

Abstract
Problem
Dendritic cell (DC)-based cancer therapies are favored approaches to stimulate anti-tumor T cells responses. Unfortunately, tolerance to tumor antigens is difficult to overcome. Biodegradable poly(lactic-co-glycolic acid) (PLGA) nanoparticles (NP) are effective reagents in the delivery of drugs and tumor-associated antigens (TAA). In this study, we assessed the capacity of a PLGA NP-based delivery system to augment CD8 T cell responses to ovarian cancer TAA.
Method of Study
Human DC were generated from blood monocytes by conventional in vitro differentiation and loaded with either soluble tumor lysate or NP/lysate conjugates (NPL). These antigen-loaded DC were then used to stimulate autologous CD8+ T cells. Cytokine production and activation markers were evaluated in the CD8+T cells.
Results
DC loading with NPL increased cytokine production by stimulated CD8 T cells and induced T cell expression of cell surface co-stimulatory molecules, typical of anti-tumor immune responses. In contrast, delivery of naked tumor lysate antigens preferentially induced a T cell profile characteristic of tolerization/exhaustion.
Conclusion
These findings indicate that delivery of TAA in NP enables DC to efficiently activate anti-tumor CD8+ T cells. PLGA NP encapsulation of tumor-derived lysate protein antigens is an encouraging new preparative methodology for DC-based vaccination meriting clinical testing.
Introduction
Ovarian carcinoma is the fifth most common malignancy affecting women in the United States and remains the cancer with the highest mortality rate among gynecological tumors. The USA incidence of ovarian carcinoma is 22,000, leading to more than 15,000 deaths annually1. This poor prognosis results largely from late diagnosis of occult malignancy, with two-thirds of patients already having advanced disease at time of presentation 2-4. Despite response rates, even in advanced ovarian carcinoma, of 73–77% to first-line therapy with platinum and Taxol, median progression-free interval is only 16–18 months and median survival only 35–38 months 5.
The appeal of novel immunological approaches is the hope that they may amplify the natural response to the tumor cells and thereby impede or prevent clinical relapse, with resulting survival benefit. Overcoming existing tumor tolerance to “tumor stem cell populations” has been suggested to be key to recurrence and chemotherapy resistance 6. Since tumor antigens are often weakly immunogenic, improved antigen (Ag) delivery would likely be advantageous for dendritic cell (DC)-based immunotherapy of solid tumors 7, including ovarian cancer, for which only a limited number of general tumor-associated antigens (TAA) have been identified 8-10.
Since an array of patient-specific antigens may be accessed through autologous tumor lysates, DC vaccines incorporating efficient presentation of antigens extracted directly from the relevant malignant cells may represent a novel approach with promise in ovarian cancer 10-12. DC are professional antigen-presenting cells (APC), which play a role in the initiation and regulation of immune responses and are central regulators of tolerance and immunity. Generation of DC vaccines is dependent on isolation of DC populations and loading them with tumor antigens, usually from lysates, peptides, exosomes, or apoptotic tumor cells 13.
While the use of tumor antigen-loaded DC to generate immune responses has been demonstrated in clinical trials in melanoma and other solid tumors, there has been little clinical evidence for the success of this strategy in ovarian cancer, although in vitro data has offered encouragement14. Hernando et al. demonstrated responses in two of six patients with ovarian cancer treated with DC pulsed with tumor antigens from apoptotic tumor cells15. One of their patients developed a tumor specific Th1 T cell response, as assessed by IFN-γ secretion. Schlienger et al., after pulsing DC with killed autologous primary ovarian tumor cells and then maturing them with CD40 ligand and TRANCE (tumor necrosis factor-related activation-induced cytokine), and identified induction of antigen-specific T cells that secreted IFN-γ upon stimulation with autologous tumor cells16. In other studies, DC pulsed with acid-eluted peptides or whole cell lysates from ovarian cancer cells induced HLA class I-restricted CTL responses against autologous ovarian tumor cells 17-19. MUC1 and HER2/neu peptide-pulsed DC vaccination in patients with advanced ovarian or breast cancer, while not generating clinical responses, did stimulate peptide-specific cytotoxic T cell responses in five of 10 patients 20. Collectively, these reports suggest that, at least in the laboratory, anti-ovarian cancer T cell responses can be augmented.
To translate in vitro evidence of anti-tumor T cell responses into clinical efficacy, loading of DC with either endogenous (DC-derived) or exogenous (DC-loaded) tumor Ag has been attempted. Endogenous Ag production within the DC has important practical limitations. Electrofusion of DC and tumor cells is laborious and time consuming21. Transduction with DNA or mRNA via viral vectors is complicated by interference with the Ag presentation machinery of DC and generation of anti-viral antibodies22.
Exogenous Ag delivery introduces other issues. External pulsing of DC by defined peptides results in only brief stimulation of T cells, because of rapid turnover of class I-peptide complexes on the cell surface. In addition, this approach can only be used in patients displaying those major histocompatibility complexes 23-27.
Tumor lysates are potentially complex enough to overcome the ability of tumors to down-regulate targeted antigens, an effect previously associated with a variety of tumor escape mechanisms 28-30. However, the use of soluble tumor lysates is confounded by the inherent instability of soluble macromolecules, poor internalization of soluble materials by DC, and inefficient cross-presentation to cytotoxic T cells (CTL) 29, 31-33.
Particulate delivery systems made from biocompatible, degradable polymers, such as poly(lactic-co-glycolic acid) (PLGA), may overcome many of the problems encountered during transfer of Ag to DC 34, 35. Encapsulation in nano/microparticles can protect Ag from degradation by proteolytic enzymes 35, 36, increase the efficiency of Ag loading 23, 25, 27 and prolong and enhance Ag release, leading to more efficient presentation of MHC-peptide complexes 27, 34, 37. Encapsulation is also associated with an improved shelf life of Ag and reduced need for adjuvants, and PLGA nanoparicles (NP) have recently been shown to be robust activators of the Nalp3 inflammasome in DC and thus capable of generating potent humoral and cellular immune responses in vivo 38, 39. PLGA NP can also target Ag delivery to critical immune cells associated with anti-tumor T cell responses 40. For all of these reasons, NP are promising reagents for the delivery of tumor-associated Ags (TAA) for DC-based cancer vaccines 41, 42. However, while peptides or single antigens encapsulated within PLGA preparations have been shown to induce specific CTL responses 43, 44, only a few studies have examined encapsulation of complex cellular material, including tumor-derived proteins 45, 46.
