

Abstract
We present a family with mild developmental delay and a duplication (6)(p22.2). Array CGH analyses revealed this 0.7 Mb duplication in all three patients, spanning candidate genes ALDH5A1, DCDC2 and KIAA0319. Results were confirmed by MLPA analysis of the dyslexia genes DCDC2 and KIAA0319. Of interest, ALDH5A1 encodes succinate semialdehyde dehydrogenase (SSADH), an enzyme responsible for γ-amino-butyric acid (GABA) degradation. Inherited deficiency of SSADH results in accumulation of the neuromodulator γ-hydroxybutyrate (GHB), which likely contributes to some aspects of the neurological phenotype of SSADH deficiency (MIM #271980). Based upon autosomal-recessive inheritance, we sequenced ALDH5A1 in all patients, which revealed no pathogenic mutations. SSADH enzyme studies in cultured white cells confirmed elevated SSADH activity, consistent with the duplication, whereas concentrations of SSA were slightly elevated in urine, suggesting oxidant stress. We speculate that the duplication (6)(p22.2) and corresponding hyperactive level of SSADH activity may have negative consequences for GABA metabolism and the role of SSADH in other metabolic sequences.
Keywords: duplication 6p22.2, SSADH, mild mental retardation
INTRODUCTION
Mild mental retardation (MR) is the most commonly occurring form of MR, accounting for ~80% of the international mental retardation population (Chapman et al., 2008). Nevertheless, only 24% of the patients with mild MR have a known etiology, as opposed to 70% of patients with severe MR (Chapman et al., 2008). Mild MR is characterized by a total IQ between 50 and 69 and generally associates with slower than normal development, which may cause minor learning difficulties and social behavioral problems (World Health Organization, 2007). Unraveling the chromosomal etiological factors of mild mental retardation is difficult, due to the subtle nature of the changes. The etiological factors may be genetic, acquired, environmental and sociocultural (Katz and Lazcano-Ponce, 2008), with genetic factors playing the largest role. Often, a copy number variation conveys the phenotype through gene-dosage effects.
Succinate semialdehyde dehydrogenase (SSADH) deficiency (MIM #271980) is a rare metabolic disorder caused by dysfunction of the SSADH enzyme in the γ-amino-butyric acid (GABA) degradation pathway. SSADH is encoded by the gene ALDH5A1 and is one of two enzymes of the GABA degradation pathway. A mutation in this gene, that renders the enzyme dysfunctional, prevents the conversion of succinic semialdehyde (SSA) to succinic acid, which in turn leads to accumulation of a by-product; γ-hydroxybutyrate (GHB) (Figure 1). GHB is a neuromodulator and increased levels in body fluids associates with mild to severe neurological defects (Gibson et al., 2003). The first patient with SSADH deficiency was reported in 1981 by Jakobs et al (Jakobs et al., 1981), and since then numerous patients with SSADH deficiency have been identified (Pearl et al., 2009), with considerable variation in phenotypic features.
Figure 1.
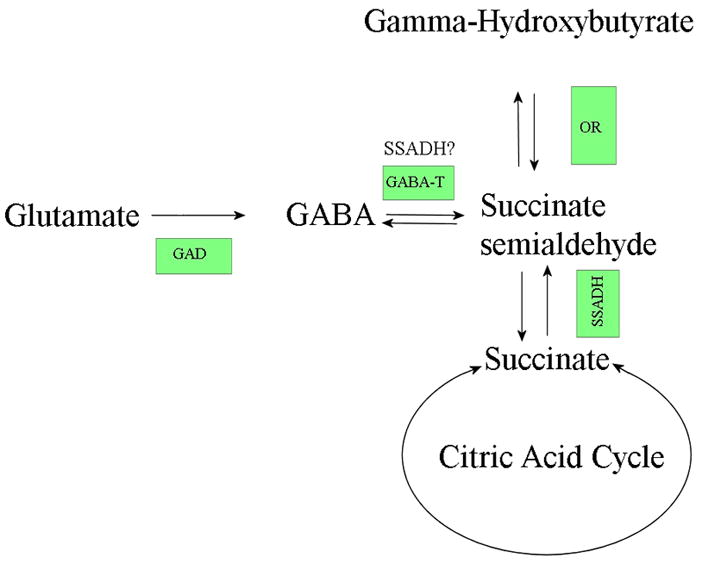
A simplified overview of the GABA degradation pathway. The boxed abbreviations indicate the enzyme responsible for conversion of the substrate at that stage of the pathway (GAD – glutamate decarboxylase; GABA-T – GABA transaminase; OR – oxidoreductase (or aldo-keto reductase); SSADH – semialdehyde dehydrogenase). It has also been suggested that SSADH may play a role in conversion of GABA to SSA (Kim et al. 2009).
