATA |
| IRS1- backward | 5′ - TTCCGAGCCAGTCTCTTCTCTA |
| IRS2- forward (AF090738) | 5′ - AGTAAACGGAGGTGGCTACA |
| IRS2- backward | 5′ - AAGCTGCTGAGAAGTCAGGT |
| GLUT4- forward (AB008453) | 5′ - ATTGGCATTCTGGTTGCCCA |
| GLUT4-backward | 5′ - GGTTCCGGATGATGTAGAGGTA |
| IGF1- forward (NM_010512) | 5′ - CTGAGCTGGTGGATGCTCTT |
| IGF1- backward | 5′ - CACTCATCCACAATGCCTGT |
| IGF1-R – forward (AF056187) | 5′ - ATTGCCTCGGAGTTGGAGAA |
| IGF1-R - backward | 5′ - AGCTGCTGCAAGTTCTGGTT |
| PPARγ- forward (NM_011146) | 5′ - CAAAGTAGAACCTGCATCTCC |
| PPARγ- backward | 5′ - CCTTCACAAGCATGAACTCC |
| PPARα- forward (NM_011144) | 5′ - CGAAGCCTACCTGAAGAACTT |
| PPARα- backward | 5′ - ATCTTGGCCACAAGCGTCTT |
| PPARβ/δ- forward (NM_011145) | 5′ - CTCGAGTATGAGAAGTGCGA |
| PPARβ/δ- backward | 5′ - CATCCGTCCAAAGCGGATAG |
| RXRγ- forward (M84819) | 5′ - TACCAGAAGTGCCTGGTCAT |
| RXRγ- backward | 5′ - TTCCACAGCAAGTTCGGCTT |
| RXRα- forward (M84817) | 5′ - GACATACGTGGAGGCAAACA |
| RXRα- backward | 5′ - AGGATGACCTGGTCGTCTAG |
| RXRβ- forward (X72017) | 5′ - CCTCCTCTCACGACTATTAG |
| RXRβ- backward | 5′ - GGACAGGAGAGACAAGAAGA |
| B2M- forward (NM_009735) | 5′ - AAGTATACTCACGCCACCCA |
| B2M- backward | 5′ - AAGACCAGTCCTTGCTGAAG |
Two different housekeeping genes were tested for this study, 18S and β-2-microglobulin (B2M). The results indicated that B2M is more appropriate to be used in this study, because its expression was very similar in GHR-KO vs. normal and also in CR vs. AL mice. Additionally, we analyzed the data after normalizing with B2M and 18S and also used raw data of the genes of interest with no normalizing to see if the amount of the matrices used in each sample was similar. This test indicated that raw data were almost identical to data after normalizing with B2M, whereas after normalizing with 18S the results were markedly different. Relative expression from RT-PCR was calculated from the equation 2A-B/2C-D (where A = Cycle Threshold [Ct] number for the gene of interest in the first control sample; B = Ct number for the gene of interest in the analyzed sample; C = Ct number for the housekeeping gene in the first control sample; D = Ct number for housekeeping gene in the analyzed sample). The first control was expressed as 1.00 by this equation, and all other samples were calculated in relation to this value. Afterwards, the results in the control group (N-AL) were averaged, and all other outputs were divided by the mean value of the relative expression in the control group to yield the fold change of the genes of interest expression compared to the control group.
Western blots
After crushing the hearts in liquid nitrogen, adding equal volume of PBS and removing 250 μl for RNA extraction, the remaining homogenate was used for protein extraction (35). Western blotting was performed using PPARγ (Cell Signalling, Beverly, MA), PPARα and PPARδ antibodies (Santa Cruz Biotechnologies, Santa Cruz, CA) and protocol described earlier (35). After stripping the membrane, a monoclonal β-actin antibody (Sigma, St. Louis, MO) was used as a control for protein loading.
Statistical Analysis
Data are expressed as mean ±SEM. To evaluate the effects of the phenotype and diet, two-way ANOVA was used followed by Fisher's PLSD. A t-test was used to evaluate the effects of diet within the phenotypes and phenotypes within diets. P<0.05 was considered significant.
RESULTS
The levels of IR, IRS1 and IRS2 mRNA in the hearts did not differ between N-AL and KO-AL animals (Fig. 1 A, B, C). Caloric restriction increased IR and IRS1 RNA levels in normal animals (P< .0193 and P< .005, respectively) and IRS1 RNA levels in KO in comparison to the corresponding AL controls (P< .02). The level of IR RNA appeared to be higher in KO mice after CR but the difference did not reach statistical significance. IRS2 expression was not affected by phenotype or by CR in either group. Similarly to the findings in 21-month-old animals, IR RNA level also did not differ between young KO and N animals. However, IRS1 and IRS2 RNA levels were increased in young KO mice in comparison to their normal siblings (P< .01 and P< .046, respectively) (Fig. 2 A, B, C). In contrast to the findings in KO animals, IR and IRS1 RNA levels in short-lived hGH transgenic mice were significantly decreased in comparison to their normal siblings (P< .0129 and P< .02, respectively) (Fig. 2 D, E). The IRS2 RNA level also appeared to be decreased in hGH mice in comparison to their normal siblings, but it did not reach a significant level (P< .514) (Fig. 2 F).
Figure 1.