We sought to determine whether PLGA NP encapsulation of whole ovarian tumor cell lysates could represent an improved method for efficient delivery of cancer-derived Ags to DC. We report that this approach stimulated a distinctive Th1/inflammatory pattern of cytokine production by autologous CD8+ T cells in classic stimulation assays. Our findings suggest that NP-mediated Ag delivery can significantly improve the ability of DC to cross-present Ag to reactive CD8+ T cells. Of special interest, this method of antigen loading empowers DC to generate in vitro anti-tumor CD8+ responses similar to those presumed necessary to overcome the existing tumor tolerance associated with ovarian cancer recurrence and metastasis, providing encouragement for potential clinical application.
Materials and Methods
Antibodies
PD-1 APC, CD4 APC-H7, and Streptavidin APC were all purchased from BD Biosciences (San Diego, CA). Biotinylated ICOS and PE CD152 (CTLA-4) were purchased from EBioscience (San Diego, CA). CD8 Percp was from Biolegend (San Diego, CA). Antibodies used for dendritic cell identification were PE CD14 and PE-Cy5 CD86, along with corresponding isotypes were also purchased from EBiosicence. HLA-DR FITC along with isotype was purchased from Beckman Coulter (Brea, CA).
Lysate preparation
Cell pellets obtained from the epithelial ovarian cancer line TARA R182 were cultured as previously described 47, 48, washed and re-suspended in ice cold PBS at concentrations ranging from 2-4 × 108/ml and subjected to four cycles of freeze-thaw cycles (with alternating liquid nitrogen and 37 °C water bath treatment) followed by sonication on ice for three 15 second intervals (Tekmar sonicator, microprobe) on ice at 38% amplitude to disrupt the cell membrane and liberate cellular proteins. Cell death and lysis were confirmed by trypan blue exclusion and hemocytometry. Lysates were spun at 12000 × g for 20 minutes at 4°C to pellet cellular debris and the supernatants were collected and used immediately or stored at -80°C until use. Protein content of the lysate preparations was measured using the Bicinchonic acid (BCA) protein assay kit (Thermo Scientific, Rockford, IL) as per the manufacturer's protocol. Absorbance at 562 nm was measured using a spectrophotometer and protein concentrations were determined by comparing absorbance readings to a standard calibration curve of bovine serum albumin (BSA) in the concentration range between 5μg/ml and 250μg/ml.
Nanoparticle fabrication
PLGA copolymers (50% lactide: 50% glycolide) with inherent viscosity (i.v.) of 0.59dL/g (∼80 kDa) (DURECT Corp., Pelham, AL) were dissolved in methylene chloride (Fisher Scientific, Pittsburgh, PA) prior to encapsulation. Polyvinyl alcohol (0.5% PVA) (Sigma-Aldrich, St. Louis, MO) was used as a stabilizer. All reagents were of analytical grade. PLGA NP were fabricated from aqueous protein solutions using the solvent evaporation method from a water/oil/water (W2/O/W1) emulsion, as described previously 46. Control NPs (blanks) were synthesized with sterile PBS replacing the aqueous protein solutions.
DC generation and isolation of CD8+ T cells
A PBMC concentrate isolated from normal control blood was collected by apheresis at the NY Blood Bank or isolated directly from Yale volunteers by standard venipuncture in heparinized tubes following informed consent. The samples were diluted 1:1 in sterile PBS, layered over Lymphocyte Separation Medium (MP Biomedicals, Solon, Ohio), and centrifuged at 2500rpm for 25 min at RT. The cells at the interface were collected and washed twice at 1500rpm for 10min at RT with sterile Hanks' BSS. Cells were counted and viability checked by Trypan blue exclusion prior to suspension in complete RPMI (RPMI 1640 plus 10% FBS, 2% Pen/Strep and 25 mM HEPES). Cells were plated in 5.0 × 106 cells/ml in 4mls into 6-well polystyrene flat-bottom cell culture plates. Monocytes were allowed to adhere for 2hrs at 37° C in a 5% CO2 atmosphere; floating cells were isolated, washed with complete RPMI, counted, and re-suspended in 90% FBS/10% DMSO prior to storage in cryovials at -80 °C for subsequent use. Adherent monocytes were incubated with complete RPMI + 0.25 ng/ml of IL-4 and 0.8 ng/ml of GM-CSF (Peprotech, Rocky Hill, NJ) with medium changes on days 1, 3 and 5. On day 6, the cells were harvested by scraping, then washed and counted. Purity of DC population was determined by flow cytometry of loss of surface expression marker CD14, and gain of HLA-DR, and CD86. CD8+ T cells were isolated from non-adherent cells by negative selection utilizing a MACS® untouched CD8+ T cell purification system (Miltenyi Biotech, Auburn, CA), as per the manufacturer's protocol. The cells were suspended in T cell medium (AimV media + 5% human Ab serum) at 0.5 × 106 cells per ml, in preparation for co-culture with DC.
DC/T cell co-culture
The effectiveness of NP-mediated TAA delivery to DC was established by assessing CD8+ T cell stimulation after DC/T cell co-culture. DC were plated in 6-well plates at 1 × 106 /ml AimV media plus 5% human Ab serum, after which they were loaded with equimolar quantities of Ag as either soluble lysate or suspended NPs (0.5 mg of NP suspended in 300 μl media) at 0.25 mg of NP per 1 × 106 DC. After 18 hrs, DC supernatants were collected and immediately stored at -80 °C for later analysis. Cells were then collected, washed extensively, and re-plated in 96-well polystyrene flat-bottom cell culture plates at a concentration of 60,000 cells/well. Autologous CD8+ T cells were added to the wells containing DC at a 1:2 DC/T cell ratio and co-culture proceeded for 3 days and cell culture supernatants were harvested and cryopreserved for later analysis. Unstimulated CD8+ T cells alone and T cells stimulated with DC loaded with no Ag or blank PLGA NP (under identical concentrations to those containing tumor lysate) acted as negative controls; experiments were done in triplicate.
