

Abstract
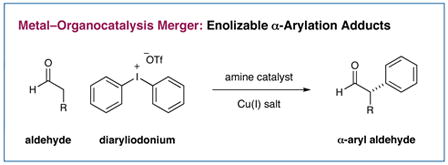
The enantioselective α-arylation of aldehydes has been accomplished using diaryliodonium salts and a combination of copper and organic catalysts. These mild catalytic conditions provide a new strategy for the enantioselective construction and retention of enolizable α-formyl benzylic stereocenters, a valuable synthon for the production of medicinal agents. As one example, this new asymmetric protocol has been applied to the rapid synthesis of (S)-ketoprofen, a commercially successful oral and topical analgesic.
Over the last decade, the catalytic union of nascent enolates with aryl coupling partners has become a mainstay transformation in organic synthesis, primarily driven by advances in transition metal catalysis.1,2 In particular, the pioneering work of Buchwald and Hartwig has provided a number of enantioselective enolate α-arylations that enable quaternary carbon formation directly adjacent to both ketone and lactone moieties.2 Slower to develop, however, have been protocols that allow the enantioselective production of enolizable α-carbonyl benzylic stereocenters (methine stereocenters), presumably due to the propensity for post-reaction racemization when elevated temperatures or basic conditions are employed.2,3

 |
(Eq 1) |
 |
(Eq 2) |
A notable exception is the seminal work of Fu and coworkers who have realized the enantioselective Negishi and Kumada coupling of α-methine bromo ketones and amides with organometallic arenes at low temperature.4 Recently, we questioned whether enolizable α-aryl aldehydes might also be enantioselectively created and retained via a novel, tandem catalysis mechanism that employs diaryliodonium salts as an arylation partner in the presence of both amine and metal catalysts.5 Herein, we describe the successful execution of these ideals and present an operationally trivial, room temperature protocol that is sufficiently mild to generate α-carbonyl benzylic stereogenicity without post-reaction racemization (eq 2).
Design Plan
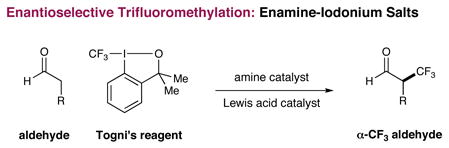
Recently, we described the enantioselective α-trifluoromethylation of aldehydes using the hypervalent Togni reagent6, in combination with an imidazolidinone catalyst and Lewis acid additives (eq 1).7 Inspired by these studies, we hypothesized that iodonium salts that incorporate aromatic groups, such as diphenyliodonium triflate, might analogously function as arylating agents with transiently generated enamines, thereby providing a new pathway for asymmetric aldehyde arylation with amine catalysts (Scheme 1). Often viewed as an electrophilic species that undergoes C–Ar bond formation via an iodonium addition/reductive elimination mechanism, diaryliodonium salts have previously been utilized for the racemic α-arylation of silyl enol ethers and silyl ketene acetals.8 As a further consideration, we recognized that these electrophilic salts are non-toxic, readily accessible, and air and moisture stable, valuable characteristics with respect to the development of an efficient organocatalytic reaction.5c
Scheme 1.

Initial Hypothesis for the α-Arylation of Aldehydes
We began our investigation using diphenyliodonium triflate, octanal, and amine catalyst 1 (Table 1). While non-catalytic reactions with enolates9 or silyl enol ethers10 often proceed without activation of the diaryliodonium salt, enamine-activated octanal failed to couple with diphenyliodonium triflate (entry 1).11 In accord with our previous trifluoromethylation studies, we next examined the use of catalytic Lewis acids that we hoped would activate the hypervalent iodine reagent towards nucleophilic attack via counterion binding.6,7 Remarkably, all metal salt additives resulted in no observable reaction (entries 2–4) with the exception of CuCl, which produced the desired α-arylation adduct in a notable 58% yield and 76% ee (entry 5). It has long been established that the arylation of carbon and oxygen based nucleophiles with diaryliodonium salts can be facilitated by the presence of catalytic copper salts, however, the mechanism of activation typically involves a Cu(I) mediated oxidative addition into the aryl-iodonium bond to generate a highly electrophilic Ar-Cu(III)-XY system.12,13 With this alternative pathway in mind (see Scheme 2), further improvements were realized when CuBr was employed (entry 6), and both yield and enantioselectivity were increased when amine catalyst 1 was replaced with catalyst 3 (entry 8). Moreover, use of trichloroacetic acid as the co-catalyst along with a mixed solvent system of toluene and diethyl ether provided excellent chemical yield and enantioselectivity (entries 9 and 10). Finally, the observed levels of efficiency and enantiocontrol were maintained with 10 mol% of catalyst 3, thereby establishing optimal conditions for further exploration.14,15
Table 1.