Previously, no reports have described the clinical effects of duplications of ALDH5A1, but, as is becoming evident, for several deletion syndromes there is a counterpart duplication syndrome with different phenotypic features. (Bruno et al. 2010) We present a family with mild developmental delay and an increased level of SSADH, likely caused by a duplication at (6)(p22.2).
MATERIAL AND METHODS
Patients
The patients are a 38 yr old mother (patient 1) and her two only children; a 19 yr old daughter (patient 2) and 18 yr old son (patient 3). The mother has a sister and brother, who are regarded as intellectually normal. The father was unavailable for further investigation.
The son was referred to Oulu University Hospital, Department of Genetics for clinical evaluation, having school difficulties related to mild developmental delay. His sister had been referred to the local clinic of mentally retarded, also for mild developmental delay. His mother also reported a history of mild intellectual problems.
Blood sample and informed consent was obtained from all three patients. Reference samples consisting of pooled blood from 4 healthy Finnish individuals, males and females respectively, were obtained from the Finnish Red Cross. DNA from patient and reference samples was extracted from whole blood according to standard protocols.
Cytogenetic Analysis
Karyotype analysis by G-banding was performed, on peripheral blood lymphocytes of all patients, according to standard protocols.
Array CGH
All patients were analyzed by array comparative genomic hybridization (array CGH) according to the manufacturer’s protocol for Human 244K oligonucleotide microarrays (Agilent Technologies, Santa Clara, CA, USA). Arrays were scanned using an Agilent scanner and data extracted with the Feature Extraction software v.9.5.3.1 and analyzed with the CGH Analytics software v.3.5.14 (hg build 18), as described previously (Siggberg et al., 2010).
SNP 6.0 array
Patient 3 was analyzed using the Genome wide Human SNP Nsp/Sty 6.0 microarray according to manufacturer protocols (Affymetrix, Santa Clara, CA, USA). Data was extracted with the Genotyping Console software v.3.0.2 using a reference model consisting of 115 Caucasian HapMap samples. Results were analyzed using the Chromosome Analysis Suite software v.1.0.
MLPA
All patients were analyzed by multiplex ligation-dependent probe amplification (MLPA) (Schouten et al., 2002) using the P150 Dyslexia MLPA kit (MRC-Holland, Amsterdam, the Netherlands) according to manufacturer’s instructions. Subsequently, MLPA amplification products were separated by capillary electrophoresis on a CEQ 8000 (Beckman Coulter, Inc., Brea, CA, USA). Data analysis was performed using Coffalyser (MRC-Holland, Amsterdam, the Netherlands)
Sequencing
All 10 exons and the adjacent splice sites of ALDH5A1 were amplified by PCR using the primers described by Akaboshi et al 2003. Sequence analysis was performed using BigDye v3.1 terminator and an ABI 3130XL (Applied Biosystems, Ijssel, The Netherlands). The obtained sequences were analyzed using the Mutation Surveyor software package (Softgenetics, State College, PA, USA).