Luminex analysis of cytokine production
To determine the levels of cytokine production following Ag loading of DC, or levels following DC co-culture with autologous CD8+ T cells, cell supernatants were analyzed in multiplex format utilizing multiplex beads for IL-1β, IL-2, IL-4, IL-6, IL-8, IL-10, IL-12(p70), IFN-γ, TNF-α, GRO-α, RANTES, MCP-1, MIP-1α, MIP-1β, GM-CSF, VEGF, and G-CSF (BioRad Laboratories; Hercules, CA) 49. Briefly, wells of a 96-well filter plate were loaded with either 50μl of prepared standard solution, or 50μl of cell-free supernatant and incubated with 1× antibody-coupled beads at ± 600 rpm for 30min in the dark at room temperature. Wells were then vacuum-washed three times with 100μl wash buffer. Samples were then incubated with 25μl of 1× biotinylated detection antibody at ± 600 rpm for 30 min at room temperature in the dark. After three additional washes, 50μl of 1× streptavidinphycoerythrin was added to each well and incubated for 10 min at ± 600 rpm at room temperature in the dark. After a final three washes, the beads were resuspended in 125μl of sheath buffer for measurement with the LUMINEX 200 (LUMINEX, Austin, TX, USA). DC alone and CD8+ T-cell alone supernatant were subtracted out for background.
Flow cytometry of CD8+ T cells following DC stimulation
Immediately following DC co-culture, T cell populations were analyzed for surface expression of known co-stimulatory molecules associated with stimulatory or inhibitory signaling. mAb recognizing the surface molecules ICOS, CTLA-4 and PD-1 were co-stained with both CD8 and CD4 specific antibodies. After staining, acquisition was performed using FACSAria cell sorter (BD Bioscience) equipped with two lasers (488nm and 635). The instrument was set for 6-color analysis using Diva software and BD com beads. A minimum of 100,000 events were acquired and results were analyzed using Flowjo (Treestar).
Results
Human monocytes cultured in GM-CSF/IL-4 differentiate into phagocytic DCs capable of engulfing large numbers of PLGA NP
To confirm that monocytes isolated from normal control blood could be differentiated into DCs capable of internalizing Ag and then cross-presenting it to reactive CD8+ T cells, CD14+ monocyte cultures were exposed to GM-CSF/IL-4 and then phenotypically characterized for classical markers of DC differentiation. As shown in figure 1A, monocyte cultures exposed to DC differentiation media became increasingly large and granular during the differentiation process as accessed by forward and side scatter. A distinct DC population, virtually absent on the day of cytokine addition (Day 0), becomes evident on Day 2 and then continues to become uniformly larger and more irregular in the presence of cytokines, with increased FSC and SSC observed by Day 4 and peaking on Day 6. Consistent with the morphological changes, we observed a differentiation process to a DC phenotype characterized by changes in cell surface expression markers as determined by flow cytometry. First we evaluated the expression of CD14, a marker of monocyte cell lineage. As shown in figure 1B, a significant decrease in CD14 expression was observed in gated cells during the cell differentiation process (days 2-6). In addition, we found an increase in the number and intensity of CD86+ as well as MHC class II+ (HLA-DR) expressing cells, a phenotype expected as cells leave the monocyte lineage and differentiate into DC. Interestingly, the expression pattern of CD86 and HLA-DR follows a similar time course as other phenotypic changes, with a major increase in the double positive CD86+/HLA-DR+ cells found at day 2 and reaching a peak of more than 70% CD86+/HLA-DR+ newly formed, immature DC, at day 6 (fig. 1C).
Fig. 1.



In vitro differentiation of isolated CD14+ monocytes to immature dendritic cells (DC). CD14+ cells were treated with GM-CSF/IL-4 for 6 days, during which CD14, HLA-DR, and CD86 expression was analyzed by flow cytometry. (A) Forward and side scatter of differentiating DC was determined on days 0, 2, 4 and 6. This same population of CD14+ monocytes lose CD14 expression over time (B), while showing increased expression of HLA-DR and CD86 (C) during conversion to DC.
In order to verify the phagocytic capacity of these cells we exposed immature DC to fluorescently labeled nanoparticles (NP) of similar size, composition and number to those used for later tumor lysate loading. As shown in figure 2, a group of morphologically distinct DC have internalized varying numbers of rhodamine-labelled NP 18hrs hours post loading, indicating these cells are indeed capable of phagocytizing NP conjugates. Visualization by phase contrast microscopy (upper panel), fluorescence (middle panel) and the overlay (bottom) indicated fluorescent particles were associated with signal in multiple focal planes, verifying that these NP were internalized and not simply adherent with the cell surface.
Fig. 2.
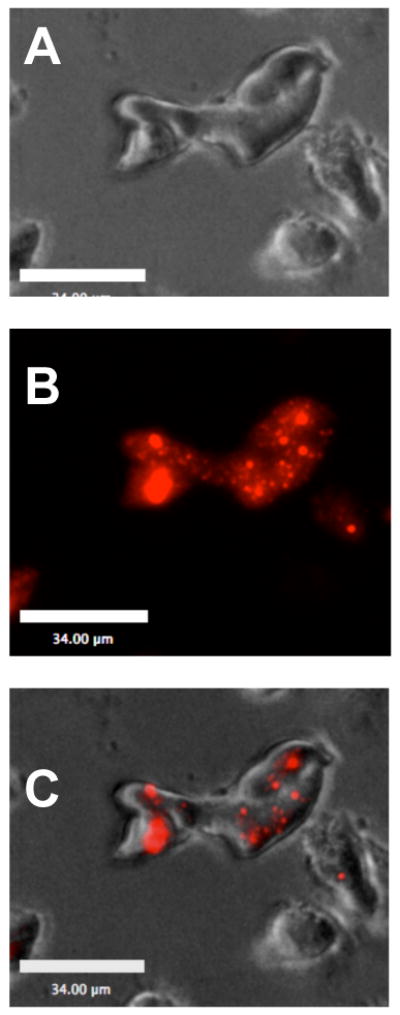
Day 6 in vitro differentiated DC avidly phagocytize PLGA nanoparticles (NP). DC were loaded with rhodamine-labeled empty NP for 18hrs at 37° C. Images were captured on Zeiss Flourescent microscope. (A) phase contrast, (B) Cy3 signal and (C) merged photo.
