

Abstract
The fatal neurodegenerative disorder Alzheimer’s disease (AD) has been linked to the formation of soluble neurotoxic oligomers of amyloid–β (Aβ) peptides. These peptides have high affinities for copper cations. Despite their potential importance in AD neurodegeneration few studies have focused on probing the Cu2+/1+ coordination environment within Aβ oligomers. Herein we present a Cu K-edge X-ray absorption spectroscopic study probing the copper–coordination environment within oligomers of Aβ(42) (sequence: DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVVIA). We find that the Cu2+ cation is contained within a square planar mixed N/O ligand environment within Aβ(42) oligomers, which is similar to the copper coordination environment of the monomeric forms of {CuIIAβ(40)} and {CuIIAβ(16)}. Reduction of the Cu2+ cation within the Aβ(42) oligomers to Cu1+ yields a highly dioxygen sensitive copper–species that contains Cu1+ in a tetrahedral coordination geometry. This can be contrasted with monomers of {CuIAβ(40)} and {CuIAβ(16)}, which contain copper in a dioxygen inert linear bis-histidine ligand environment [Shearer and Szalai, J. Am. Chem. Soc., 2008, 130, 17826]. The biological implications of these findings are discussed.
Alzheimer’s disease (AD) is one of the leading causes of senile dementia. It has been estimated that over the next two decades the number of AD patients will double leading to a major health crisis in the developed world.1 Furthermore, AD is not merely a disease of the elderly as a small but significant number of patients acquire early on-set AD before the age of 60.2 Key to the development of AD is the formation of small peptides, amyloid–β (Aβ) peptides, from the processing of the neuronal membrane amyloid precursor protein (APP). These Aβ peptides are the key constituent of insoluble extracellular plaques, which are found postmortem in the majority of AD victims. Variable processing of the APP produces Aβ peptides of different lengths; the 40 and 42 residue long peptides (Aβ(40) and Aβ(42)) are the largest constituents of extracellular plaques. Despite the prevalence of extracellular plaques in AD victims it is the oligomers of Aβ, which are the precursors of plaques, that have been identified as the most potent neurotoxins in AD. There is a high correlation between dementia and the presence of these oligomers.3 Both Aβ monomers, oligomers, and plaques have a high affinity for redox–active transition metal cations. For example, Cu2+ concentrations within extracellular plaques have been found in the high micromolar range,4 while Cu2+/1+ Kd values from Aβ monomers have been reported in the femtomolar range.5,6 Thus, it has been speculated that redox–active copper cations may be involved in AD etiology via the initiation of oxidative stress.7 To gain insight into the nature of the copper coordination environment within Aβ oligomers we present X–ray absorption and EPR spectra probing the coordination environment of Cu2+/1+ within Aβ(42) oligomers (Aβ(42) sequence: DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVVIA).
SDS solubilized oligomers of Aβ(42) were prepared at pH = 7.2 according to the methods of Yu et al.8 Three different treatments were considered: oligomers without Cu2+ added, oligomers with one equivalent of Cu2+ per Aβ(42) added prior to oligomer formation, and oligomers with one equivalent of Cu2+ per Aβ(42) added after oligomer formation. SDS–PAGE analysis and Thioflavin T dye-binding assays9 indicate that the addition of Cu2+ either before or after formation of the oligomers does not disrupt their formation (supporting information). We note that a small amount of flocculant solid forms over time, suggesting the onset of fibril formation. TEM studies to characterize this solid are ongoing.
X–band EPR spectroscopy was first used to investigate these Cu2+ loaded Aβ(42) oligomers (with copper added both before and after incubation) (Figure 1). These spectra were compared to the EPR spectrum of {CuIIAβ(40)},10 which contains all of the copper coordinating residues within Aβ. We find that the three EPR spectra are nearly identical with g∥ ~ 2.274 and A∥ ~ 164 ± 4 G in the oligomer samples. The major difference is that the oligomeric Cu2+ loaded Aβ(42) spectra have hyperfine splitting that is broadened relative to {CuIIAβ(40)}. Overall these data suggest a similar square–planar (N/O)4 Cu2+ coordination environment within the monomers vs. oligomers.10,11
Fig. 1.
