

Abstract
The XP-E DNA damage binding protein (DDB2) is involved in early recognition of global genome DNA damage during DNA nucleotide excision repair (NER). We found that skin fibroblasts from 4 newly reported XP-E patients with numerous skin cancers and DDB2 mutations had slow repair of 6-4 photoproducts (6-4PP) and markedly reduced repair of cyclobutane pyrimidine dimers (CPD). NER proteins (XPC, XPB, XPG, XPA, and XPF) co-localized to CPD and 6-4PP positive regions immediately (< 0.1h) after localized UV irradiation in cells from the XP-E patients and normal controls. While these proteins persist in normal cells, surprisingly, within 0.5h these repair proteins were no longer detectable at the sites of DNA damage in XP-E cells. Our results indicate that DDB2 is not required for the rapid recruitment of NER proteins to sites of UV photoproducts or for partial repair of 6-4PP but is essential for normal persistence of these proteins for CPD photoproduct removal.
INTRODUCTION
Xeroderma pigmentosum (XP) is a rare recessive disorder with hypersensitivity to sun exposure, leading to a more than 1000-fold increase in UV-induced skin cancer in association with defective nucleotide excision repair (NER) (1). XP is genetically heterogeneous with 7 NER complementation groups designated XP-A to XP-G and an XP variant form with deficiency in trans-lesion synthesis by polymerase η. Cells from XP-E patients show a mild hypersensitivity to killing by UV and a variable level of unscheduled DNA synthesis that is about 50% of normal but increases with increasing UV dose (2–4).
UV causes formation of major DNA photoproducts: cyclobutane pyrimidine dimers (CPD) and pyrimidine-6-4-pyrimidone photoproducts (6-4PP). In normal human cells NER efficiently recognizes and removes these photoproducts. NER is comprised of two distinct sub-pathways, global genome NER (GG-NER) and transcription-coupled NER (TC-NER), which differ in damage recognition but share the same core mechanism (5). TC-NER, which removes lesions from the transcribed strand of active genes, is initiated by stalling of an elongating RNA polymerase II that acts to recruit the NER machinery. The damage recognition steps of GG-NER, which removes lesions from the overall genome and non-transcribed DNA strands are not well understood. The earliest damage recognition factor in GG-NER is DDB2, which is involved in the repair of UV photoproducts, especially CPD (6,7), and is part of the UV-DDB-ubiquitin ligase complex (6,7). The XPC-RAD23B-CEN2 complex is a later initiator of GG-NER (5,8) with translocation to DNA damage sites mediated by DDB2 (9). XP-E cells have been reported to have reduced repair of CPD but nearly normal repair of 6-4 photoproducts (10–13).
UV-damaged DNA binding protein (UV-DDB) is a heterodimer of DDB1 (p127) and DDB2 (p48) (5,8) which binds with high affinity to DNA damaged by UV. XP-E patients have mutations in the DDB2 gene which inactivate DDB activity resulting in the loss of UV-DDB binding (11,14–19). Only 11 XP-E patients in 9 families have been reported (18). We examined the rate of removal of photoproducts from 4 recently identified XP-E patients who had large numbers of skin cancers (18). Using a local UV irradiation technique combined with fluorescent antibody labeling (12,20–22) we also investigated the time course of recruitment and removal of NER proteins at sites of DNA damage.
MATERIALS AND METHODS
Cell lines, culture conditions and DNA/RNA isolation
Normal (AG13153) and XP-E XP408BE (GM01389) primary skin fibroblasts were obtained from the Human Genetic Mutant Cell Repository, Camden, NJ. XP-E siblings XP37BE (KR04156) and XP66BE (KR04158) fibroblasts (18) were established at the Human Genetic Mutant Cell Repository from skin biopsies sent from NIH. XP1GO culture was established at the Department of Dermatology, Goettingen, Germany. The cells were grown in Dulbecco’s modified Eagle medium containing 40mM glutamine and 10% fetal bovine serum in an 8% CO2 humidified incubator at 37°C.
ELISA
Confluent human normal and XP-E fibroblasts were UV-irradiated with 10 J/m2 UVC (254 nm) and incubated for various time periods. An enzyme-linked immunosorbent assay (ELISA) with TDM-2 and 64M-2 monoclonal antibodies was used to measure 6-4PP and CPD repair as described (23).