Expression of genes related to insulin signaling in cardiac muscle tissue of normal (N) and growth hormone receptor/binding protein knockout (GHR-KO) mice fed ad libitum (AL) or subjected to 30% caloric restriction (CR). The data from real-time PCR were normalized by housekeeping gene β-2-microglobulin (B2M) and expressed as the relative expression. Means ± SE. a,b,c – values that do not share the same letter in the superscript are statistically significant (P<0.05)
(A) IR, (B) IRS1, (C) IRS2, (D) IGF1, (E) IGF1R
Figure 2.

Expression of genes related to insulin signaling in cardiac muscle tissue of normal (N) and growth hormone receptor/binding protein knockout (GHR-KO) mice fed ad libitum at the age of three months (left column), and of normal (N) and human growth hormone (hGH) transgenic mice fed ad libitum at the age of 9 months (right column). The data from real-time PCR were normalized by housekeeping gene β-2-microglobulin (B2M) and expressed as the relative expression. Means ± SE. a,b – values that do not share the same letter in the superscript are statistically significant (P<0.05)
(A) IR - KO, (B) IRS1 - KO, (C) IRS2- KO, (D) IR-hGH, (E) IRS1-hGH, (F) IRS2- hGH
Interestingly, cardiac IGF1 RNA levels did not differ between 21-month-old N-AL and KO-AL mice, but CR increased the level of this molecule in normal animals (P< .0117) (Fig. 1 D) with no effect in knockouts. However, the IGF1 RNA level was decreased in the heart tissue of young KO mice (P< .01), while it was not changed in hGH transgenic mice in comparison to their controls (3 A, B). IGF1R RNA was increased in KO-AL mice in comparison to N-AL controls (P< .0374) (Fig. 1 E). IGF1R RNA was also increased in N-CR animals as compared to N-AL mice (P< .0427). Young KO mice were also characterized by elevated level of IGF1R (P<0.0024), while in hGH transgenic mice IGF1R was decreased in comparison to their normal controls (P< .01) (Fig. 3 C, D).
Figure 3.

Expression of IGF1 and IGF1R mRNA in cardiac muscle of normal (N) and GHR/BP knockout (KO) mice fed ad libitum at the age of 3 months old, and of normal and human growth hormone transgenic (hGH) mice at the age of 9 months. The data from real-time PCR were normalized by the housekeeping gene β-2-microglobulin (B2M). Means ± SEM. a,b – values that do not share the same letter in the superscript are significantly different (P<0.05)
(A) IGF1 - KO (B) IGF1 – hGH, (C) IGF1R- KO, (D) IGF1R - hGH
Although cardiac GLUT4 RNA levels did not differ between N-AL and KO-AL mice, the GLUT4 protein level in the heart was decreased in the KO-AL as compared to N-AL mice (P< .0012) (Fig. 4 A, B). Caloric restriction increased GLUT 4 RNA level in both normal and KO mice (P< .046 and P< .034, respectively), but increased GLUT4 protein levels only in KO-CR animals in comparison to KO-AL mice (P< .0012). GLUT4 RNA levels did not differ between normal and KO hearts at the age of 3 months, and were decreased in hGH mice in comparison to normal siblings (P< .0098) (Fig. 4 C, D).
Figure 4.

Expression of GLUT4 gene in cardiac muscle of normal (N) and GHR/BP knockout (KO) mice fed ad libitum (AL) or subjected to 30% caloric restriction (CR). (A) GLUT4 RNA expression, (B) GLUT4 protein level
Expression of GLUT4 mRNA in cardiac muscle of normal (N) and GHR/BP knockout (KO) mice fed ad libitum at the age of 3 months, and of normal and human growth hormone transgenic (hGH) mice at the age of 9 months. (C) GLUT4 - KO, (D) GLUT4 – hGH. The data from real-time PCR were normalized by the housekeeping gene β-2-microglobulin (B2M). Equal loading of protein for Western blots was verified using β-actin. Means ± SEM. a,b,c – values that do not share the same letter in the superscript are significantly different (P<0.05)
PPARγ RNA levels appeared to be elevated in KO-AL in comparison to N-AL and also in both normal and KO animals after CR, but neither the effect of phenotype alone, nor the effects of CR alone reached statistical significance. A combination of GHR disruption and CR lead to a significant increase of PPARγ mRNA levels (KO-CR vs. N-AL) (Fig 5 A). At the PPARγ protein level, there was also no significant difference between the phenotypes, but numerical differences suggested a trend opposite to that detected at the RNA level, namely a reduction rather than an increase in KO-AL in comparison to N-AL. PPARγ protein levels were significantly increased in KO-CR mice in comparison to KO-AL animals (P<0.028) (Fig 5 B). Furthermore, caloric restriction increased PPARα expression at both RNA and protein levels in KO-CR in comparison to KO-AL mice (P<0.0053 and P<0.018, respectively) (Fig. 5 C, D). The levels of PPARα protein were decreased in the hearts of KO-AL mice in comparison to normal animals (P< 0.0119) with no corresponding changes in PPARα RNA levels. The effects of phenotype on cardiac PPARβ/δ expression were, in general, opposite at the RNA and protein levels. The PPARβ/δ RNA was increased in KO-AL mice in comparison to their normal siblings (P<0.002), but the protein product level was decreased in KO-AL in comparison to N-AL mice (P<0.0023) (Fig. 5 E, F). Caloric restriction increased the level of PPARβ/δ protein only in KO animals (P< 0.0147), while a corresponding trend in the levels of PPARβ/δ RNA was not statistically significant. In young animals PPARγ RNA levels did not differ between KO and normal mice (Fig. 6A). PPARα and PPARβ/δ RNA levels were significantly increased in the hearts of young KO mice as compared to normal siblings (P<0.0339 and P<0.048, respectively) (Fig. 6 B, C). For both PPARα and PPARβ/δ RNA, short living hGH mice showed alterations opposite to those found in KO mice, namely down regulation rather than increase in comparison to their normal siblings (P<0.0127 and P<0.023, respectively) (Fig. 6 E, F). The levels of PPARγ RNA were also reduced in hGH mice (P< 0.0023) (Fig. 6 D).