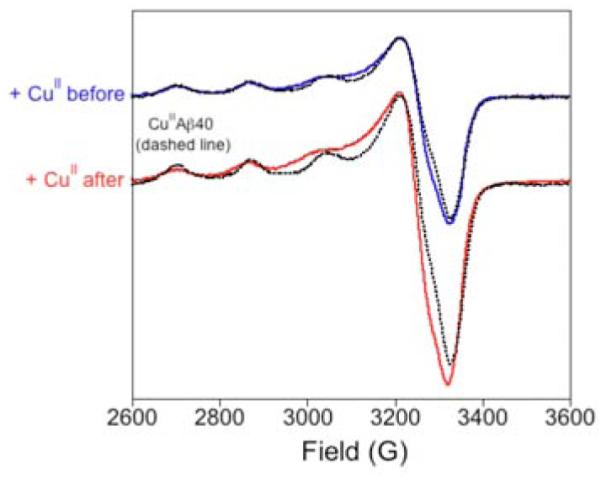
X–band EPR spectra of Cu2+ loaded Aβ(42) oligomers and {CuIIAβ(40)}.
These Cu2+ loaded Aβ(42) oligomers were subsequently investigated by copper K-edge X-ray absorption spectroscopy. The XANES region of the copper K-edge spectrum (Figure 2) was nearly identical to that observed for the {CuIIAβ(16)} and {CuIIAβ(40)} monomers previously reported,10 and is consistent with square-planar Cu2+. The only significant difference in the XANES of the monomers vs. the oligomer is that an edge feature previously observed in the monomer spectrum at ~8986 eV is blurred into the edge of the oligomer spectrum. This may suggest a slightly different coordination environment for Cu2+ within the Aβ(42) oligomers relative to the monomers. It was also noted that, unlike the monomers, the Cu2+ cation within the Aβ(42) oligomers was not prone to photoreduction in the X-ray beam even after prolonged exposure to the beam.10
Fig. 2.
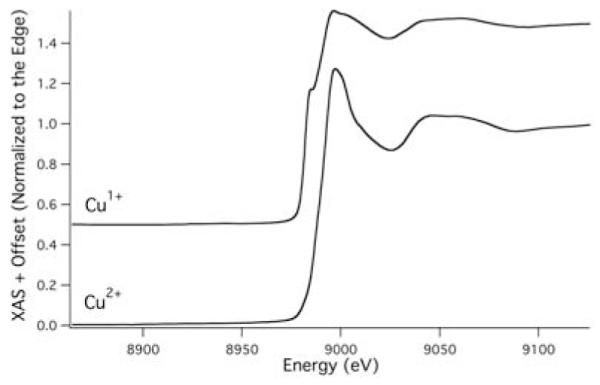
XANES region of the Cu K-edge spectrum of copper loaded Aβ(42) oligomers before (Cu2+) and after (Cu1+) first ascorbate reduction.
The EXAFS region of the copper K-edge spectrum for Cu2+ loaded Aβ(42) oligomers (Figure 3) was best modeled with Cu2+ in a four coordinate mixed N/O ligand environment. Based on a multiple scattering analysis, a best fit to the EXAFS data was obtained using three imidazole donors (from histidine residues) and one additional nitrogen or oxygen ligand per Cu2+ cation (Figure 4). Two of these imidazole donors are included in one shell, while the third was significantly different and was included in a different shell. Although consistent with an EXAFS analysis of monomers of {CuIIAβ(40)} by Stellato et al.,12 this 3-imidazole 1-N/O donor copper–coordination environment is different than our previous analysis of both {CuIIAβ(16)} and {CuIIAβ(40)} monomers.10 In that study we obtained a best fit to the EXAFS data for Cu2+ containing Aβ monomers using only two histidine imidazole donors and two additional N/O ligands. That 2 N/O 2 imidazole solution was consistent with our own and others’ previous work on Cu2+ containing Aβ monomers.11,13,14 Although a 2 imidazole 2 N/O solution to the EXAFS data for the Cu2+ loaded Aβ(42) oligomers could be located, this yielded a significant decrease in the quality of the fits to the experimental data, and yielded unrealistic Debye-Waller values for the shorter imidazole shell (see supporting information). We therefore excluded this as a valid model for the copper coordination environment for the oligomers.
Fig. 3.