Immunofluorescence assay
Cytoplasmic bead labeled XP and normal cells were grown in mixed culture, irradiated with 100 J/m2 UVC (254 nm) through an isopore polycarbonate filter (Millipore) with 5 μm pores, incubated for various times and processed for localization of NER proteins by immunofluorescence as described previously (9,12,20–22). Fluorescence images were obtained using a LSM 510 confocal microscope (Zeiss).
RESULTS
Reduced repair of CPD and 6-4PP in XPE cells
We performed ELISA to measure the removal of the 6-4PP and CPD photoproducts in XP-E and normal cells at various times after UV irradiation in order to determine the DNA repair kinetics. The clinical symptoms and mutations in the DDB2 gene in these 4 newly identified XP-E patients were reported previously (18). The hallmarks of all 4 patients, XP1GO, siblings XP37BE and XP66BE, and XP408BE, were that they did not experience acute burning on minimal sun exposure and developed several hundreds of skin cancers. Cells from all 4 XP-E patients showed slow repair of 6-4 PP. In normal cells, almost 97% of 6-4PP were removed at 3h post-UV irradiation, whereas cells from the XP-E patients removed 62~66% of 6-4PP (Fig. 1A). However, by 24 h the repair of 6-4PP was not significantly different in the XP-E and normal cells.
Figure 1.
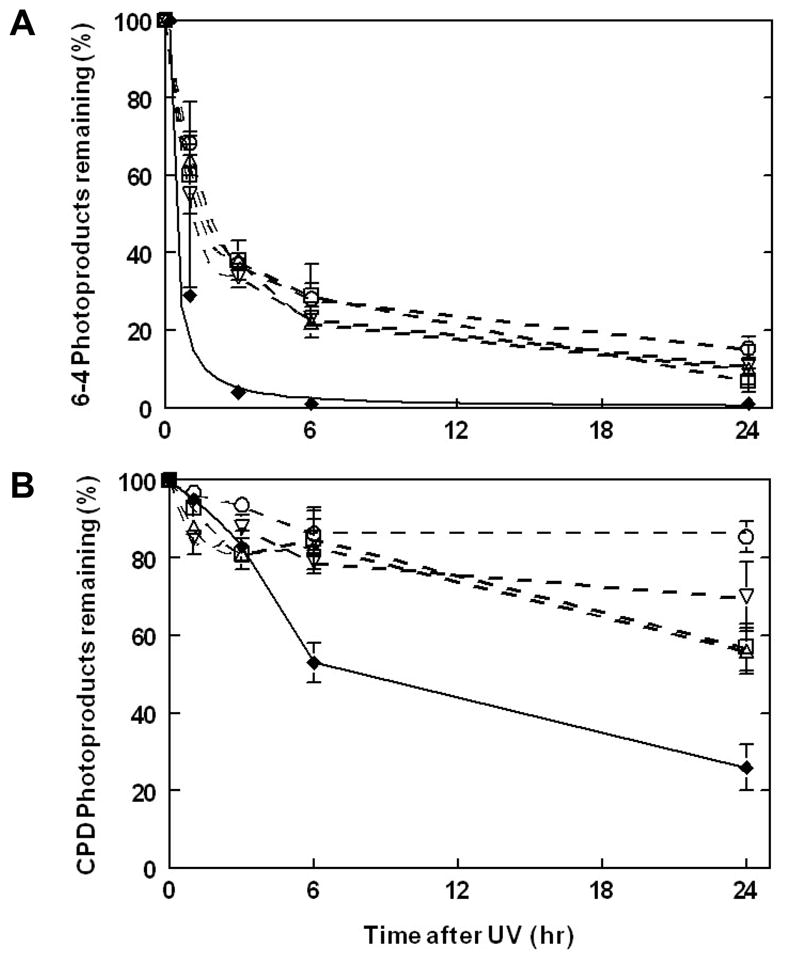
Reduced repair of photoproducts in XP-E cells. XP-E cells were treated with UVC (10 J/m2) and incubated for up to 24 h. The removal of (A) 6-4PP and (B) CPD was measured with an ELISA assay. Results represent mean values ± sem of at least n=3 experiments. Normal cells (AG13145) - closed diamonds ◆; XPD cells: XP408BE –open squares □, XP37BE – open vertical triangles ▵, XP66BE – open inverted triangles ▽, XP1GO – open circles ○.