NP-mediated delivery of ovarian cancer cell lysates to DC leads to a fundamentally different, and inflammatory biased, cytokine production pattern when compared to delivery of lysates in their soluble form
Our next objective was to determine whether the means of delivering tumor antigens (Ag) to DC affected their processing, response and cross-presentation of tumor Ag to CD8+ T cells. Cultured day 6 immature DC were loaded for 18 hrs with equimolar quantities of total tumor protein in either soluble Ag form (soluble lysate) or lysate encapsulated in PLGA nanospheres (NPL) and the cellular supernatants collected and analyzed for differential DC cytokine expression. As controls, non-loaded DC and DC loaded with a “blank” (non-Ag containing) PLGA nanospheres were prepared in parallel to control for spontaneous DC cytokine production, and production linked only to non-specific NP stimulation, respectively. First we characterized the DC response to the different Ag delivery modalities prior to exposure to T cells. As shown in figure 3, NP-mediated delivery of tumor lysates (NPL) led to a unique DC cytokine production profile characterized by significantly higher levels of cytokines and chemokines including IL-1β, IL-6, IFNγ, TNFα, G-CSF and RANTES. Virtually none of these cytokines were measurably produced in DC incubated with either the soluble cell lysate or NP alone. In contrast, DC loaded with soluble cell lysate showed consistently higher levels of IL-10 and GM-CSF in several of the donors (data not shown). Analyzed as a whole, the differential production of these molecules by DC would strongly suggest an “inflammatory” or Th1-biased cytokine profile when the tumor lysate is delivered via nanoparticles.
Fig. 3.

Cytokine/chemokine production profile of DC following delivery of ovarian cancer lysates in soluble or NP-associated form. Ovarian cancer lysate encapsulated in NP activates DC to produce pro-inflammatory cytokines such as IL-1β, IL-6, IFNγ, TNFα, G-CSF and RANTES, an effect not associated with soluble Ag delivery or non-specific NP stimulation [media= DC only, NP= DC loaded with empty nanoparticle; L= DC loaded with equimolar soluble lysate; NPL= DC loaded with NP-encapsulated lysate].
Increased stimulation of CD8+ T cell responses is associated with NP-mediated Ag delivery
To determine whether the substantial differences observed in DC cytokine production following NP-mediated Ag delivery would also be reflected in the capacity of these DC to stimulate Ag-specific T cells, additional experiments were undertaken to use identically-loaded DC populations as stimulators in co-culture reactions with autologous CD8+ T cells. First, DC populations were loaded with identical Ag forms and controls as described above, washed and cultured with donor-derived purified CD8+ T cells at DC/T cell ratios of 1:2 for 3 days. Identical cultures without CD8+ T cell addition were run in parallel and DC-derived cytokine production subtracted to determine the precise cytokine contribution of reactive T cells. As shown in figure 4a, CD8+ T cells from co-cultures with DC exposed to NPL had a significantly up-regulated production pattern of the pro-inflammatory cytokines IL-1β, IL-6, IL-8, IFNγ and RANTES which were specifically and characteristically produced by this group. IL-12 was also consistently over-expressed in some but not all individuals (data not shown).
Fig. 4.


Differential cytokine profile of CD8+ T cells following co-culture with Ag-loaded DC. DC were loaded with different Ag forms [NP=empty nanoparticle; L= lysate; NPL= NP-lysate] and co-cultured with autologous CD8 T-cells for 72 hrs, after which cell-free conditioned media (CM) was analyzed for cytokine production. (A) CD8 T cells co-cultured with DC loaded with NPL produce a pro-inflammatory/Th1 cytokine profile characterized by specific induction of IL-1β, IL-6, IFNγ, TNFα, G-CSF and RANTES. (B) CD8 T cells co-cultured with DC loaded with soluble lysate produce a weakly Th2 or potentially tolergenic profile.
A different and significantly contrasting pattern was observed in CD8+ T cells co-cultured with DC loaded with soluble lysates. As shown in figure 4b, CD8 T cell stimulation following co-culture with DC loaded with soluble lysates produces a cytokine pattern which would be considered weakly Th2 or tolerogenic in nature. Delivery of soluble Ag lead to low but consistent T cell production of IL-4, GM-CSF and IL-2; cytokines not produced at measurable levels in association with NPL DC delivery. Taken together, these results indicate that stimulation capacity following delivery of an identical Ag source, i.e., tumor cell lysates, can be strongly influenced by the methodology used to introduce that Ag to DC, and subsequently these loaded DC are driven to respond to the Ag source in extremely varied ways, with contrasting cytokine production profiles and T cell stimulation capacities.
T cells co-cultured with NPL-loaded DCs differentially express co-stimulatory molecules
Next we determined whether the differential stimulation capacity of the NPL delivery system could also lead to changes in phenotype of CD8+ T cells following DC co-culture. To accomplish this, CD8+ T cell populations were co-stained for CD8 and the co-stimulator molecules associated with T cell tolerization/exhaustion (PD-1, CTLA-4) or T cell positive stimulation (ICOS) following DC exposure. As shown in figure 5, DC loaded with NPL (fig. 5- column four) produced a T cell expression pattern characterized by significantly increased cell surface ICOS with virtually no change in PD-1 or CTLA-4 expression. In contrast, DC stimulation following loading with soluble tumor lysates (fig. 5-column three), led to increased expression of PD-1 and CTLA-4, hallmarks of T cells which have received negative co-stimulation, with little change in ICOS expression. This expression pattern provides additional data supporting the differential T cell stimulation capacity of NP-mediated Ag delivery, and is consistent with a model in which NP-mediated Ag delivery drives increased DC stimulation and improved Ag cross-presentation to reactive CD8+ T cells.
Fig. 5.