Enzyme and metabolite studies
Lymphoblasts were developed from isolated polymorphonuclear cells in whole blood and immortalization using Epstein-Barr Virus (EBV). For assay of SSADH, the conversion of substrate succinic semialdehyde was monitored via the stoichiometric reduction of NAD+ to NADH using spectrofluorometry (Gibson et al 1991). Urine samples of all patients were analyzed for GHB and SSA content employing a stable-isotope dilution assay (d6-GHB or 13C4-SSA as internal standard) with combined gas chromatography-mass spectrometry.
RESULTS
Clinical presentation
All patients in this family (patients 1, 2 and 3) manifested mild developmental delays. In patient 3 neuropsychological examinations, using the Wechsler Intelligence Scale for Children (WISC) test, revealed more pronounced deficiency in non-verbal tests. Overall performance in A Developmental NEuroloPSYcological Assesement (NEPSY) test were subnormal – clear problems with visuospatial and -constructive functions, language and communication was poor, visual attentiveness showed poor visual deduction and visuomotor precision, and poor focus and attention abilities were noted. At 10 years of age (patient 3) maturity levels varied from the level of a 6 year old and up in some tasks. Early motor milestones were within normal limits. The mother (patient 1) and the daughter (patient 2) have similar features as patient 3, and additionally have shown atypical attacks of loss of consciousness and the mother had anticonvulsive therapy at the age of 7–12 years, because of atypical seizures, whereas EEG did not reveal evidence for epileptic activity.
Molecular analyses
The clinical presentation of the patients suggested a chromosomal rearrangement and therefore cytogenetic analysis was performed. This revealed a seemingly balanced inversion of chromosome 1 in all patients. Patients 1 and 2 had the karyotype 46, XX, inv(1)(p31.2p34.3) and patient 3 46, XY, inv(1)p31.2p35), his inversion being slightly larger than his relatives.
To confirm the balanced state of the inversion, array CGH was performed by 244K oligonucleotide array CGH analysis of all patients as well as SNP 6.0 array analysis of patient 3. Results indicated that the inversion on chromosome 1 was balanced, as no chromosomal aberrations were detected in the breakpoint area. As the inversion was seen also in seemingly healthy persons in the family (data not shown), it is unlikely to be associated with the clinical features observed in the patients.
Array CGH analysis revealed identical 0.7 Mb duplications at chromosome 6p22.2 in all three patients, with an average log2 ratio of 0.5, indicating gain of one copy. This aberration has not been reported in the Database of Genomic Variants (DGV), the Database of Chromosomal Imbalance and Phenotype in Humans using Ensemble Resources (DECIPHER), or the European Cytogeneticists Association Register of Unbalanced Chromosome Aberrations (ECARUCA) databases, nor had we seen it before in other Finnish patients we have studied.
The duplication ranges from basepair positions 24293550 to 25057643 (probes A_16_P17480886 to A_16_P37551683, hg 18) (Figure 2). It covers the genes DCDC2, MRS2L, GPLD1, ALDH5A1, KIAA0319, TTRAP, ACOT13, C6orf62, GMNN and C6orf32 and additionally contains 5 areas of unknown content. Gene annotations were cross-checked with the Ensemble and the USCS genome browsers.
Figure 2.

A representative Array CGH plot of all patients. Red dots indicate a gain, in this case with an average log2-ration of +0.5. The shaded area indicates the calculated duplicated area according to the ADAM2-algorithm.
MLPA using the P150 Dyslexia kit was performed to analyze the two dyslexia genes DCDC2 and KIAA0319. The results confirmed that all three family members carried a partial duplication of DCDC2 and a complete duplication of KIAA0319 (Figure 3).
Figure 3.

MLPA results of patient 3 using the dyslexia kit. On the x-axis are the names of the probes in the P150 Dyslexia MLPA kit (MRC-Holland, Amsterdam, the Netherlands), targeting the exons of genes; ROBO2, ROBO1, NRSN1, DCDC2, KAAG1 and KIAA0319 as well as a control probe. On the y-axis are the allelic ratio, 1 representing normal two copies, and 1.5 representing one additional copy. A one copy duplication is indicated in KIAA0319 and DCDC2.