It is well established that the rate of CPD repair in normal cells is markedly slower than that of 6-4PP repair (10,11,21,24–26). About 74 % of the CPDs were removed by 24h post-UV irradiation in the normal cells. In contrast only 22~44% of the CPD were removed from the XP-E cells (Fig. 1B).
At sites of focal UV damage, NER proteins localize rapidly in both normal and XPE cells but fail to persist in XPE cells
To visualize repair of DNA photoproducts, we performed fluorescent immunostaining at various time points after local UV- irradiation (Fig. 2A). XP-E cells (XP37BE; 2 μm beads) and normal cells (0.8 μm beads) on the same coverslips were treated with UV irradiation through isopore polycarbonate filters and stained with antibodies for CPD or for 6-4PP along with antibodies for XPC protein. Foci of immunofluorescence were not present in the unirradiated cells (first column). Foci of CPD (second row) and 6-4PP (bottom row) immunofluorescence were observed immediately (<0.1 hr) after UV exposure (second column) in the normal (red arrows) and XP-E (yellow arrows) cells. CPD and 6-4PP staining was visible at 0.5 h after UV exposure (third column) in the normal and XP-E cells. At 3 h post-UV exposure (fourth column) CPD were still observed in the normal and the XP-E cells. In contrast, 6-4 PP were not visible in the normal cells but were still detectable in the XP-E cells at this time. At 24 h post-UV CPD were still present in the XP-E cells. This is in good agreement with our repair results in Fig. 1.
Figure 2.

Recruitment and redistribution of NER proteins after focal UV irradiation in normal and XP-E (XP37BE) cells. Normal cells (0.8 μm beads – red arrows) and XP-E cells (2 μm beads – yellow arrows) were co-cultured and irradiated with 100 J/m2 UVC, fixed, permeabilized, and double stained with antibodies specific for XP proteins along with antibodies for CPD or for 6-4PP at different time points after irradiation. Cells were visualized with Alexa Fluor R 568 goat anti-mouse IgG conjugate. (A) Time course of XPC, CPD and 6-4PP recruitment and redistribution. (B) Localization of XPB, XPG, XPA and XPF proteins at 0.1 h and 0.5 h after focal UV.
We then investigated the recruitment of the NER proteins to sites of photodamage. Immediately (<0.1h) after UV-irradiation, normal and XP-E cells showed XPC protein staining that co-localized with the CPD and 6-4PP photoproduct foci (Fig. 2A, top row and third row). The NER proteins XPB, XPG, XPA, and XPF, also rapidly co-localized to the CPD positive regions immediately after localized UV irradiation in cells from both the normal donor and the XP-E patient (Fig. 2B). Similar results were observed with XPD protein (data not shown). There were no significant differences between normal and XP-E cells immediately after UV irradiation demonstrating that UV-DDB was not required for the rapid recruitment of NER proteins.
At 0.5 h the staining of NER proteins XPC, XPB, XPG, XPA, and XPF, was not detectable in most of the XP-E cells but was visible in the normal cells (Fig. 2A and B). Counts of >100 nuclei per slide indicated that at <0.1 h post-UV 75–80% of the nuclei were positive for XPC staining in the normal and the XP-E cells. At 0.5 h post-UV the XPC staining persisted in 75–80% of the normal cells but was only detectible in about 5% of the XP-E cells. This is similar to the low level or absence of post-UV XPC localization at 0.5 h in XPE cells reported by others (9,12). There was a similar low frequency of XPB, XPG, XPA, and XPF positive XP-E cells at 0.5 h post-UV. At 24 h post-UV the NER proteins were not detectible in the normal or XP-E cells at sites of persistent CPD staining (data not shown).