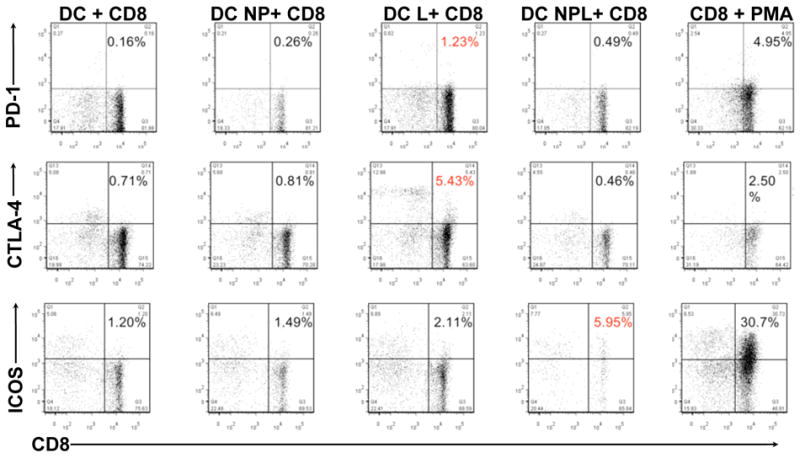
Differential expression of CD8+ T cell activation markers following co-culture with NPL-loaded DC. 72 hrs post-exposure to NPL DC or control Ag forms, CD8+ cells exposed to NPL-loaded DC express markers of positive activation, such as ICOS, while failing to up-regulate negative co-stimulators such as PD-1 and CTLA-4 (panel four). Conversely, CD8+ cells exposed to lysate-loaded DC express increased cell surface inhibitory molecules such as PD-1 and CTLA-4, but comparably little ICOS (panel three). Positive control CD8+ T cells stimulated non-specifically with PMA/ionomycin show strongly increased expression of all markers (panel five), while control unloaded DC (panel one) and DC loaded with empty NP (panel two) show little or no up-regulation [DC +CD8= unloaded DC in co-culture, DC NP +CD8= DC loaded with empty NP, DC L + CD8= soluble lysate-loaded DC, DC NPL +CD8= NPL-loaded DC, CD8 + PMA= T cells non-specifically stimulated with PMA/ionomycin].
Discussion
We report here that loading of DC with NP/Ag conjugates stimulates distinctive and potentially superior anti-ovarian cancer Ag CD8 T cell responses, when compared to conventional approaches. For example, delivery of an identical cell lysate to DC in a soluble, non-particulate form, promoted secretion of weakly Th2 or tolerogenic cytokines, which in turn caused CD8+ T cells to display inhibitory cell surface molecules commonly associated with T cell tolerance or exhaustion. Delivery by NP, by contrast, led to an inflammatory/Th1 biased cytokine production pattern, and led to T cell expression of co-stimulatory molecules indicative of positive stimulation and CD8+ T cell activation.
Since the DC and CD8+ T cells involved in these studies were derived from normal volunteer blood, the responses we observed suggest that the stimulatory Ag may be shared by both ovarian cancer cells and normal tissues. This phenomenon has been previously identified in melanoma, where a large number of the Ags targeted by immunotherapy are reactive with CTL from both cancer patients and normal individuals 50, 51. It is possible, if not likely, that had the DC and CD8 populations been isolated from ovarian cancer patients, rather than normal subjects, T cell responses to autologous tumor might have been even more vigorous. It will now be important to similarly test for such patient enhanced responses. Yet, these initial findings raise the possibility that administration of tumor NPL, with or without a DC intermediate, may potentially be a new and potent methodology to break tolerance to existing TAA in the tumor micro-environment of ovarian cancer patients. We hypothesize that the responses noted here in normal individuals could have direct relevance to the immune modification associated with tumor growth and progression in these cancer patients. Since it is well-established that tumors are capable of inducing a tolerogenic state which disables existing anti-tumor immune responses through a series of cellular and cytokine-based escape mechanisms 49, 52, 53; methodologies which overcome this state and “reawaken” significant cytolytic T cell responses to tolerized antigens would be critical step forward in the successful design of next-generation immunotherapeutic vaccines.
Additional efforts to generate and clone Ag-specific T cell lines from ovarian tumor-bearing individuals following NPL loading of DC is also a potentially promising avenue for TAA identification in epithelial ovarian cancer, since very few tumor–relevant Ags have been characterized, severely hampering traditional immunotherapy efforts. Utilizing the increased efficiency of NPL reagents in the stimulation of anti-tumor T cells from ovarian cancer patients, it may be possible to identify a larger, and more diverse, population of reactive T cells useful in a variety of complementary therapies, including autologous T cell transfer and CD8+ TCR transduction 54. Therefore, further efforts to use NPL in Ag identification in ovarian cancer are an active area of interest in our group.
While the overall numbers of CD8+T cells responding in an Ag-specific manner in this assay are relatively low (∼1-5% of T cells, see fig. 5), these totals are actually quite significant when considering these are tumor-associated Ags being delivered to normal control DC and responding T cells. In the only other major malignancy, melanoma, in which tumor–specific T cells have been stimulated from normal control blood, the authors reported CD8+ precursor frequencies 1000-fold lower than those reported here (in the range of 0.001% of total) for Ag-specific CD8+ cells reactive to specific tumor associated peptide epitopes from proteins such as gp100, tyrosinase and Mart 136, 55-58. The higher frequency of apparently Ag-specific T cells observed here is perhaps a reflection of the increased Ag complexity available when targeting whole tumor lysates, with their panoply of tumor associated proteins vs. single epitopes from individual TAA, which were repetitively stimulated from extremely small precursor populations in the melanoma system.
In summary, this study shows that NP-mediated delivery of whole tumor lysates produces a distinctive DC and Ag-specific T cell stimulation pattern which is characterized by significant increases in inflammatory cytokine production and expression of positive co-stimulatory molecules. This pattern differs from that associated with delivery of tumor lysate Ags in their soluble form to DC, a methodology previously utilized in a variety of DC loading vaccination schema, which initiated little or no stimulation and appears to weakly skew T cell responses towards cytokines which could promote regulatory T cell (Treg) induction or anergy. These differential responses are likely linked to a fundamentally different mechanism by which DC process engulfed particulate Ag forms, and the response of the DC endocytic compartment to PLGA NP.