The ALDH5A1 gene of patient 1 was sequenced for mutations. Results showed no mutations in the gene. However, 6 heterozygous polymorphisms were detected throughout the gene, which all showed a ratio of 2:1 for each to these polymorphisms suggesting the presence of three alleles, in accordance with the array CGH and SNP analysis results.
Cells derived from all patients were analyzed to measure the level of SSADH enzyme activity. Results show increased SSADH activity for all three (Table I), which was associated with normal urinary levels of GHB (Table II) and increased levels of urinary SSA (Table III).
Table I.
Succinic semialdehyde dehydrogenase activity in patient lymphocytes. Normal levels range from 278–598 pmol/min/mg protein. Each patient has increased activity.
| Patient | SSADH activity (pmol/min/mg protein) |
|---|---|
| 1 | 1110 |
| 2 | 913 |
| 3 | 634 |
Table II.
Urine concentrations of GHB in patient samples. Normal levels range from 0.09–5.0 mmol/mol creat. Each patient has normal levels of GHB.
| Patient | GHB levels (mmol/mol creat) |
|---|---|
| 1 | 0.42 |
| 2 | 0.28 |
| 3 | 0.29 |
Table III.
Urine concentrations of SSA in patient samples. Normal levels range from 0.2–0.6 mmol/mol creat. Each patient has increased levels of SSA.
| Patient | SSA levels (mmol/mol creat) |
|---|---|
| 1 | 2.06 |
| 2 | 0.99 |
| 3 | 0.80 |
DISCUSSION
We believe that the most important finding in this family is the duplication (6)(p22.2). There are reports of an interstitial 6p deletion syndrome with a 2.2 Mb critical region at (6)(p22.3). The common clinical phenotype of this syndrome includes developmental delay, brain-, heart-, and kidney defects, eye abnormalities, short neck, craniofacial malformations, hypotonia, and clinodactyly or syndactyly (Davies et al., 1999; Zirn et al., 2008; Bremer et al., 2009). The duplication at (6)(p22.2) in our patients lies outside this critical region and no deletions have been described in this area. The (6)(p22.2) duplication covers ten genes, among those DCDC2 and KIAA0319 that have been strongly linked to developmental dyslexia, and TTRAP and ACOT13 are suggested as candidate genes for this trait (Cope et al., 2005; Luciano et al., 2007).
Since mild developmental delay (without clear mental retardation) was present in all family members, especially in relation to non-verbal tasks and vision-spatial conceptualization (a phenotype suggestive of SSADH deficiency, although less severe), we hypothesized that the strongest candidate gene in the duplicated area was ALDH5A1, encoding the GABA degradative enzyme SSADH (Figure 1). Loss of SSADH enzyme function leads to elevations both of GABA (an inhibitory neurotransmitter) and GHB (with its own neuromodulatory properties) in physiological fluids, and both species are believed to contribute to various aspects of the neurological phenotype observed in patients (Pearl et al 2009). The clinical manifestations of SSADH deficiency vary from mild to severe neurological symptomatology, often related to the residual level of functional enzyme activity. In general, the most severe form of the disease associates with completely absent SSADH activity. It has however been suggested that some residual enzyme function activity (i.e. haploinsufficiency) might cause milder symptoms (Dervent et al 2004). The mild clinical presentation in all family members suggested that we might detect haploinsufficiency or very mild mutations in the SSADH gene, which turned out not to be the case.