Evaluation of Metal Additives and Amine Catalysts
 | ||||
|---|---|---|---|---|
| entry | amine·HX (mol %) | metal salt (mol %) | yield (%)a | ee (%)b |
| 1 | 1· TFA (20 mol%) | None | 0 | -- |
| 2 | 1· TFA (20 mol%) | Sc(OTf)3 (10 mol%) | 0 | -- |
| 3 | 1· TFA (20 mol%) | Sm(OTf)3(10 mol%) | 0 | -- |
| 4 | 1· TFA (20 mol%) | Zn(OTf)2(20 mol%) | 0 | -- |
| 5 | 1· TFA (20 mol%) | CuCl (10 mol%) | 58 | 76 |
| 6 | 1· TFA (20 mol%) | CuBr(10 mol%) | 63 | 76 |
| 7 | 2· TFA (20 mol%) | CuBr(10 mol%) | 47 | 43 |
| 8 | 3· TFA (20 mol%) | CuBr(10 mol%) | 74 | 87 |
| 9 | 3· TCA (20 mol%) | CuBr(10 mol%) | 90 | 84 |
| 10c | 3· TCA (10 mol%) | CuBr(10 mol%) | 90 | 92 |
GC yield.
Determined by chiral HPLC analysis of the corresponding alcohol, absolute configuration determined by chemical correlation or by analogy.
Performed with 2:1 toluene/Et2O.
Scheme 2.
Revised Mechanism for the Aldehyde α-Arylation
As outlined in Table 2, this α-arylation strategy is tolerant to aldehydic coupling partners that incorporate a wide range of functional groups, including arenes and olefins (entries 2 and 3, 84–87%, 94% ee) as well as ethers, esters, and carbamates (entries 4–7, 74–81%, 91–93% ee). Notably, sterically demanding substrates (R = piperidinyl or isopropyl) are also accommodated with minimal impact on yield or enantiocontrol (entries 7 and 8, 68–81%, 90–93% ee). In addition, enantiopure β-chiral aldehydes can also be used for the diastereoselective construction of both syn- and anti-α,β-disubstituted products (entries 9 and 10, 67–81%, 20:1 to 5:1 dr). While a matched and mismatched outcome is observed in each case, respectively, it is interesting to note that the induced stereochemistry in both examples is dominated by the catalyst architecture.
Table 2.
 | |||
|---|---|---|---|
| 1 |
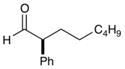 90% yield, 92% ee |
2 |
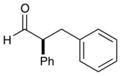 87% yield, 94% ee |
| 3d |
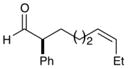 84% yield, 94% ee |
4e,f |
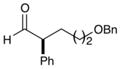 77% yield, 91% ee |
| 5 |
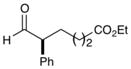 80% yield, 93% ee |
6 |
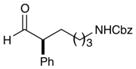 74% yield, 93% ee |
| 7e |
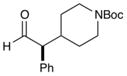 81% yield, 93% ee |
8e |
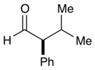 68% yield, 90% ee |
| 9 |
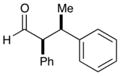 81% yield, >20:1 dr |
10d,f,g |
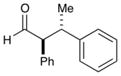 67% yield, 5:1 dr |
Absolute configuration assigned by chemical correlation or by analogy.
Isolated yield of the corresponding alcohol.15
Enantiomeric excess determined by chiral HPLC analysis of the corresponding alcohol.
1:1 toluene/Et2O.
20 mol% 3·TCA.
20 mol% CuBr.
40 mol% 3·TCA.
We have also found that a broad range of aryl and heteroaryl rings can be enantioselectively coupled with aldehyde-derived enamines in this tandem-catalysis reaction (Table 3). While symmetrical diaryliodonium triflates can be successfully employed in this context, we have found that the elegant approach of Gaunt to generate Ar-Cu(III)-XY systems from non-symmetrical aryl-mesityl reagents is preferred for reasons of practicality.12a–c,16 Notably, electron-poor (entries 1–4, 6–7), as well as electron-rich arenes (entries 5, 8–10), transfer readily with excellent levels of enantiocontrol (≥91% ee). Moreover, a broad range of para- and meta-substituted aryl rings with diverse steric and electronic properties (ethers, esters, nitro groups, and halides) can be readily exploited in this protocol (entries 2–4, 6–10, 67–91%, 91–93% ee). It is important to note, however, that ortho-substitution has thus far been restricted to small groups such as fluorine (entry 7, 67%, 91% ee). In addition to phenyl rings, we were delighted to find that heteroaromatics of an electron-rich (entry 11, 81%, 92% ee) or electron-poor nature (entry 12, 70%, 93% ee) are suitable coupling partners, a valuable finding with respect to potential medicinal chemistry applications.