Unlike DC pinocytosis of soluble proteins, NP-associated Ag appear to access the MHC class I compartment in the cytoplasm directly, providing an Ag reservoir for prolonged and enhanced Ag presentation, as previously reported by our group and others 27, 38. This is a possible explanation for our observed NP association with highly efficient T cell stimulation, characterized by T cell production of inflammatory or Th1-biased cytokines, and generation of cytolytic T cells expressing positive co-stimulator molecules indicative of potentially potent Ag-specific responses.
To that end, we propose a model for DC antigen presentation that highlights the differences in stimulatory capacity of different Ag forms (figure 6). We suggest that sub-optimal Ag delivery to DC, like that associated with soluble delivery of TAA, will lead to inefficient Ag presentation by loaded DC and induction of failed or tolerogenic T cell responses (fig. 6, left). These responses would be characterized by DC production of cytokines such as IL-10 and IL-4, and T cell expression of negative co-stimulator molecules such as PD-1 and CTLA-4. NP-mediated delivery of tumor-associated proteins (fig 6, right), by contrast, would be expected to be inherently stimulatory to DC, as NP phagocytosis leads to activation of the Nalp3 inflammasome and characteristic production of the inflammatory cytokines such as IL-1β, IL-6, IL-8, IFNγ and RANTES by DC and Ag-specific CD8+ T cells.
Fig. 6.

Model of differential T cell stimulation following NP-mediated TAA delivery. DC loaded with sub-optimal antigen forms (such as soluble tumor lysate- left panel) result in T cell stimulation reactions which lead to anergy or tolerance, and are unable to overcome negative signals in the tumor micro-environment. NP-mediated delivery of Ag, in contrast, leads to DC activation and production of pro-inflammatory/Th1 cytokines which can activate quiescent or tolerized anti-tumor T cells (right panel), potentially overcoming tumor tolerance. NPL-mediated Ag delivery would therefore represent an new and promising methodology for increasing the potency of DC-based anti-tumor vaccination schema in ovarian cancer.
It should be noted that that while some degree of intra-donor differences existed between DC cultured from six normal individuals tested, this distinctive inflammatory pattern was uniformly associated with NP-mediated Ag delivery from 100% of donors. This indicates that a reproducible and characteristic DC stimulation profile could reasonably be expected from both normal and potentially tumor-bearing individuals, an observation with direct relevance to the design of DC-based vaccination protocols in the future.
Acknowledgments
This study was supported in part by grants from NCI/NIH RO1CA127913, RO1CA118678, The Janet Burros Memorial Foundation, The Sands Family Foundation, The Brozman Foundation and the Discovery To Cure Research Program.
References
- 1.Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 2.Einhorn N. Ovarian cancer. Early diagnosis and screening. Hematol Oncol Clin North Am. 1992;6:843–850. [PubMed] [Google Scholar]
- 3.Pearce CL, Near AM, Van Den Berg DJ, et al. Validating genetic risk associations for ovarian cancer through the international Ovarian Cancer Association Consortium. Br J Cancer. 2009;100:412–420. doi: 10.1038/sj.bjc.6604820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Onnis A. The management of ovarian cancer: an update. Eur J Gynaecol Oncol. 1997;18:157–160. [PubMed] [Google Scholar]
- 5.Ozols RF. Chemotherapy for ovarian cancer. Semin Oncol. 1999;26:34–40. [PubMed] [Google Scholar]
- 6.Alvero AB, Chen R, Fu HH, et al. Molecular phenotyping of human ovarian cancer stem cells unravels the mechanisms for repair and chemoresistance. Cell Cycle. 2009;8:158–166. doi: 10.4161/cc.8.1.7533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dougan M, Dranoff G. Immune therapy for cancer. Annu Rev Immunol. 2009;27:83–117. doi: 10.1146/annurev.immunol.021908.132544. [DOI] [PubMed] [Google Scholar]
- 8.Odunsi K, Sabbatini P. Harnessing the immune system for ovarian cancer therapy. Am J Reprod Immunol. 2008;59:62–74. doi: 10.1111/j.1600-0897.2007.00560.x. [DOI] [PubMed] [Google Scholar]
- 9.Matsuzaki J, Qian F, Luescher I, et al. Recognition of naturally processed and ovarian cancer reactive CD8+ T cell epitopes within a promiscuous HLA class II T-helper region of NY-ESO-1. Cancer Immunol Immunother. 2008;57:1185–1195. doi: 10.1007/s00262-008-0450-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Santin AD, Hermonat PL, Ravaggi A, et al. Induction of human papillomavirus-specific CD4(+) and CD8(+) lymphocytes by E7-pulsed autologous dendritic cells in patients with human papillomavirus type 16- and 18-positive cervical cancer. J Virol. 1999;73:5402–5410. doi: 10.1128/jvi.73.7.5402-5410.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Santin AD, Bellone S, Underwood LJ, et al. Novel immunotherapeutic strategies in gynecologic oncology. Dendritic cell-based immunotherapy for ovarian cancer. Minerva Ginecol. 2002;54:133–144. [PubMed] [Google Scholar]
- 12.Cannon MJ, Santin AD, O'Brien TJ. Immunological treatment of ovarian cancer. Curr Opin Obstet Gynecol. 2004;16:87–92. doi: 10.1097/00001703-200402000-00015. [DOI] [PubMed] [Google Scholar]
- 13.Santin AD, Bellone S, Roman JJ, et al. Therapeutic vaccines for cervical cancer: dendritic cell-based immunotherapy. Curr Pharm Des. 2005;11:3485–3500. doi: 10.2174/138161205774414565. [DOI] [PubMed] [Google Scholar]