Previous reports have documented hyperactive enzyme function associated with human chromosomal duplications, for example in acid phosphatase, beta-glucururonidase and HK1 activities (Larson et al 1982; Danesino et al 1981; Dallapiccola et al 1979). SSADH, however, is unique in catalyzing both GABA degradation and the oxidation of 4-hydroxynonenal (4-HNE), an carbonyl product of lipid peroxidation formed during oxidant stress (Murphy et al 2003). Lipids, abundant in neural tissue (e.g., ceramides, cerebrosides, etc) are sensitive to reactive oxygen species (ROS) damage (Luczaj et al 2010), and these products (e.g., 4-HNE, malondialdehyde) may initiate additional downstream damage to various biomacromolecules. We, and others, have demonstrated increased SSA, itself a carbonyl, in brain tissue from SSADH-deficient mice that associates with significantly decreased glutathione levels (Gibson et al 2006; Sauer et al 2007). These data suggest ongoing oxidative damage in the murine model, and presumably in patients, and the detection of increased urinary SSA levels in our patients (Table III) is consistent with a state of oxidant stress. Moreover, Kim et al (2009) has shown that SSADH is sensitive to the intracellular redox potential, excluding SSA from the enzyme active site under oxidizing conditions and allowing entry when the environment is more reduced. Accordingly, even hyperexpressed SSADH activity (due to gene duplication) in an oxidized environment may result in increased SSA, but it remains paradoxical that there was no associated increase in GHB. However, the enzymes involved in SSA metabolism (SSADH, and aldo-keto reductase 7a2 (AKR7a2), generating GHB from SSA) are expressed in different compartments, the former in mitochondria and the latter cytosolic. Thus, either different pools of SSA exist in these cellular compartments or there is a shuttle system between compartments for SSA that remains to be identified. Measuring GHB, GABA and 4-HNE concentrations in other body fluids of our patients might shed further light on the outcome of increased SSADH activity, but such samples were unavailable from our patients.
It remains to be determined whether some of the genes in the (6)(p22.2) duplication we have identified manifest a dosage effect and are sensitive to overproduction. Nonetheless, it would seem likely that the dup(6)(p22.2) is somehow clinically relevant to the etiology of mild developmental delay in this family, an observation which can only be further examined in additional families and patients with this duplication.
Acknowledgments
We thank the family for participating in this study. This study was kindly funded by Rinnekoti research foundation (LS), Medicinska Understödsföreningen Liv och hälsa rf (LS), Nylands Nation (LS), the State Appropriations of Helsinki and Uusimaa Hospital district (LS and SK), and NIH NS40270 and HD58553 (KMG)
References
- Akaboshi S, Hogema BM, Novelletto A, Malaspina P, Salomons GS, Maropoulos GD, Jakobs C, Grompe M, Gibson KM. Mutational spectrum of the succinate semialdehyde dehydrogenase (ALDH5A1) gene and functional analysis of 27 novel disease-causing mutations in patients with SSADH deficiency. Hum Mutat. 2003;22:442–450. doi: 10.1002/humu.10288. [DOI] [PubMed] [Google Scholar]