Table 3.
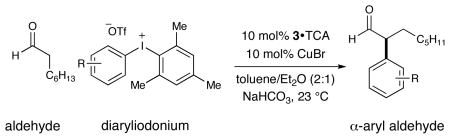 | ||
|---|---|---|
1d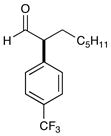 95% yield, 94% ee |
2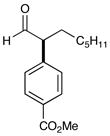 90% yield, 91% ee |
3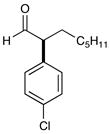 86% yield, 93% ee |
4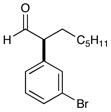 91% yield, 92% ee |
5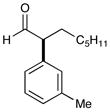 92% yield, 91% ee |
6e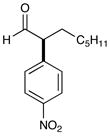 70% yield, 93% ee |
7d,f,g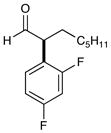 67% yield, 91% ee |
8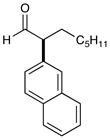 87% yield, 91% ee |
9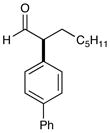 87% yield, 92% ee |
10d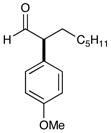 88% yield, 91% ee |
11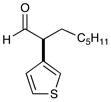 81% yield, 92% ee |
12 f,h,i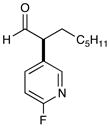 70% yield, 93% ee |
Absolute configuration assigned by chemical correlation or analogy.
Isolated yield of the corresponding alcohol.
Enantiomeric excess determined by chiral HPLC analysis of the corresponding alcohol.
Symmetrical diaryliodonium triflate was used.
Using 20 mol% 3·TCA.
Using 40 mol% 3·TCA.
Using 20 mol% CuBr.
Using 1:1 toluene/Et2O.
Using 5 mol% CuBr.
Mechanistic Considerations
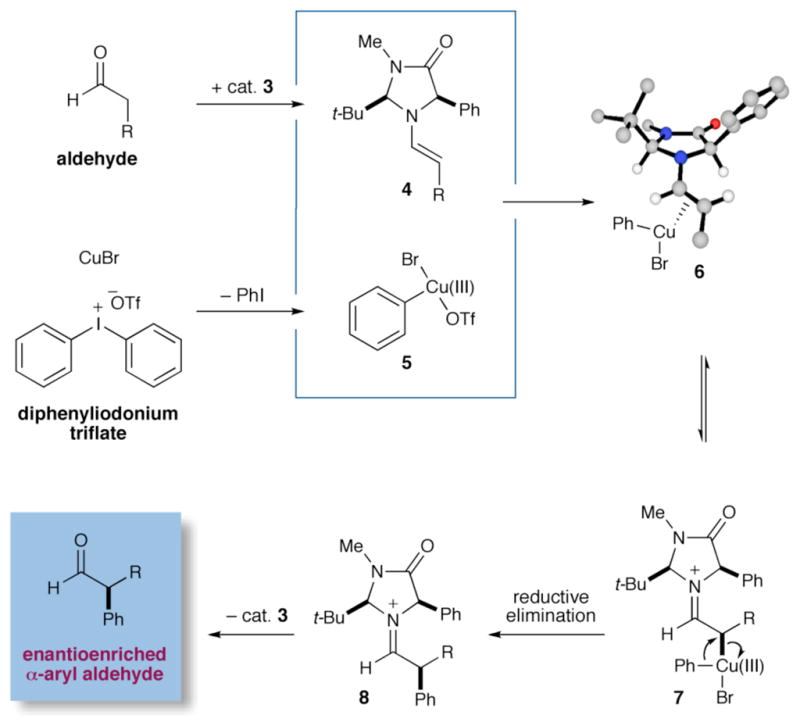
As illustrated in Scheme 2, we propose that this tandem-catalysis17 pathway begins with concurrent formation of both the activated chiral enamine 4 (from condensation of amine catalyst 3 with the aldehyde substrate) and the electron-deficient aryl-Cu(III) species 5, (arising from oxidative addition of Cu(I) into the C–I bond of the diaryliodonium triflate system). We expect that the highly electrophilic Cu(III)-aryl system 5 will rapidly coordinate to the Re face of the enamine (Si face shielded by the phenyl substituent of 3, see 618) to form the π-Cu complex 6. Rapid bond isomerization will subsequently lead to the η1-iminium 7 organocopper, which upon reductive elimination should release the optically enriched α-aryl iminium 8 and reconstitute the Cu(I)Br catalyst. Hydrolysis of iminium 8 will subsequently regenerate the organocatalyst partner 3 along with the desired α-aryl aldehyde product.