Figure 5.

Expression of PPARs superfamily genes in cardiac muscle tissue of normal (N) and GHR/BP knockout (KO) mice fed ad libitum (AL) or subjected to 30% caloric restriction (CR). The data from real time PCR were normalized by the housekeeping gene β-2-microglobulin (B2M). Equal loading of protein for Western blots was verified using β-actin. Means ± SEM. a,b,c – values that do not share the same letter in the superscript are significantly different (P<0.05)
(A) PPARγ RNA expression, (B) PPARγ protein level, (C) PPARα RNA expression, (D) PPARα protein level, (E) PPARβ/δ RNA expression, (F) PPARβ/δ protein level
Figure 6.

Expression of PPARs superfamily genes in cardiac muscle tissue of normal (N) and growth hormone receptor/binding protein knockout (GHR-KO) mice fed ad libitum at the age of three months (left column), and of normal (N) and human growth hormone (hGH) transgenic mice fed ad libitum at the age of 9 months (right column). The data from real-time PCR were normalized by housekeeping gene β-2-microglobulin (B2M) and expressed as the relative expression. Means ± SE. a,b – values that do not share the same letter in the superscript are statistically significant (P<0.05)
(A) PPARγ - KO, (B) PPARα - KO, (C) PPARβ/δ- KO, (D) PPARγ-hGH, (E) PPARα- hGH, (F) PPARβ/δ- hGH
The levels of RXRγ, RXRα and RXRβ/δ mRNA were significantly increased in KO-AL mice in comparison to N-AL animals (P< .0273, P< .0113, and P< .0314, respectively) (Fig. 7 A, B, C). However, the only significant effect of CR on RXR expression was a decrease in RXRγ mRNA levels in KO-CR as compared to KO-AL mice (Fig. 7). RXRα and RXRβ/δ RNA levels in young KO mice were also increased in comparison to their normal controls (P< .0097 and P< .0375, respectively) with no differences in RXRγ RNA levels (Fig. 8 A, B, C). The levels of RXRγ, RXRα and RXRβ/δ RNA were significantly decreased in hGH transgenic mice in comparison to their normal siblings (P< .0177, P< .021 and P< .024, respectively) (Fig. 8 D, E, F).
Figure 7.
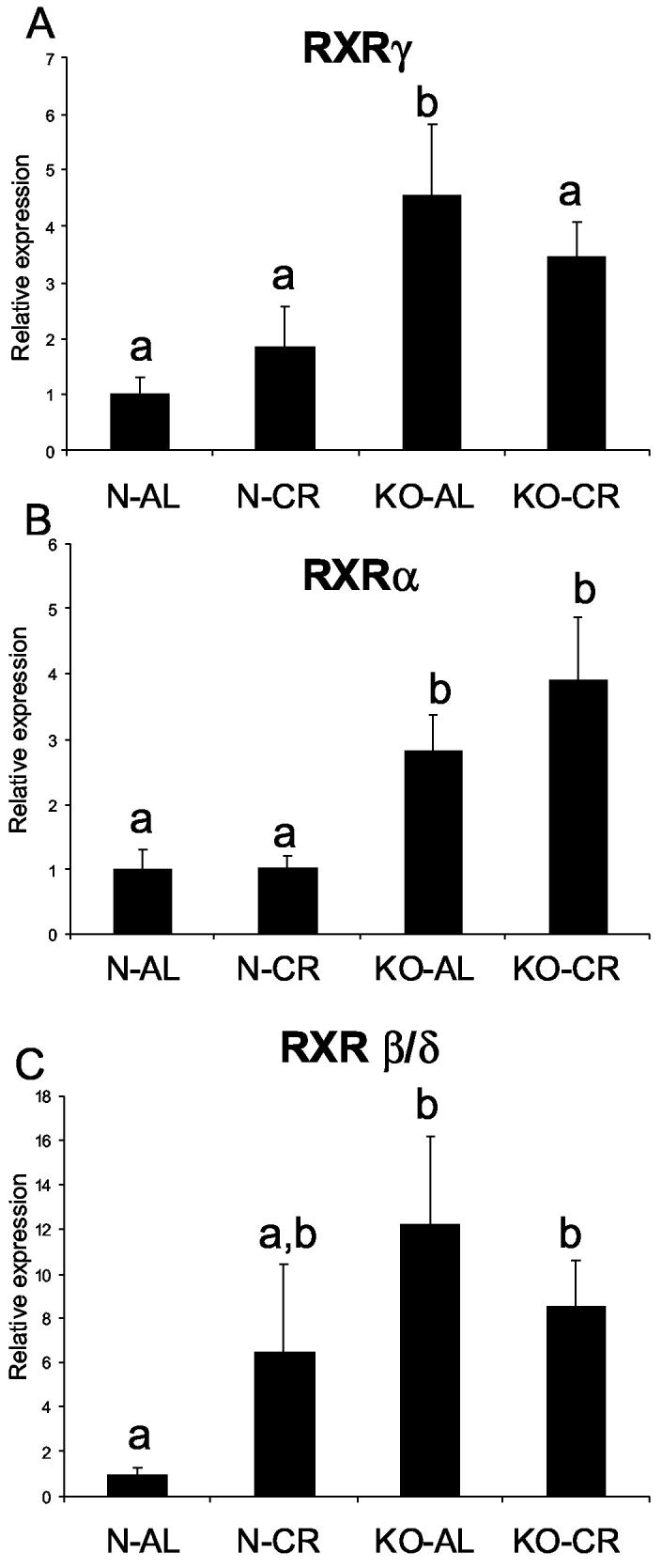
Expression of RXR genes in cardiac muscle tissue of normal (N) and GHR/BP knockout (KO) mice fed ad libitum (AL) or subjected to 30% caloric restriction (CR). The data from real time PCR were normalized by housekeeping gene β-2-microglobulin (B2M). Means ± SEM. a,b,c – values that do not share the same letter in the superscript are significantly different (P<0.05). (A) RXRγ, (B) RXRα, (C) RXRβ/δ
Figure 8.