- Bremer A, Schoumans J, Nordenskjold M, Anderlid BM, Giacobini M. An interstitial deletion of 7.1Mb in chromosome band 6p22.3 associated with developmental delay and dysmorphic features including heart defects, short neck, and eye abnormalities. Eur J Med Genet. 2009;52:358–362. doi: 10.1016/j.ejmg.2009.06.002. [DOI] [PubMed] [Google Scholar]
- Bruno D, Anderlid BM, Lindstrand A, van Ravenswaaij-Arts C, Ganesamoorthy D, Lundin J, Martin CL, Douglas J, Nowak C, Adam M, Kooy R, Van der Aa N, Reyniers E, Vandeweyer G, Stolte-Dijkstra I, Dijkhuizen T, Yeung A, Delatycki M, Borgström B, Thelin L, Cardoso C, van Bon B, Pfundt R, de Vries B, Wallin A, Amor D, James P, Slater H, Schoumans J. Further molecular and clinical delineation of co-locating 17p13.3 microdeletions and microduplications that show distinctive phenotypes. J Med Genet. 2010;47:299–311. doi: 10.1136/jmg.2009.069906. [DOI] [PubMed] [Google Scholar]
- Chapman DA, Scott KG, Stanton-Chapman TL. Public health approach to the study of mental retardation. Am J Ment Retard. 2008;113:102–116. doi: 10.1352/0895-8017(2008)113[102:PHATTS]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Cope N, Harold D, Hill G, Moskvina V, Stevenson J, Holmans P, Owen MJ, O’Donovan MC, Williams J. Strong evidence that KIAA0319 on chromosome 6p is a susceptibility gene for developmental dyslexia. Am J Hum Genet. 2005;76:581–591. doi: 10.1086/429131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallapiccola B, Chessa L, Vignetti P, Ferrante E, Gandini E. Increased HK1 activity levels in the red cells of a patient with a de novo trisomy 10p: t(Y;10)(p11; p12) Hum Genet. 1979;50:45–9. doi: 10.1007/BF00295588. [DOI] [PubMed] [Google Scholar]
- Danesino C, Gimelli G, Cuoco C, Ciccone MO. Triplex gene dosage effect of beta-glucuronidase and possible assignment to band q22 in a partial duplication 7q. Hum Genet. 1981;56:371–3. doi: 10.1007/BF00274695. [DOI] [PubMed] [Google Scholar]
- Davies AF, Mirza G, Sekhon G, Turnpenny P, Leroy F, Speleman F, Law C, van Regemorter N, Vamos E, Flinter F, Ragoussis J. Delineation of two distinct 6p deletion syndromes. Hum Genet. 1999;104:64–72. doi: 10.1007/s004390050911. [DOI] [PubMed] [Google Scholar]
- Dervent A, Gibson KM, Pearl PL, Salomons GS, Jakobs C, Yalcinkaya C. Photosensitive absence epilepsy with myoclonias and heterozygosity for succinic semialdehyde dehydrogenase (SSADH) deficiency. Clin Neurophysiol. 2004;115:1417–22. doi: 10.1016/j.clinph.2004.01.002. [DOI] [PubMed] [Google Scholar]
- Gibson KM, Gupta M, Pearl PL, Tuchman M, Vezina LG, Snead OC, 3rd, Smit LM, Jakobs C. Significant behavioral disturbances in succinic semialdehyde dehydrogenase (SSADH) deficiency (gamma-hydroxybutyric aciduria) Biol Psychiatry. 2003;54:763–768. doi: 10.1016/s0006-3223(03)00113-6. [DOI] [PubMed] [Google Scholar]
- Gibson KM, Gupta M, Senephansiri H, Jansen EEW, Montine TJ, Hyland K, Switzer RC, Snead OC, Jakobs C. Diseases of Neurotransmission, from bench to bed. In: Hoffmann GF, editor. Symposia Proceedings. SPS Verlagsgesellschaft mbH; Heilbronn, Germany: 2006. [Google Scholar]
- Gibson KM, Lee CF, Chambliss KL, Kamali V, Francois B, Jaeken J, Jakobs C. 4-hydroxybutyric aciduria: application of a fluorometric assay to the determination of succinic semialdehyde dehydrogenase activity in extracts of cultured human lymphoblasts. Clin Chim Acta. 1991;196:219–221. doi: 10.1016/0009-8981(91)90076-o. [DOI] [PubMed] [Google Scholar]