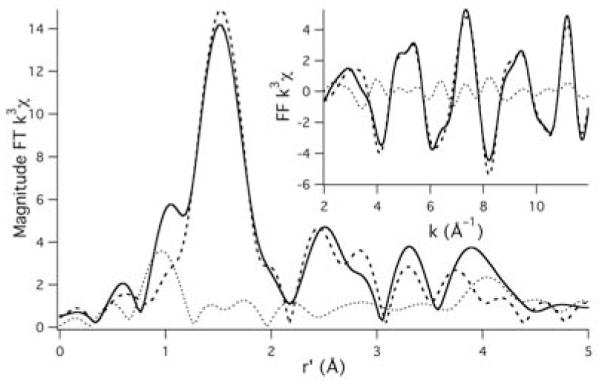
Magnitude FT k3 EXAFS spectrum for Cu2+ loaded Aβ(42) oligomers. The solid spectrum is the experimental data, the thick dashed spectrum is the best fit to the experimental data, and the thin dotted line is the difference spectrum. The inset depicts the FF k3 EXAFS spectrum (FT from 2.0 to 12.0Å−1; backtransformed from 1.0 to 4.0 Å). Best fits to the data: N–shell (n = 1.0; r = 1.939(4)Å; σ2 = 0.005(2)Å2); 1st imidizole–shell (n = 2.0; r = 1.96(2)Å; σ2 = 0.007(3)Å2; θ = 3(2)°; ψ = 48(2)°); 2nd imidizole–shell (n = 1.0; r = 2.03(2)Å; σ2 = 0.004(2)Å2; θ = 27(3)°; ψ = 51(11)°); Eo = 8985.2 eV; GOF = 0.67.
Fig. 4.
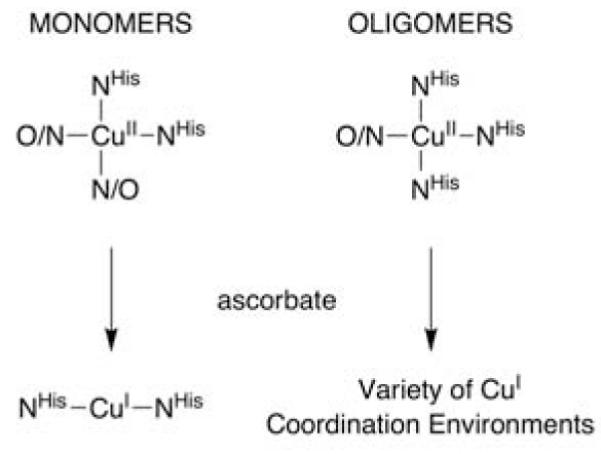
Copper coordination motifs within Aβ monomers and oligomers.
Reduction of the Cu2+ centers in copper-loaded Aβ(42) oligomers was effected by the addition of two equivalents of ascorbate to the XAS sample under anaerobic conditions. The XANES spectrum of the resulting species was consistent with the reduction of the copper–center to the +1 oxidation state as signified by the −2.1(2) eV shift in the energy of the edge (Figure 2).15 The edge region of the XANES for reduced copper–loaded Aβ(42) oligomers is most consistent with a 4-coordinate (i.e. tetrahedral) Cu1+ center.15 This is in stark contrast to the XANES spectrum of both {CuIAβ(16)} and {CuIAβ(40)} monomers, which is consistent with a linear 2-ccordinate Cu1+ coordination environment (Figure 4).10 Thus, on the basis of the XANES spectra alone it is apparent that there is a dramatic difference in Cu1+ coordination environments within Aβ monomers vs. Aβ(42) oligomers. This was confirmed by a examination of the EXAFS region of the reduced copper–loaded Aβ(42) oligomers. An analysis of the EXAFS region yielded only nonsensical refinements, and is most easily rationalized by considering that Cu1+ is contained in a mixture of coordination motifs.
The time it takes for the reoxidation of Cu1+-loaded Aβ(42) oligomers is consistent with tetrahedral Cu1+ as well. Unlike both {CuIAβ(16)} and {CuIAβ(40)} monomers, which require 16+ hours of air oxidation to achieve reoxidation of the copper–center back to Cu2+,10 the reoxidation of the reduced copper-loaded Aβ(42) oligomers by air was complete upon warming of the XAS sample. The XANES and EXAFS spectra of the resulting reoxidized copper-loaded Aβ(42) oligomers were nearly identical to what was observed prior to oxidation (supporting information). In contrast the XANES spectrum of the re-reduced copper-loaded Aβ(42) oligomer, although consistent with tetrahedral Cu1+, is considerably different than that observed upon reduction by ascorbate the first time. Thus, it seems reasonable to suggest that a variety of structurally ill-defined redox active Cu1+ coordination motifs are found within Cu1+ containing Aβ(42) oligomers.