- 14.Bondurant KL, Crew MD, Santin AD, et al. Definition of an immunogenic region within the ovarian tumor antigen stratum corneum chymotryptic enzyme. Clin Cancer Res. 2005;11:3446–3454. doi: 10.1158/1078-0432.CCR-04-2043. [DOI] [PubMed] [Google Scholar]
- 15.Hernando JJ, Park TW, Kubler K, et al. Vaccination with autologous tumour antigen-pulsed dendritic cells in advanced gynaecological malignancies: clinical and immunological evaluation of a phase I trial. Cancer Immunol Immunother. 2002;51:45–52. doi: 10.1007/s00262-001-0255-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schlienger K, Chu CS, Woo EY, et al. TRANCE- and CD40 ligand-matured dendritic cells reveal MHC class I-restricted T cells specific for autologous tumor in late-stage ovarian cancer patients. Clin Cancer Res. 2003;9:1517–1527. [PubMed] [Google Scholar]
- 17.Santin AD, Bellone S, Ravaggi A, et al. Induction of ovarian tumor-specific CD8+ cytotoxic T lymphocytes by acid-eluted peptide-pulsed autologous dendritic cells. Obstet Gynecol. 2000;96:422–430. doi: 10.1016/s0029-7844(00)00916-9. [DOI] [PubMed] [Google Scholar]
- 18.Santin AD, Hermonat PL, Ravaggi A, et al. In vitro induction of tumor-specific human lymphocyte antigen class I-restricted CD8 cytotoxic T lymphocytes by ovarian tumor antigen-pulsed autologous dendritic cells from patients with advanced ovarian cancer. Am J Obstet Gynecol. 2000;183:601–609. doi: 10.1067/mob.2000.107097. [DOI] [PubMed] [Google Scholar]
- 19.Zhao X, Wei YQ, Peng ZL. Induction of T cell responses against autologous ovarian tumors with whole tumor cell lysate-pulsed dendritic cells. Immunol Invest. 2001;30:33–45. doi: 10.1081/imm-100103689. [DOI] [PubMed] [Google Scholar]
- 20.Brossart P, Wirths S, Stuhler G, et al. Induction of cytotoxic T-lymphocyte responses in vivo after vaccinations with peptide-pulsed dendritic cells. Blood. 2000;96:3102–3108. [PubMed] [Google Scholar]
- 21.Shu S, Zheng R, Lee WT, et al. Immunogenicity of dendritic-tumor fusion hybrids and their utility in cancer immunotherapy. Crit Rev Immunol. 2007;27:463–483. doi: 10.1615/critrevimmunol.v27.i5.50. [DOI] [PubMed] [Google Scholar]
- 22.Shu S, Cohen P. Tumor-dendritic cell fusion technology and immunotherapy strategies. J Immunother. 2001;24:99–100. [PubMed] [Google Scholar]
- 23.Shen H, Ackerman AL, Cody V, et al. Enhanced and prolonged cross-presentation following endosomal escape of exogenous antigens encapsulated in biodegradable nanoparticles. Immunology. 2006;117:78–88. doi: 10.1111/j.1365-2567.2005.02268.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shen LF, Zhang YD, Shen HJ, et al. Liver targeting and the delayed drug release of the nanoparticles of adriamycin polybutylcyanoacrylate in mice. Chin Med J (Engl) 2006;119:1287–1293. [PubMed] [Google Scholar]
- 25.Ludewig B, Junt T, Hengartner H, et al. Dendritic cells in autoimmune diseases. Curr Opin Immunol. 2001;13:657–662. doi: 10.1016/s0952-7915(01)00275-8. [DOI] [PubMed] [Google Scholar]
- 26.Ludewig B, Bonilla WV, Dumrese T, et al. Perforin-independent regulation of dendritic cell homeostasis by CD8(+) T cells in vivo: implications for adaptive immunotherapy. Eur J Immunol. 2001;31:1772–1779. doi: 10.1002/1521-4141(200106)31:6<1772::aid-immu1772>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 27.Ludewig B, McCoy K, Pericin M, et al. Rapid peptide turnover and inefficient presentation of exogenous antigen critically limit the activation of self-reactive CTL by dendritic cells. J Immunol. 2001;166:3678–3687. doi: 10.4049/jimmunol.166.6.3678. [DOI] [PubMed] [Google Scholar]
- 28.Greeneltch KM, Schneider M, Steinberg SM, et al. Host immunosurveillance controls tumor growth via IFN regulatory factor-8 dependent mechanisms. Cancer Res. 2007;67:10406–10416. doi: 10.1158/0008-5472.CAN-07-1228. [DOI] [PubMed] [Google Scholar]
- 29.Stewart TJ, Abrams SI. Altered immune function during long-term host-tumor interactions can be modulated to retard autochthonous neoplastic growth. J Immunol. 2007;179:2851–2859. doi: 10.4049/jimmunol.179.5.2851. [DOI] [PubMed] [Google Scholar]
- 30.Stewart TJ, Greeneltch KM, Lutsiak ME, et al. Immunological responses can have both pro- and antitumour effects: implications for immunotherapy. Expert Rev Mol Med. 2007;9:1–20. doi: 10.1017/S1462399407000233. [DOI] [PubMed] [Google Scholar]
- 31.Lin ML, Zhan Y, Villadangos JA, et al. The cell biology of cross-presentation and the role of dendritic cell subsets. Immunol Cell Biol. 2008;86:353–362. doi: 10.1038/icb.2008.3. [DOI] [PubMed] [Google Scholar]
- 32.Bode U, Lorchner M, Ahrendt M, et al. Dendritic cell subsets in lymph nodes are characterized by the specific draining area and influence the phenotype and fate of primed T cells. Immunology. 2008;123:480–490. doi: 10.1111/j.1365-2567.2007.02713.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Toyokawa H, Nakao A, Bailey RJ, et al. Relative contribution of direct and indirect allorecognition in developing tolerance after liver transplantation. Liver Transpl. 2008;14:346–357. doi: 10.1002/lt.21378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scholl I, Boltz-Nitulescu G, Jensen-Jarolim E. Review of novel particulate antigen delivery systems with special focus on treatment of type I allergy. J Control Release. 2005;104:1–27. doi: 10.1016/j.jconrel.2004.12.020. [DOI] [PubMed] [Google Scholar]
- 35.Roth-Walter F, Scholl I, Untersmayr E, et al. Mucosal targeting of allergen-loaded microspheres by Aleuria aurantia lectin. Vaccine. 2005;23:2703–2710. doi: 10.1016/j.vaccine.2004.11.052. [DOI] [PubMed] [Google Scholar]