- Jakobs C, Bojasch M, Monch E, Rating D, Siemes H, Hanefeld F. Urinary excretion of gamma-hydroxybutyric acid in a patient with neurological abnormalities. the probability of a new inborn error of metabolism. Clin Chim Acta. 1981;111:169–178. doi: 10.1016/0009-8981(81)90184-4. [DOI] [PubMed] [Google Scholar]
- Katz G, Lazcano-Ponce E. Intellectual disability: Definition, etiological factors, classification, diagnosis, treatment and prognosis. Salud Publica Mex. 2008;50(Suppl 2):s132–41. doi: 10.1590/s0036-36342008000800005. [DOI] [PubMed] [Google Scholar]
- Kim YG, Lee S, Kwon OS, Park SY, Lee SJ, Park BJ, Kim KJ. Redox-switch modulation of human SSADH by dynamic catalytic loop. EMBO J. 2009;28:959–68. doi: 10.1038/emboj.2009.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson LM, Bruce AW, Saumur JH, Wasdahl WA. Further evidence by gene dosage for the regional assignment of erythrocyte acid phosphatase (ACP1) and malate dehydrogenase (MDH1) loci on chromosome 2p. Clin Genet. 1982;22:220–5. doi: 10.1111/j.1399-0004.1982.tb01437.x. [DOI] [PubMed] [Google Scholar]
- Luciano M, Lind PA, Duffy DL, Castles A, Wright MJ, Montgomery GW, Martin NG, Bates TC. A haplotype spanning KIAA0319 and TTRAP is associated with normal variation in reading and spelling ability. Biol Psychiatry. 2007;62:811–817. doi: 10.1016/j.biopsych.2007.03.007. [DOI] [PubMed] [Google Scholar]
- Luczaj W, Moniuszko A, Rusak M, Pancewicz S, Zajkowska J, Skrzydlewska E. Lipid peroxidation products as potential bioindicators of Lyme arthritis. Eur J Clin Microbiol Infect Dis. 2011;30:415–422. doi: 10.1007/s10096-010-1102-0. [DOI] [PubMed] [Google Scholar]
- Murphy TC, Amarnath V, Gibson KM, Picklo MJ., Sr Oxidation of 4-hydroxy-2-nonenal by succinic semialdehyde dehydrogenase (ALDH5A) J Neurochem. 2003;86:298–305. doi: 10.1046/j.1471-4159.2003.01839.x. [DOI] [PubMed] [Google Scholar]
- Pearl PL, Gibson KM, Cortez MA, Wu Y, Carter Snead O, 3rd, Knerr I, Forester K, Pettiford JM, Jakobs C, Theodore WH. Succinic semialdehyde dehydrogenase deficiency: Lessons from mice and men. J Inherit Metab Dis. 2009;32:343–352. doi: 10.1007/s10545-009-1034-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer SW, Kölker S, Hoffmann GF, Ten Brink HJ, Jakobs C, Gibson KM, Okun JG. Enzymatic and metabolic evidence for a region specific mitochondrial dysfunction in brains of murine succinic semialdehyde dehydrogenase deficiency (Aldh5a1−/− mice) Neurochem Int. 2007 Mar;50(4):653–9. doi: 10.1016/j.neuint.2006.12.009. Epub 2007 Jan 13. [DOI] [PubMed] [Google Scholar]
- Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002 Jun 15;30(12):e57. doi: 10.1093/nar/gnf056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siggberg L, Ala-Mello S, Jaakkola E, Kuusinen E, Shcuit R, Kohlhase-Brugues J, Böhm D, Ignatius J, Knuutila S. Array CGH in clinical diagnostics of mental retardation – A study of 150 finnish patients. Am J Med Genet Part A. 2010;152A:1398–1410. doi: 10.1002/ajmg.a.33402. [DOI] [PubMed] [Google Scholar]
- World Health Organization . WHO ICD-10, 10th revision. chapter V. 2007. International Statistical Classification of Diseases and related health problems; pp. F70–F79. [Google Scholar]
- Zirn B, Hempel M, Hahn A, Neubauer B, Wagenstaller J, Rivera-Brugues N, Strom TM, Kohler A. Polyneuropathy, scoliosis, tall stature, and oligodontia represent novel features of the interstitial 6p deletion phenotype. Am J Med Genet A. 2008;146A:2960–2965. doi: 10.1002/ajmg.a.32536. [DOI] [PubMed] [Google Scholar]