Expression of RXR genes in cardiac muscle tissue of normal (N) and growth hormone receptor/binding protein knockout (GHR-KO) mice fed ad libitum at the age of three months (left column), and of normal (N) and human growth hormone (hGH) transgenic mice fed ad libitum at the age of 9 months (right column). The data from real-time PCR were normalized by housekeeping gene, β-2-microglobulin (B2M) and expressed as the relative expression. Means ± SE. a,b – values that do not share the same letter in the superscript are statistically significant (P<0.05)
(A) RXRγ - KO, (B) RXRα - KO, (C) RXRβ/δ- KO, (D) RXRγ-hGH, (E) RXRα-hGH, (F) RXRβ/δ- hGH
DISCUSSION
In the present study, long-lived GHR/GHBP knockout (KO) mice have been subjected to long-term 30% CR. We have previously reported that this regimen of CR produces the expected reduction in body weight in both KO and normal mice (35). In the same study, CR down regulated circulating IGF1 levels in normal animals, while the extremely low levels of circulating IGF1 in both KO-AL and KO-CR animals were below the detection limits of the employed ELISA kit used in these studies (35). Moreover, CR significantly decreased blood glucose in both phenotypes, with no significant difference between KO and N mice fed ad libitum (N-AL 189.4±9.6, N-CR 139.7±14.1, KO-AL 155.0±14.1, and KO-CR 74.8±11.5, mg/dl) (35). It was also shown that the insulin level was significantly decreased in normal and KO animals after CR, while KO-AL animals had the expected reduction of insulin as compared to N-AL mice. (N-AL 4.2±0.7, N-CR 1.4±0.2, KO-AL 0.9±0.1, and KO-CR 0.4±0.1, ng/ml) (35). The genotype (KO vs. N) and CR-induced increase in insulin sensitivity suggested by these findings was supported by the results of homeostasis model analysis (HOMA) (6). Studies in worms, flies and mice (including GHR-KO, Ames dwarfs and Snell dwarfs), as well as widely examined effects of CR in various animal species, strongly indicate insulin signaling as an important pathway in the control of aging (5).
Insulin Receptor
The first gene in the insulin signaling pathway is IR. In the heart muscle tissue, expression of this gene was not affected by the phenotype at either of the ages examined, with no differences between KO-AL and N-AL mice. This resembled our findings in skeletal muscle from the same animals (7). However, in the short-lived animals overexpressing the hGH, IR RNA levels were decreased. This could suggest that in old and young KO animals, IRs in either the heart or the skeletal muscle are unlikely to contribute to enhanced whole-body insulin sensitivity, while up regulation of this molecule in the liver of KO animals (7) can be related to improved whole-body insulin action. However, increased levels of IR in normal animals after CR and a similar trend in KO-CR mice, together with reduced expression of this gene in aged hGH mice, suggest that CR can protect insulin action in the hearts from the effects of aging. We base this suggestion on the fact that RT-PCR used in this study measures relative expression. Thus the findings would seem to imply that CR does not cause an increase in IR RNA level in the hearts of old KO and normal mice, but in both N-AL and KO-AL the IR RNA levels declines with age, resembling the findings in aged hGH transgenic mice. Thus it is tempting to speculate that CR maintains the content of IR at a level similar to that measured in the young mice and perhaps delays aging of the heart in these animals.
Insulin Receptor Substrate 1 and Insulin Receptor Substrate 2
The expression of IRS1 and IRS2 was apparently not affected by the phenotype in old KO animals when compared to normal controls thus resembling the findings on IR expression. However, in the hearts of young KO mice, IRS1 and IRS2 RNA levels were increased in comparison to their normal siblings. This suggests that IR expression is reduced with age in KO-AL to the level measured in N-AL mice, and it is likely the level of these messages continues to decline with age, as observed in the aged hGH mice. Significantly increased levels of IRS1 RNA in N-CR and KO-CR in comparison to their AL siblings can suggest that CR protects the hearts from aging. This suggestion is indirectly supported by the observations in young KO animals and in aged hGH mice.