- 36.Kovacsovics-Bankowski M, Rock KL. A phagosome-to-cytosol pathway for exogenous antigens presented on MHC class I molecules. Science. 1995;267:243–246. doi: 10.1126/science.7809629. [DOI] [PubMed] [Google Scholar]
- 37.Grant EP, Michalek MT, Goldberg AL, et al. Rate of antigen degradation by the ubiquitin-proteasome pathway influences MHC class I presentation. J Immunol. 1995;155:3750–3758. [PubMed] [Google Scholar]
- 38.Demento S, Steenblock ER, Fahmy TM. Biomimetic approaches to modulating the T cell immune response with nano- and micro- particles. Conf Proc IEEE Eng Med Biol Soc. 2009;2009:1161–1166. doi: 10.1109/IEMBS.2009.5332625. [DOI] [PubMed] [Google Scholar]
- 39.Demento SL, Eisenbarth SC, Foellmer HG, et al. Inflammasome-activating nanoparticles as modular systems for optimizing vaccine efficacy. Vaccine. 2009;27:3013–3021. doi: 10.1016/j.vaccine.2009.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brannon-Peppas L, Blanchette JO. Nanoparticle and targeted systems for cancer therapy. Adv Drug Deliv Rev. 2004;56:1649–1659. doi: 10.1016/j.addr.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 41.Waeckerle-Men Y, Gander B, Groettrup M. Delivery of tumor antigens to dendritic cells using biodegradable microspheres. Methods Mol Med. 2005;109:35–46. doi: 10.1385/1-59259-862-5:035. [DOI] [PubMed] [Google Scholar]
- 42.Waeckerle-Men Y, Groettrup M. PLGA microspheres for improved antigen delivery to dendritic cells as cellular vaccines. Adv Drug Deliv Rev. 2005;57:475–482. doi: 10.1016/j.addr.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 43.Partidos CD, Vohra P, Jones D, et al. CTL responses induced by a single immunization with peptide encapsulated in biodegradable microparticles. J Immunol Methods. 1997;206:143–151. doi: 10.1016/s0022-1759(97)00102-6. [DOI] [PubMed] [Google Scholar]
- 44.Jones DH, Partidos CD, Steward MW, et al. Oral delivery of poly(lactide-co-glycolide) encapsulated vaccines. Behring Inst Mitt. 1997:220–228. [PubMed] [Google Scholar]
- 45.Ren JM, Zou QM, Wang FK, et al. PELA microspheres loaded H. pylori lysates and their mucosal immune response. World J Gastroenterol. 2002;8:1098–1102. doi: 10.3748/wjg.v8.i6.1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Solbrig CM, Saucier-Sawyer JK, Cody V, et al. Polymer nanoparticles for immunotherapy from encapsulated tumor-associated antigens and whole tumor cells. Mol Pharm. 2007;4:47–57. doi: 10.1021/mp060107e. [DOI] [PubMed] [Google Scholar]
- 47.Kamsteeg M, Rutherford T, Sapi E, et al. Phenoxodiol-an isoflavon analogue-induces apoptosis in chemo-resistant ovarain cancer cells. Oncogene. 2003;22:2611–2620. doi: 10.1038/sj.onc.1206422. [DOI] [PubMed] [Google Scholar]
- 48.Chen R, Alvero AB, Silasi DA, et al. Regulation of IKKbeta by miR-199a affects NF-kappaB activity in ovarian cancer cells. Oncogene. 2008;27:4712–4723. doi: 10.1038/onc.2008.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen R, Alvero AB, Silasi DA, et al. Inflammation, cancer and chemoresistance: taking advantage of the toll-like receptor signaling pathway. Am J Reprod Immunol. 2007;57:93–107. doi: 10.1111/j.1600-0897.2006.00441.x. [DOI] [PubMed] [Google Scholar]
- 50.Jager E, Ringhoffer M, Arand M, et al. Cytolytic T cell reactivity against melanoma-associated differentiation antigens in peripheral blood of melanoma patients and healthy individuals. Melanoma Res. 1996;6:419–425. doi: 10.1097/00008390-199612000-00003. [DOI] [PubMed] [Google Scholar]
- 51.Jager E, Ringhoffer M, Dienes HP, et al. Granulocyte-macrophage-colony-stimulating factor enhances immune responses to melanoma-associated peptides in vivo. Int J Cancer. 1996;67:54–62. doi: 10.1002/(SICI)1097-0215(19960703)67:1<54::AID-IJC11>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 52.Chen R, Alvero AB, Silasi DA, et al. Cancers take their Toll--the function and regulation of Toll-like receptors in cancer cells. Oncogene. 2008;27:225–233. doi: 10.1038/sj.onc.1210907. [DOI] [PubMed] [Google Scholar]
- 53.Mor G, Straszewski S, Kamsteeg M. Role of the Fas/Fas ligand system in female reproductive organs: survival and apoptosis. Biochem Pharmacol. 2002;64:1305. doi: 10.1016/s0006-2952(02)01267-4. [DOI] [PubMed] [Google Scholar]
- 54.Rosenberg SA, Dudley ME. Adoptive cell therapy for the treatment of patients with metastatic melanoma. Curr Opin Immunol. 2009;21:233–240. doi: 10.1016/j.coi.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kuo YR, Huang CW, Goto S, et al. Alloantigen-pulsed host dendritic cells induce T-cell regulation and prolong allograft survival in a rat model of hindlimb allotransplantation. J Surg Res. 2009;153:317–325. doi: 10.1016/j.jss.2008.05.034. [DOI] [PubMed] [Google Scholar]
- 56.Klarquist J, Barfuss A, Kandala S, et al. Melanoma-associated antigen expression in lymphangioleiomyomatosis renders tumor cells susceptible to cytotoxic T cells. Am J Pathol. 2009;175:2463–2472. doi: 10.2353/ajpath.2009.090525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kaka AS, Shaffer DR, Hartmaier R, et al. Genetic modification of T cells with IL-21 enhances antigen presentation and generation of central memory tumor-specific cytotoxic T-lymphocytes. J Immunother. 2009;32:726–736. doi: 10.1097/CJI.0b013e3181ad4071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Michaeli Y, Denkberg G, Sinik K, et al. Expression hierarchy of T cell epitopes from melanoma differentiation antigens: unexpected high level presentation of tyrosinase-HLA-A2 Complexes revealed by peptide-specific, MHC-restricted, TCR-like antibodies. J Immunol. 2009;182:6328–6341. doi: 10.4049/jimmunol.0801898. [DOI] [PubMed] [Google Scholar]