DISCUSSION
Slower 6-4PP and markedly reduced CPD repair in XP-E cells
We found a slower repair of 6-4PP in these newly reported XP-E cells (Fig. 1A) similar to that reported by Itoh et al (11) and Hwang et al (10) for other XP-E patients. Moser et al (12) reported a normal rate of removal of 6-4PP in XP-E cells. The repair of UV damage in XP-E cells has been reported to increase with increasing UV dose (2,4). The markedly reduced CPD repair (Fig. 1B) is in agreement with the report by Hwang et al (10). Interestingly, Itoh et al (11) reported an XPE cell line (Oops1) from a patient with multiple skin cancers that had slow repair of 6-4PP but normal repair of CPD. This may be related to the C-terminal DDB2 nonsense mutation in this cell line. These results indicate that the NER complex can function to some extent in the absence of DDB2 activity and implies that, like DDB2 (27), XPC predominately binds to 6-4PP (28), but does not efficiently recognize CPD’s in vivo (12). UV-DDB appears to promote recruitment of XPC to DNA sites containing UV photolesions (12). However, the process is not completely understood since partial correction of an XPC defective cell line with a 5′truncated XPC cDNA resulted in normal repair of CPD and defective repair of 6-4PP (29).
Rapid localization of NER proteins to photoproducts does not require UV-DDB
In agreement with previous studies we found that the NER proteins (XPC, XPB, XPG, XPA, and XPF) are rapidly recruited to CPD or 6-4PP photolesions in normal and XP-E cells (Fig. 2A) (12,20,21). This was also observed by Moser et al (12) except they reported a slightly lower level of XPC protein accumulation in XP-E cells compared to normal cells. This may be due to the lower UV dose used (30 vs. 100 J/m2), the fact that XP-E cells exhibit greater levels of repair (measured by unscheduled DNA synthesis (UDS)) with increasing dose of UV (2,4), and that UDS at early times after UV mainly reflects repair of 6-4PP. UV-DDB has a greater binding affinity and specificity than XPC for UV-induced photoproducts (8,28,30). UV induced transient immobilization of fluorescently labeled XPC, indicating its involvement in NER, is regulated in a biphasic manner depending on the number of 6-4PP and the relative abundance of UV-DDB molecules. In normal cells at low UV doses the UV-DDB dependent pathway predominates over direct recognition by XPC(13). Reducing the amount of UV-DDB by siRNA concomitantly reduced the immobilization of XPC after low dose UV (13). In contrast, XPC directly recognizes 6-4PP levels greatly exceeding the relative number of available UV-DDB molecules (5,8,13). In the absence of XPC, NER proteins cannot be detected at either type of DNA damage (Table 1) (22,31). This suggests that XPC, but not UV-DDB, is essential for the rapid recruitment of NER proteins at sites of DNA photodamage.
Table 1.
Time course of recruitment and redistribution of NER proteins and photoproducts in normal, XP-E, XP-C, and XP-B deficient cells after focal UV irradiation
| Normal Cells† | XP-E Cells† | XP-C Cells†† | XP-B Cells††† | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0.1 hr | 0.5 hr | 24 hr | 0 hr | 0.5 hr | 24 hr | 0 hr | 0.5 hr | 24 hr | 0.1 hr | 0.5 hr | 24 hr | |
| XPC | +++ | + | − | +++ | − | − | − | − | − | +++ | +++ | +++ |
| XPB | +++ | ++ | − | +++ | − | − | − | − | − | − | − | − |
| XPD | +++ | ++ | − | +++ | − | − | − | − | − | − | +++ | +++ |
| XPG | +++ | ++ | − | +++ | − | − | − | − | − | +++ | +++ | +++ |
| XPA | +++ | ++ | − | +++ | − | − | − | − | − | +++ | +++ | +++ |
| XPF | +++ | ++ | − | +++ | − | − | − | − | − | − | − | +++ |
| CPD | +++ | +++ | + | +++ | +++ | ++ | +++ | +++ | ++ | +++ | +++ | +++ |
| 6-4PP | +++ | + | − | +++ | ++ | + | +++ | +++ | ++ | +++ | +++ | ++ |
Persistence of NER proteins requires UV-DDB
In normal cells NER proteins can be visualized at the site of UV damage at 0.5 h and are no longer observed at 24 h (Fig. 2, Table 1) (12,20,21). In contrast, XPB or XPD defective cells continue to show persistence of NER proteins at sites of unrepaired DNA damage at 24 h (20,21) (Table 1). We found that the NER proteins XPC, XPB, XPG, XPA, and XPF were already redistributed from sites of local UV damage in XP-E (XP37BE) cells at 0.5 h. This is in agreement with previous reports assessing XPC recruitment in XP408BE cells (9), in GM01389h Tert cells (12) and with mutated XP37BE protein translocation to UV-damaged DNA (32).