One of us has shown that Cu1+ is the thermodynamically preferred oxidation state of copper within Aβ monomers.6 Also, in a recent study concerning the structure of Cu1+ co-ordinated monomers of Aβ(40) and Aβ(16) we noted that the sluggish O2 reactivity of these copper adducts would preclude significant oxidative damage resulting from such species. These two findings are both at odds with the supposition that redox active copper coordinated to Aβ peptides are responsible for the oxidative damage observed in AD patients.7 This study, in contrast, demonstrates that copper coordination within Aβ oligomers does produce a highly O2 reactive Cu1+ center. If such a situation occurs in vivo then it is likely that significant oxidative damage could result from the rapid redox cycling of copper in the O2 rich reducing environment of the extracellular fluid. We are currently investigating the properties of these copper–loaded oligomers and how they pertain to AD processes.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (J.S.: P20 RR-016464) and the Alzheimers Association (V.A.S.: IIRG-07-5821). XAS studies were supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-98CH10886.
Footnotes
Electronic Supplementary Information (ESI) available: Experimental procedures for the production of SDS stabilized amyloid-β oligomers, spectroscopic experimental procedure, alternate fits to the EXAFS data for the Cu2+ loaded Aβ(42) oligomers and additional spectra. See DOI: 10.1039/b000000x/
References
- 1.Ferri CP, Prince M, Brayne C, Brodaty H, Fratiglioni L, Ganguli M, Hall K, Hasegawa K, Hendrie H, Huang Y, Jorm A, Mathers C, Menezes PR, Rimmer E, Scazufca M. Lancet. 2005;35:352. doi: 10.1016/S0140-6736(05)67889-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Raman A, Lin X, Suri M, Hewitt M, Constantinescu CS, Phillips MF. J. Neuro. Sci. 2007;260:78. doi: 10.1016/j.jns.2007.04.013. [DOI] [PubMed] [Google Scholar]
- 3.Ono K, Condron MM, Teplow DB. Proc. Natl. Acad. Sci. USA. 2009;106:14745. doi: 10.1073/pnas.0905127106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lovell MA, Robertson JD, Teesdale WJ, Campbell JL, Markesbery WR. J. Neurol. Sci. 1998;38:7609. doi: 10.1016/s0022-510x(98)00092-6. [DOI] [PubMed] [Google Scholar]
- 5.Faller P, Hureau C. Dalton Trans. 2009:1080. doi: 10.1039/b813398k. [DOI] [PubMed] [Google Scholar]
- 6.Feaga HA, Maduka RC, Foster M, Szalai VA. in revision. [DOI] [PubMed]
- 7.Hung YH, Bush AI, Cherny RA. J. Biol. Inorg. Chem. 2010;15:61. doi: 10.1007/s00775-009-0600-y. [DOI] [PubMed] [Google Scholar]
- 8.Yu L, Edalji R, Harlan JE, Holzman TF, Lopez AP, Labkovsky B, Hillen H, Barghorn S, Ebert U, Richardson PL, Miesbauer L, Solomon L, Bartley D, Walter K, Johnson RW, Hajduk PJ, Olejniczak ET. Biochemistry. 2009;48:1870. doi: 10.1021/bi802046n. [DOI] [PubMed] [Google Scholar]
- 9.Barghorn S, Nimmrich V, Striebinger A, Krantz C, Keller P, Janson B, Bahr M, Schmidt M, Bitner RS, Harlan J, Barlow E, Ebert U, Hillen H. J. Neurochem. 2005;95:834. doi: 10.1111/j.1471-4159.2005.03407.x. [DOI] [PubMed] [Google Scholar]
- 10.Shearer J, Szalai VA. J. Am. Chem. Soc. 2008;130:17826. doi: 10.1021/ja805940m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Drew SC, Noble CJ, Masters CL, Hanson GR, Barnham KJ. J. Am. Chem. Soc. 2009;131:1195. doi: 10.1021/ja808073b. [DOI] [PubMed] [Google Scholar]
- 12.Stellato F, Menestrina G, Serra MD, Potrich C, Tomazzolli R, Meyer-Klaucke W, Morante S. Eur. Biophys. J. 2006:340. doi: 10.1007/s00249-005-0041-7. [DOI] [PubMed] [Google Scholar]
- 13.Karr JW, Akintoye H, Kaupp LJ, Szalai VA. Biochemistry. 2005;44:5478. doi: 10.1021/bi047611e. [DOI] [PubMed] [Google Scholar]
- 14.Kowalik-Jankowska T, Ruta M, Wisniewska K, Lankiewica L. J. Inorg. Biochem. 2003;95:270. doi: 10.1016/s0162-0134(03)00128-4. [DOI] [PubMed] [Google Scholar]
- 15.Kau L-S, Spira-Solomon DJ, Penner-Hahn JE, Hodgson KO, Solomon EI. J. Am. Chem. Soc. 1987;109:6433. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.