To further demonstrate the value of this new α-arylation strategy, we focused upon the development of a unique and expeditious route to ketoprofen, an oral and topical non-steroidal anti-inflammatory drug.19 As shown in detail in Scheme 3, implementation of our standard arylation conditions at 0 °C with propionaldehyde and iodonium 9 (prepared in two steps from 3-aminobenzophenone, see Supporting Information), followed by in situ aldehyde oxidation, successfully furnished (S)-ketoprofen in 71% yield and 92% ee (a total of three chemical operations from commercial materials).
Scheme 3.

Rapid Enantioselective Synthesis of (S)-Ketoprofen
In conclusion, we have developed a new technology that allows the direct and enantioselective α-arylation of aldehydes using a combination of metal and organic catalysis. The value of this transformation has been highlighted via an expedient synthesis of (S)-ketoprofen, a well-established and commercially successful medicinal agent. Further investigations into the mechanism of this transformation are ongoing in our laboratory.
Supplementary Material
Acknowledgments
Financial support was provided by NIHGMS (R01 GM078201-05) and kind gifts from Merck and Bristol-Myers Squibb. A.E.A. thanks the Natural Sciences and Engineering Research Council of Canada (NSERC) for a predoctoral fellowship.
Footnotes
Supporting Information Available. Experimental procedures and spectral data are provided (53 pages, pdf). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For racemic α-arylation of carbonyl compounds, see: Muratake H, Nakai H. Tetrahedron Lett. 1999;40:2355.Martín R, Buchwald SL. Angew Chem, Int Ed. 2007;46:7236. doi: 10.1002/anie.200703009.Vo GC, Hartwig JF. Angew Chem, Int Ed. 2008;47:2127. doi: 10.1002/anie.200705357.
- 2.For asymmetric α-arylation of carbonyl compounds, see: García-Fortanet J, Buchwald SL. Angew Chem, Int Ed. 2008;47:8108. doi: 10.1002/anie.200803809.Taylor AM, Altman RA, Buchwald SL. J Am Chem Soc. 2009;131:9900. doi: 10.1021/ja903880q.Liao X, Weng Z, Hartwig JF. J Am Chem Soc. 2008;130:195. doi: 10.1021/ja074453g.
- 3.(a) Conrad JC, Kong J, Laforteza BN, MacMillan DWC. J Am Chem Soc. 2009;131:11640. doi: 10.1021/ja9026902. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Alemán J, Cabrera S, Maerten E, Overgaard J, Jørgensen KA. Angew Chem, Int Ed. 2007;46:5520. doi: 10.1002/anie.200701207. [DOI] [PubMed] [Google Scholar]
- 4.(a) Dai X, Strotman NA, Fu GC. J Am Chem Soc. 2008;130:3302. doi: 10.1021/ja8009428. [DOI] [PubMed] [Google Scholar]; (b) Lundin PM, Esquivias J, Fu GC. Angew Chem, Int Ed. 2009;48:154. doi: 10.1002/anie.200804888. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lou S, Fu GC. J Am Chem Soc. 2010;132:1264. doi: 10.1021/ja909689t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Lundin PM, Fu GC. J Am Chem Soc. 2010;132:11027. doi: 10.1021/ja105148g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.For reviews on diaryliodonium salts, see Merritt EA, Olofsson B. Angew Chem, Int Ed. 2009;48:9052. doi: 10.1002/anie.200904689.Varvoglis A. The Organic Chemistry of Polycoordinated Iodine. VCH Publishers; New York: 1992. Grushin VV. Chem Soc Rev. 2000;29:315.