Model for NER protein accumulation and persistence in normal and XP-E cells
Immediately after localized UV irradiation NER proteins rapidly accumulate in normal and XP-E cells indicating that DDB2 is not required for this process (Figure 3). XPC can bind 6-4PP directly while binding to CPD requires preliminary binding of UV-DDB2 to allow XPC to access the lesion (5,12,13,28,32). XPC is ubiquitinated by a UV-DDB2-containing complex in the normal cells resulting in increased binding to photoproducts (5,8,33). The 6-4PPs are excised and the persistent XPC and downstream NER proteins can – in a generally slower mode of action - excise the CPD due to the increased binding. In contrast, in the XP-E cells by 0.5 h the XPC and downstream NER proteins do not persist (or fall to an undetectable level) especially at CPD sites, possibly as a result of failure of UV-DDB2-mediated ubiquitination of XPC.
Figure 3.

Schematic diagram of early and late global genome nucleotide excision repair in cells from normal and XP-E patients (see text for details).
Acknowledgments
This research was supported in part by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health. This research was supported in part by the Deutsche Krebshilfe and the Deutsche Forschungsgemeinschaft (to S.E.). We thank Drs. Kiyoji Tanaka, Osaka University and Leon Mullenders, Leiden University for insightful comments. We thank the patients for their participation.
References
- 1.Bradford PT, Goldstein AM, Tamura D, Khan SG, Ueda T, Boyle J, Oh KS, Imoto K, Inui H, Moriwaki SI, Emmert S, Pike KM, Raziuddin A, Plona TM, DiGiovanna JJ, Tucker MA, Kraemer KH. Cancer and neurologic degeneration in xeroderma pigmentosum: long term follow-up characterises the role of DNA repair. J Med Genet. 2010 doi: 10.1136/jmg.2010.083022. jmg.2010.083022 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Weerd-Kastelein EA, Keijzer W, Bootsma D. A third complementation group in xeroderma pigmentosum. Mutat Res. 1974;22:87–91. doi: 10.1016/0027-5107(74)90013-x. [DOI] [PubMed] [Google Scholar]
- 3.Inui H, Oh KS, Nadem C, Ueda T, Khan SG, Metin A, Gozukara E, Emmert S, Slor H, Busch DB, Baker CC, DiGiovanna JJ, Tamura D, Seitz CS, Gratchev A, Wu WH, Chung KY, Chung HJ, Azizi E, Woodgate R, Schneider TD, Kraemer KH. Xeroderma pigmentosum-variant patients from America, Europe, and Asia. J Invest Dermatol. 2008;128:2055–2068. doi: 10.1038/jid.2008.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kraemer KH, De Weerd-Kastelein EA, Robbins JH, Keijzer W, Barrett SF, Petinga RA, Bootsma D. Five complementation groups in xeroderma pigmentosum. Mutat Res. 1975;33:327–340. doi: 10.1016/0027-5107(75)90208-0. [DOI] [PubMed] [Google Scholar]
- 5.Sugasawa K. Regulation of damage recognition in mammalian global genomic nucleotide excision repair. Mutat Res. 2010;685:29–37. doi: 10.1016/j.mrfmmm.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 6.Scharer OD, Campbell AJ. Wedging out DNA damage. Nat Struct Mol Biol. 2009;16:102–104. doi: 10.1038/nsmb0209-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scrima A, Konickova R, Czyzewski BK, Kawasaki Y, Jeffrey PD, Groisman R, Nakatani Y, Iwai S, Pavletich NP, Thoma NH. Structural basis of UV DNA-damage recognition by the DDB1-DDB2 complex. Cell. 2008;135:1213–1223. doi: 10.1016/j.cell.2008.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sugasawa K. UV-induced ubiquitylation of XPC complex, the UV-DDB-ubiquitin ligase complex, and DNA repair. J Mol Histol. 2006;37:189–202. doi: 10.1007/s10735-006-9044-7. [DOI] [PubMed] [Google Scholar]