The effects of CR and phenotype on IR, IRS1 and IRS2 RNA levels in cardiac muscle are opposite to those detected in the skeletal muscle, which was characterized by down regulation of IR, IRS1 and IRS2 after CR in both normal and knockout animals (7). They also differ from the findings in the liver, where no changes were observed after CR (7). We can conclude that the effects of the phenotype and CR on the expression of these insulin-related genes are organ specific and that effects of CR in different organs can be opposite.
Insulin-like Growth Factor-1 and Insulin-like Growth Factor-1-Receptor
The lack of difference in the cardiac expression of IGF1 RNA between long-lived KO and normal animals was surprising in view of the extremely low levels of circulating IGF1 (non-detectable by Elisa kit) and IGF1 mRNA in the liver of KO animals in comparison to their normal siblings (7). Down regulation of IGF1 biosynthesis in these animals related to the disrupted action of GH clearly does not apply to all organs. A previous study which used an independently derived line of GHR (−/−) mutant mice showed that hepatic and kidney IGF1 RNA levels were decreased without changes in IGF1 expression in other organs including the heart (40). The levels of IGF1 RNA in the hippocampus of Ames dwarf mice do not differ from the levels measured in normal siblings, although Ames dwarf mice are characterized by suppressed hepatic IGF1 expression and circulating IGF1 levels (41). However, the cardiac expression of IGF1 in young KO mice was decreased in comparison to their normal siblings, which is closer to the findings in the skeletal muscle (7). Interestingly, IGF1R RNA levels were elevated in KO-AL mice in comparison to N-AL and also in young, 3-month-old KO mice, while hGH transgenic mice had reduced level of IGF1R message. Because IGF1R can activate IRS1 and IRS2 just like IR, this can explain higher levels of these substrates in young KO mice in which IR RNA levels were not elevated (42). Study of IGF1R transgenic mice showed that over expressed IGF1R can cause enlargement of the heart (42). In GHR-KO mice examined in the present study, elevated level of IGF1R in the cardiac muscle may compensate for the lack of GH action and thus allow normal heart development. In hGH transgenic animals, the decreased IGF1R was likely due to high levels of GH. Moreover, the expression of IRS1 and IRS2 in aged hGH transgenic mice was down regulated, probably because of low level of IR and IGF1R.
Glucose Transporter 4
Increased RNA expression of the GLUT4 gene in normal and KO animals after CR and the corresponding changes of GLUT4 protein in KO-CR animals suggest improved glucose transport in cardiac muscle in animals that had limited availability of food. This effect is the opposite of what we observed in the skeletal muscle of CR animals (7). Results obtained in young KO and normal mice indicate that GLUT4 RNA expression does not change with age. However, GH transgenic animals had significantly reduced levels of GLUT4 RNA, which indicates that an excess of GH can decrease glucose transport in cardiac muscle.
Peroxisome Proliferator-Activated Receptor γ
In the present study, the only significant effect of CR on PPARγ expression was at the protein level in KO mice. PPARγ RNA expression was not altered in young KO animals. Thus it is difficult to arrive at any conclusions concerning the possible role of cardiac PPARγ in mediating the responses to GH resistance or CR. However, these data clearly indicate that cardiac muscle responds differently than skeletal muscle with respect to this gene. In the skeletal muscle, both CR and GHR knockout caused down regulation of PPARγ gene expression (6). Concerning the possible cardio-protective effects of PPARγ agonists, decreased level of this nuclear receptor in aged hGH transgenic animals together with a trend for PPARγ RNA to be higher in old N-CR and KO-CR mice in comparison to their AL controls may imply anti-aging protection in the hearts of CR animals.
Peroxisome Proliferator-activated Receptor α
Chronic enhancement of PPARα action in the heart was believed to cause myocardial lipid accumulation and other aspects of heart pathology associated with diabetes (27). Thus decreased PPARα protein level in KO-AL in comparison to N-AL hearts could be related to enhanced insulin sensitivity of these animals, and could be suspected of protecting them from diabetic conditions. However, the levels of PPARα RNA and protein were increased in KO-CR animals in comparison to KO-AL, and PPARα RNA level also appeared to be elevated in N-CR mice when compared to N-AL. This indicates that CR may maintain the level of PPARα in the cardiac muscle of old N and KO animals, thus resembling the situation in the heart of 3-month-old KO mice that have elevated levels of PPARα RNA in comparison to normal controls. Moreover, the effects of CR on the expression of this nuclear receptor were opposite to the changes detected in aged hGH transgenic mice, in which PPARα expression was reduced. Perhaps CR maintains the expression of PPARα RNA in old animals at a level similar to those measured in 3-month-old mice.