- 6.(a) Kieltsch I, Eisenberger P, Togni A. Angew Chem, Int Ed. 2007;46:754. doi: 10.1002/anie.200603497. [DOI] [PubMed] [Google Scholar]; (b) Koller R, Stanek K, Stolz D, Aardoom R, Niedermann K, Togni A. Angew Chem, Int Ed. 2009;48:4332. doi: 10.1002/anie.200900974. [DOI] [PubMed] [Google Scholar]
- 7.Allen AE, MacMillan DWC. J Am Chem Soc. 2010;132:4986. doi: 10.1021/ja100748y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Zhdankin V. Top Curr Chem. 2003;224:99. [Google Scholar]; (b) Koser GF. Top Curr Chem. 2003;224:137. [Google Scholar]; (c) Merrit EA, Olofsson B. Synthesis. 2011:517. [Google Scholar]
- 9.Beringer FM, Forgione PS, Yudis MD. Tetrahedron. 1960;8:49.Beringer FM, Galton SA, Huang SJ. J Am Chem Soc. 1962;84:2819.Beringer FM, Forgione PS. Tetrahedron. 1963;19:739.Beringer FM, Forgione PS. J Org Chem. 1963;28:714.Beringer FM, Daniel WJ, Galton SA, Rubin G. J Org Chem. 1966;31:4315.Chen Z, Jin Y, Stang PJ. J Org Chem. 1987;52:4115.Hampton KG, Harris TM, Hauser CR. J Org Chem. 1964;29:3511.Gao P, Portoghese PS. J Org Chem. 1995;60:2276.Oh CH, Kim JS, Jung HH. J Org Chem. 1999;64:1338.For asymmetric examples, see: Ochiai M, Kitagawa Y, Takayama N, Takaoka Y, Shiro M. J Am Chem Soc. 1999;121:9233.Aggarwal VK, Olofsson B. Angew Chem, Int Ed. 2005;44:5516. doi: 10.1002/anie.200501745.
- 10.Reactions with silyl enol ethers require the diaryliodonium fluoride in the absence of other additives, see: Chen K, Koser GF. J Org Chem. 1991;56:5764.Iwama T, Birman VB, Kozman SA, Rawal VH. Org Lett. 1999;1:673. doi: 10.1021/ol990759j.
- 11.At 90 °C only 4% of product is observed in the absence of any metal salt. This is consistent with reports from Kuehne where cyclohexanone-derived enamines failed to react effectively with diaryliodonium salts (≤8%), see: Kuehne ME. J Am Chem Soc. 1962;84:837.
- 12.For examples of copper-catalyzed arylation reactions, see: Phipps RJ, Grimster NP, Gaunt MJ. J Am Chem Soc. 2008;130:8172. doi: 10.1021/ja801767s.Phipps RJ, Gaunt MJ. Science. 2009;323:1593. doi: 10.1126/science.1169975.Duong HA, Gilligan RE, Cooke ML, Phipps RJ, Gaunt MJ. Angew Chem, Int Ed. 2010;50:463. doi: 10.1002/anie.201004704.Ryan JH, Stang PJ. Tetrahedron Lett. 1997;38:5061.
- 13.For studies on the mechanism of copper-catalyzed arylation, see: Lockhart TP. J Am Chem Soc. 1983;105:1940.Beringer FM, Geering EJ, Kuntz I, Mausner M. J Phys Chem. 1956;60:141.
- 14.The addition of NaHCO3 is necessary to remove the trifluoromethanesulfonic acid that is formed during the reaction.
- 15.The isolated α-formyl adducts are generally stable at room temperature without significant racemization. 2-Phenyloctanal was isolated with 90% ee through direct purification by flash chromatography (see Supporting Information). Rechecking the enantiopurity of the 2-phenyloctanal after 3 weeks confirmed the initial 90% ee (i.e. no erosion in optical purity).
- 16.Sanford and coworkers also used this approach in the Pd-catalyzed arylation of C-H bonds, see: Kalyani D, Deprez NR, Desai LV, Sanford MS. J Am Chem Soc. 2005;127:7330. doi: 10.1021/ja051402f.Deprez NR, Sanford MS. J Am Chem Soc. 2009;131:11234. doi: 10.1021/ja904116k.
- 17.For a review on the merger of organocatalysis and transition metal-catalysis for tandem catalytic activation, see: Zhong C, Shi X. Eur J Org Chem. 2010:2999.
- 18.DFT calculations for the enamine were performed with the use of B3LYP/6-311+G(2d,2p)//B3LYP/6-31G(d). The arylcopper(III) species was not included in the DFT calculation.
- 19.Mack DJ, Brichacek M, Plichta A, Njardarson JT Department of Chemistry, University of Arizona. [accessed Jan 2011];Top 200 Pharmaceutical Products by Worldwide Sales in 2009. http://cbc.arizona.edu/njardarson/group/top-pharmaceuticals-poster.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.