- 9.Wang QE, Zhu Q, Wani G, Chen J, Wani AA. UV radiation-induced XPC translocation within chromatin is mediated by damaged-DNA binding protein, DDB2. Carcinogenesis. 2004;25:1033–1043. doi: 10.1093/carcin/bgh085. [DOI] [PubMed] [Google Scholar]
- 10.Hwang BJ, Ford JM, Hanawalt PC, Chu G. Expression of the p48 xeroderma pigmentosum gene is p53-dependent and is involved in global genomic repair. Proc Natl Acad Sci U S A. 1999;96:424–428. doi: 10.1073/pnas.96.2.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Itoh T, Mori T, Ohkubo H, Yamaizumi M. A newly identified patient with clinical xeroderma pigmentosum phenotype has a non-sense mutation in the DDB2 gene and incomplete repair in (6-4) photoproducts. J Invest Dermatol. 1999;113:251–257. doi: 10.1046/j.1523-1747.1999.00652.x. [DOI] [PubMed] [Google Scholar]
- 12.Moser J, Volker M, Kool H, Alekseev S, Vrieling H, Yasui A, van Zeeland AA, Mullenders LH. The UV-damaged DNA binding protein mediates efficient targeting of the nucleotide excision repair complex to UV-induced photo lesions. DNA Repair (Amst) 2005;4:571–582. doi: 10.1016/j.dnarep.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 13.Nishi R, Alekseev S, Dinant C, Hoogstraten D, Houtsmuller AB, Hoeijmakers JH, Vermeulen W, Hanaoka F, Sugasawa K. UV-DDB-dependent regulation of nucleotide excision repair kinetics in living cells. DNA Repair (Amst) 2009;8:767–776. doi: 10.1016/j.dnarep.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 14.Chu G, Chang E. Xeroderma pigmentosum group E cells lack a nuclear factor that binds to damaged DNA. Science. 1988;242:564–567. doi: 10.1126/science.3175673. [DOI] [PubMed] [Google Scholar]
- 15.Itoh T, Linn S, Ono T, Yamaizumi M. Reinvestigation of the classification of five cell strains of xeroderma pigmentosum group E with reclassification of three of them. J Invest Dermatol. 2000;114:1022–1029. doi: 10.1046/j.1523-1747.2000.00952.x. [DOI] [PubMed] [Google Scholar]
- 16.Itoh T, Linn S. XP43TO, previously classified as xeroderma pigmentosum Group E, should be reclassified as xeroderma pigmentosum variant. J Invest Dermatol. 2001;117:1672–1674. doi: 10.1046/j.0022-202x.2001.01619.x. [DOI] [PubMed] [Google Scholar]
- 17.Keeney S, Eker AP, Brody T, Vermeulen W, Bootsma D, Hoeijmakers JH, Linn S. Correction of the DNA repair defect in xeroderma pigmentosum group E by injection of a DNA damage-binding protein. Proc Natl Acad Sci U S A. 1994;91:4053–4056. doi: 10.1073/pnas.91.9.4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oh KS, Emmert S, Tamura D, DiGiovanna JJ, Kraemer KH. Multiple Skin Cancers in Adults with Mutations in the XP-E (DDB2) DNA Repair Gene. J Invest Dermatol. 2010 doi: 10.1038/jid.2010.352. jid2010352 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rapic-Otrin V, Navazza V, Nardo T, Botta E, McLenigan M, Bisi DC, Levine AS, Stefanini M. True XP group E patients have a defective UV-damaged DNA binding protein complex and mutations in DDB2 which reveal the functional domains of its p48 product. Hum Mol Genet. 2003;12:1507–1522. doi: 10.1093/hmg/ddg174. [DOI] [PubMed] [Google Scholar]
- 20.Boyle J, Ueda T, Oh KS, Imoto K, Tamura D, Jagdeo J, Khan SG, Nadem C, DiGiovanna JJ, Kraemer KH. Persistence of repair proteins at unrepaired DNA damage distinguishes diseases with ERCC2 (XPD) mutations: cancer-prone xeroderma pigmentosum vs. non-cancer-prone trichothiodystrophy. Hum Mutat. 2008;29:1194–1208. doi: 10.1002/humu.20768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oh KS, Imoto K, Boyle J, Khan SG, Kraemer KH. Influence of XPB helicase on recruitment and redistribution of nucleotide excision repair proteins at sites of UV-induced DNA damage. DNA Repair (Amst) 2007;6:1359–1370. doi: 10.1016/j.dnarep.2007.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Volker M, Mone MJ, Karmakar P, van Hoffen A, Schul W, Vermeulen W, Hoeijmakers JH, van Driel R, van Zeeland AA, Mullenders LH. Sequential assembly of the nucleotide excision repair factors in vivo. Mol Cell. 2001;8:213–224. doi: 10.1016/s1097-2765(01)00281-7. [DOI] [PubMed] [Google Scholar]