Peroxisome Proliferator-activated Receptor β/δ
PPARβ/δ RNA levels were significantly higher while the protein levels were lower in long-lived KO mice in comparison to their normal siblings. This lack of correspondence between the data from Western blot and real-time PCR may reflect regulation at the post-transcriptional level, or differential stability of RNA and/or protein in normal and KO mice. Although, the function of PPARβ/δ in the heart is not known, it was shown that this nuclear receptor is expressed at a level similar to PPARα, suggesting the possibility that it may control the expression of cardiac genes (27). In our study, PPARβ/δ protein was markedly increased in KO-CR in comparison to KO-AL mice, with a suggestive (though not statistically significant) trend in the same direction at the RNA level. These findings closely resemble the effects of GH resistance and CR on the levels of PPARα RNA and protein. Similar to PPARα and PPARβ/δ expression levels in old KO mice, young KO had increased level of PPARβ/δ, while in the short-living hGH animals the level of PPARα RNA was significantly decreased.
Retinoid X receptors
The RNA expression of RXRγ, RXRα and RXRβ/δ was increased in KO-AL mice in comparison to their normal siblings, which can imply increased potential to form heterodimers with PPARs, thus improving the actions of PPARs superfamily receptors in the heart muscle. Alterations in the expression of all three RXRs in young KO vs. young normal mice closely corresponded to the changes observed in the corresponding PPARs. In hGH transgenic mice, RXRγ, RXRα and RXRβ/δ were decreased in comparison to their normal siblings, which mimicked the changes observed at the levels of PPARs.
Summary
In our study of genes involved in the initial steps of insulin action and PPARs nuclear receptor superfamily in the heart, we found that both the GHR gene knockout and the CR treatment that extend lifespan in mice serve as regulators of cardiac expression of these genes. Genotype and diet-induced alterations in the message levels and protein products of these genes in the heart were very different from the changes in the expression of the same genes in the skeletal muscle and liver described previously in the same animals. We found significant increase in the expression of analyzed genes in one or both phenotypes after CR. This outcome, taken together with the up regulation in young KO mice and suppression in short-lived hGH animals, suggests that CR may be protecting the hearts of these animals from the aging processes.
Acknowledgments
This project was supported by NIA, AG 19899 and U19 AG023122, by the Ellison Medical Foundation, and by the SIU Geriatrics Medicine and Research Initiative. JJK is supported, in part, by the State of Ohio's Eminent Scholar Program that includes a gift from Milton and Lawrence Goll, by DiAthegen, LLC, and by NIA AG19889. The authors would like to thank Dr. Grzegorz K. Przybylski for assistance with RT-PCR data analysis, Dr. Jeffrey A. Chesky for helpful comments on the manuscript and Marty Wilson for laboratory assistance. We would also like to thank Anna Masternak and Steve Sandstrom for help editing the manuscript.
REFERENCES
- 1.Zhou Y, Xu BC, Maheshwari HG, He L, Reed M, Lozykowski M, Okada S, Wagner TE, Cataldo LA, Coshigano K, Baumann G, Kopchick JJ. A mammalian model of Laron syndrome produced by targeted disruption of the mouse growth hormone receptor/binding protein gene (the Laron mouse) Proc. Natl. Acad. Sci. USA. 1997;94:13215–13220. doi: 10.1073/pnas.94.24.13215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu J-L, Coschigano KT, Robertson K, Lipsett M, Guo Y, Kopchick JJ, Kumar U, Liu YL. Disruption of growth hormone receptor gene causes diminished pancreatic islet size and increased insulin sensitivity in mice. Am J Physiol Endocrinol Metab. 2004;287:E405–E413. doi: 10.1152/ajpendo.00423.2003. [DOI] [PubMed] [Google Scholar]
- 3.Coschigano KT, Clemmons D, Bellush LL, Kopchick JJ. Assesment of growth parameters and lifespan of GHR/BP gene disrupted mice. Endocrinology. 2000;141:2608–2613. doi: 10.1210/endo.141.7.7586. [DOI] [PubMed] [Google Scholar]
- 4.Weindruch R, Sohal RS. Seminars in medicine of the Beth Israel Deaconess Medical Center. Caloric intake and aging. N Engl J Med. 1997;337:986–994. doi: 10.1056/NEJM199710023371407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tatar M, Bartke A, Antebi A. The Endocrine Regulation of Aging by Insulin-like Signals. Science. 2003;299:1346–1351. doi: 10.1126/science.1081447. [DOI] [PubMed] [Google Scholar]
- 6.Masternak MM, Al-Regaiey KA, Del Rosario Lim MM, Bonkowski MS, Panici JA, Przybylski GK, Bartke A. Caloric Restriction Results in Decreased Expression of Peroxisome Proliferator-Activated Receptors (PPARs) Superfamily in muscle of Normal and Long-Lived GHR-KO Mice. J Gerontol A Biol Sci Med Sci. 2005;60:1238–45. doi: 10.1093/gerona/60.10.1238. [DOI] [PubMed] [Google Scholar]
- 7.Masternak MM, Al-Regaiey KA, Del Rosario Lim MM, Jiminez-Ortega V, Panici JA, Bonkowski MS, BArtke A. Effects of Caloric Restriction on Insulin Pathway Gene Expression in the Skeletal Muscle and Liver of Normal and Long-Lived GHR-KO Mice. Experimental Gerontology. 2005;40:679–684. doi: 10.1016/j.exger.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 8.Masternak MM, Al-Regaiey KA, Del Rosario Lim MM, Jimenez-Ortega V, Panici JA, Bonkowski MS, Kopchick JJ, Bartke A. Effects of caloric restriction and GH resistance on the expression level of Peroxisome proliferators-activated receptors (PPARs) superfamily in the liver of normal and long-lived GHR-KO mice. J Gerontol A Biol Sci Med Sci. 2005;60:1394–1398. doi: 10.1093/gerona/60.11.1394. [DOI] [PubMed] [Google Scholar]
- 9.Brownsey RW, Boone AN, Allard MF. Actions of insulin on the mammalian heart: metabolism, pathology and biochemical mechanisms. Cardiovascular Research. 1997;34:3–24. doi: 10.1016/s0008-6363(97)00051-5. [DOI] [PubMed] [Google Scholar]
- 10.Swan JW, Anker SD, Walton C, Godsland IF, Clark AL, Leyva F, Stevenson JC, Coats AJ. Insulin resistance in chronic heart failure: relation to severity and etiology of heart failure. J Am Coll Cardiol. 1997;30:527–532. doi: 10.1016/s0735-1097(97)00185-x. [DOI] [PubMed] [Google Scholar]
- 11.Shiojima I, Yefremashvili M, Luo Z, Kureshi Y, Takahashi A, Tao J, Rosenzweig A, Khan CR, Abel ED, Walsh K. Akt signaling mediates postnatal hearth growthin response to insulin and nutritional status. The Journal of Biological Chemistry. 2002;40:37670–37677. doi: 10.1074/jbc.M204572200. [DOI] [PubMed] [Google Scholar]
- 12.Long P, Nguyen Q, Thurow C, Broderick TL. Caloric restriction restores the cardioprotective effects of preconditioning in the rat heart. Mechanism of Ageing and Development. 2002;123:1411–1413. doi: 10.1016/s0047-6374(02)00068-4. [DOI] [PubMed] [Google Scholar]
- 13.Backer JM, Myers MG, Jr, Shoelson SE, Chin DJ, Sun XJ, Miralpeix M, Hu P, Margolis B, Skolnik EY, Schlessinger J, White MF. Phosphatidylinositol 3′-kinase is activated by association with IRS-1 during insulin stimulation. EMBO Journal. 1992;11:3469–3479. doi: 10.1002/j.1460-2075.1992.tb05426.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun XJ, Wang LM, Zhang Y, Yenush L, Myers MJ, Glasheen E. Role of IRS-2 in insulin and cytokine signalling. Nature. 1995;377:173–177. doi: 10.1038/377173a0. [DOI] [PubMed] [Google Scholar]
- 15.Czech MP, Corvera S. Signaling mechanisms that regulate glucose transport. J. Biol. Chem. 1999;274:1865–1868. doi: 10.1074/jbc.274.4.1865. [DOI] [PubMed] [Google Scholar]
- 16.Thorens B, Charron MJ, Lodish HF. Molecular physiology of glucose transporters. Diabetes Care. 1990;13:209–218. doi: 10.2337/diacare.13.3.209. [DOI] [PubMed] [Google Scholar]
- 17.Brown-Borg HM, Borg KE, Meliska CJ, Bartke A. Dwarf mice and the ageing process. Nature. 1996;384:33. doi: 10.1038/384033a0. [DOI] [PubMed] [Google Scholar]
- 18.Flurkey K, Papaconstantinou J, Miller RA, Harrison DE. Lifespan extension and delayed immune and collagen aging in mutant mice with defects in growth hormone production. Proc Natl. Acad. Sci. U.S.A. 2001;98:6736–6741. doi: 10.1073/pnas.111158898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coshigano KT, Clemmons D, Bellush LL, Kopchick JJ. Assessment of growth parameters and lifespan of GHR/BP gene-disrupted Mice. Endocrinology. 2000;141:2608–2613. doi: 10.1210/endo.141.7.7586. [DOI] [PubMed] [Google Scholar]
- 20.Coshigano KT, Holland AN, Riders ME, List EO, Flyvbjerg A, Kopchick JJ. Deletion, but not antagonism, of the mouse growth hormone receptor results in severely decreased body weights, insulin, and insulin-like growth factor I levels and increased Lifespan. Endocrinology. 2003;144:3799–3810. doi: 10.1210/en.2003-0374. [DOI] [PubMed] [Google Scholar]
- 21.Holzenberger M, et al. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature. 2003;421:182–187. doi: 10.1038/nature01298. [DOI] [PubMed] [Google Scholar]
- 22.Moller DE, Berger JP. Role of PPARs in the regulation of obesity-related insulin sensitivity and inflammation. International Journal of Obesity. 2003;27:S17–S21. doi: 10.1038/sj.ijo.0802494. [DOI] [PubMed] [Google Scholar]
- 23.Knouff C, Auwerx J. PPARγ calls for activation in moderation: lessons from genetics and pharmacology. Endocrine Reviews. 2004;25:899–918. doi: 10.1210/er.2003-0036. [DOI] [PubMed] [Google Scholar]