- 23.Thoms KM, Kuschal C, Oetjen E, Mori T, Kobayashi N, Laspe L, Boeckmann L, Schon MP, Emmert S. Cyclosporin A, but not everolimus, inhibits DNA repair mediated by calcineurin: Implications for tumorigenesis under immunosuppression. Exp Dermatol. 2010 doi: 10.1111/j.1600-0625.2010.01213.x. [DOI] [PubMed] [Google Scholar]
- 24.Mitchell DL, Haipek CA, Clarkson JM. (6-4)Photoproducts are removed from the DNA of UV-irradiated mammalian cells more efficiently than cyclobutane pyrimidine dimers. Mutat Res. 1985;143:109–112. doi: 10.1016/s0165-7992(85)80018-x. [DOI] [PubMed] [Google Scholar]
- 25.Thomas DC, Okumoto DS, Sancar A, Bohr VA. Preferential DNA repair of (6-4) photoproducts in the dihydrofolate reductase gene of Chinese hamster ovary cells. J Biol Chem. 1989;264:18005–18010. [PubMed] [Google Scholar]
- 26.Mitchell DL, Brash DE, Nairn RS. Rapid repair kinetics of pyrimidine(6-4)pyrimidone photoproducts in human cells are due to excision rather than conformational change. Nucleic Acids Res. 1990;18:963–971. doi: 10.1093/nar/18.4.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Treiber DK, Chen Z, Essigmann JM. An ultraviolet light-damaged DNA recognition protein absent in xeroderma pigmentosum group E cells binds selectively to pyrimidine (6- 4) pyrimidone photoproducts. Nucleic Acids Res. 1992;20:5805–5810. doi: 10.1093/nar/20.21.5805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Batty D, Rapic’-Otrin V, Levine AS, Wood RD. Stable binding of human XPC complex to irradiated DNA confers strong discrimination for damaged sites. J Mol Biol. 2000;300:275–290. doi: 10.1006/jmbi.2000.3857. [DOI] [PubMed] [Google Scholar]
- 29.Emmert S, Kobayashi N, Khan SG, Kraemer KH. The xeroderma pigmentosum group C gene leads to selective repair of cyclobutane pyrimidine dimers rather than 6-4 photoproducts. Proc Natl Acad Sci U S A. 2000;97:2151–2156. doi: 10.1073/pnas.040559697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sugasawa K, Okuda Y, Saijo M, Nishi R, Matsuda N, Chu G, Mori T, Iwai S, Tanaka K, Tanaka K, Hanaoka F. UV-induced ubiquitylation of XPC protein mediated by UV-DDB-ubiquitin ligase complex. Cell. 2005;121:387–400. doi: 10.1016/j.cell.2005.02.035. [DOI] [PubMed] [Google Scholar]
- 31.Khan SG, Oh KS, Emmert S, Imoto K, Tamura D, DiGiovanna JJ, Shahlavi T, Armstrong N, Baker CC, Neuburg M, Zalewski C, Brewer C, Wiggs E, Schiffmann R, Kraemer KH. XPC initiation codon mutation in xeroderma pigmentosum patients with and without neurological symptoms. DNA Repair (Amst) 2009;8:114–125. doi: 10.1016/j.dnarep.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luijsterburg MS, Goedhart J, Moser J, Kool H, Geverts B, Houtsmuller AB, Mullenders LH, Vermeulen W, van Driel R. Dynamic in vivo interaction of DDB2 E3 ubiquitin ligase with UV-damaged DNA is independent of damage-recognition protein XPC. J Cell Sci. 2007;120:2706–2716. doi: 10.1242/jcs.008367. [DOI] [PubMed] [Google Scholar]
- 33.Takedachi A, Saijo M, Tanaka K. DDB2 complex-mediated ubiquitylation around DNA damage is oppositely regulated by XPC and Ku and contributes to the recruitment of XPA. Mol Cell Biol. 2010;30:2708–2723. doi: 10.1128/MCB.01460-09. [DOI] [PMC free article] [PubMed] [Google Scholar]