- 24.Petersen KF, Krssak M, Inzucchi S, Cline GW, Dufour S, Shulman GI. Mechanism of troglitazone action in type 2 diabetes. Diabetes. 2000;49:827–831. doi: 10.2337/diabetes.49.5.827. [DOI] [PubMed] [Google Scholar]
- 25.Kraegen EW, James DE, Jenkins AB, Chisholm DJ, Storlien LH. A potent in vivo effect of ciglitazone on muscle insulin resistance induced by high fat feeding of rats. Metabolism. 1989;38:1089–1093. doi: 10.1016/0026-0495(89)90045-0. [DOI] [PubMed] [Google Scholar]
- 26.Zierath JR, Ryder JW, Doebber T, Woods J, Wu M, Ventre J, Li Z, McCrary C, Berger J, Zhang B, Moller DE. Role of skeletal muscle in thiazolidinedione insulin sensitizer (PPARgamma agonist) action. Endocrinology. 1998;139:5034–5041. doi: 10.1210/endo.139.12.6364. [DOI] [PubMed] [Google Scholar]
- 27.Schoonjans K, Martin G, Staels B, Auwerex J. Peroxisome proliferator-activated receptors, orphans with ligands and functions. Curr. Opin. Lipidol. 1997;8:159–166. doi: 10.1097/00041433-199706000-00006. [DOI] [PubMed] [Google Scholar]
- 28.Kelly DP. PPARs of the heart Three is a Crowd. Circ. Res. 2003;92:482–484. doi: 10.1161/01.RES.0000064382.46274.95. [DOI] [PubMed] [Google Scholar]
- 29.Peters JM, Hennuyer N, Staels B, Fruchart JC, Fievet C, Gonzalez FJ, Auwerex J. Alterations in lipoprotein metabolism in peroxisome proliferators-activated receptor α-deficient mice. The Journal of Biological Chemistry. 1997;272:27307–27312. doi: 10.1074/jbc.272.43.27307. [DOI] [PubMed] [Google Scholar]
- 30.Michalik L, Desvergene B, Tan NS, Basu-Modak S, Escher P, Rieusset J, Peters JM, Kaya G, Gonzalez FJ, Zakany J. Impaired skin wound healing in peroxisome proliferators-activated receptor (PPAR)α and PPARβ mutant mice. J. Cell Biol. 2001;154:799–814. doi: 10.1083/jcb.200011148. at al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tan NS, Michalik L, Noy N, Yasmin R, Pacot C, Heim M, Fluhmann B, Desvergene B, Wahli W. Critical roles of PPARβ/δ in keratinocyte response to inflammation. Genes Dev. 2001;15:3263–3277. doi: 10.1101/gad.207501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang YX, Lee C, Tiep S, Yu RT, Ham J, Kang H, Evans RM. Peroxisome-proliferator-activated receptor δ activates fat metabolism to prevent obesity. Cell. 2003;113:159–170. doi: 10.1016/s0092-8674(03)00269-1. [DOI] [PubMed] [Google Scholar]
- 33.IJpenberg A, Tan NS, Gelman L, Kersten S, Seydoux J, Xu J, Metzerg D, Canapel L, Chambon P, Wahli W, Desvergene B. In vivo activation of PPAR target genes by RXR homodimers. The EMBO Journal. 2004;23:2083–2091. doi: 10.1038/sj.emboj.7600209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mattison JA, Wright C, Bronson RT, Roth GS, Ingram DK, Bartke A. Studies of aging in Ames dwarf mice: effects of caloric restriction. J. Am. Aging Assoc. 2000;23:9. doi: 10.1007/s11357-000-0002-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Al-Regaiey K, Masternak MM, Bonkowski M, Sun L, Bartke A. Long-lived growth hormone receptor knockout mice: Interaction of reduced IGF-1/insulin signaling and caloric restriction. Endocrinology. 2005;146:851–860. doi: 10.1210/en.2004-1120. [DOI] [PubMed] [Google Scholar]
- 36.Yun JS, Wagner TE. Study of human growth hormone transgenic mice: Female reproductive system. 10th Korea Symp Sci Technol. 1987;3-1:279–282. [Google Scholar]
- 37.Bartke A, Steger RW, Hodges S, Yun J, Wagner T. Infertility in transgenic female mice with human growth hormone expression. J Exp Zool. 1988;248:121–124. doi: 10.1002/jez.1402480116. [DOI] [PubMed] [Google Scholar]
- 38.Selden RF, Yun JS, Moore DD, Rowe ME, Malia MA, Wagner TE, Goodman HM. Glucocorticoid regulation of human growth hormone expression in transgenic mice and transiently transfected cells. J Endocrinol. 1989;122:49–60. doi: 10.1677/joe.0.1220049. [DOI] [PubMed] [Google Scholar]
- 39.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 40.Lupu F, Terwilliger JD, Lee K, Segre GV, Efstratiadis A. Roles of growth hormone and insulin-like growth factor 1 in mouse postnatal growth. Developmental Biology. 2001:141–162. doi: 10.1006/dbio.2000.9975. [DOI] [PubMed] [Google Scholar]
- 41.Sun LY, Al-Regaiey K, Masternak MM, Wang J, Bartke A. Local expression of GH and IGF-1 in the hippocampus of GH-deficient long-lived mice. Neurobiol Aging. 2005;26(6):929–37. doi: 10.1016/j.neurobiolaging.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 42.McMullen JR, Shioi T, Huang W, Zhang L, Tarnavski O, Bisping E, Schinke M, Kong S, Sherwood MC, Brown J, Riggi L, Kang PM, Izumo S. The Insulin-like Growth Factor 1 Receptor Induces Physiological Heart Growth via the Phosphoinositide 3-Kinase(p110α) Pathway. J. Biol. Chem. 2004;279(Issue 6):4782–4793. doi: 10.1074/jbc.M310405200. [DOI] [PubMed] [Google Scholar]