

Abstract
Alternative pre-mRNA splicing is a major mechanism by which the proteomic diversity of eukaryotic genomes is amplified. Much akin to neuropsychiatric disorders themselves, alternative splicing events can be influenced by genetic, developmental, and environmental factors. Here we review the evidence that abnormalities of splicing may contribute to the liability toward these disorders. First, we introduce the phenomenon of alternative splicing and describe the processes involved in its regulation. We then review the evidence for specific splicing abnormalities in a wide range of neuropsychiatric disorders, including psychotic disorders (schizophrenia), affective disorders (bipolar disorder and major depressive disorder), suicide, substance abuse disorders (cocaine abuse and alcoholism), and neurodevelopmental disorders (autism). Next, we provide a theoretical reworking of the concept of “gene-focused” epidemiologic and neurobiologic investigations. Lastly, we suggest potentially fruitful lines for future research that should illuminate the nature, extent, causes, and consequences of alternative splicing abnormalities in neuropsychiatric disorders.
Keywords: alternative splicing, mRNA, pre-mRNA, RNA processing, spliceosome
Introduction
Anthony and Gallo (2010) recently reviewed the contributions of splicing abnormalities and other RNA-processing events to neurological and neurodegenerative disorders; however, a decade has passed since the role of alternative splicing in neuropsychiatric disorders was reviewed by Grabowski and Black (2001). Much has changed in the last decade, including our understanding of the core splicing machinery and its interacting protein and ribonucleoprotein partners, as well as the ubiquity and importance of alternative splicing and the technological advances that facilitate its detection and quantification. Thus, we provide here a brief overview of the phenomenon of alternative splicing and a review of the evidence for splicing abnormalities in neuropsychiatric disorders, including several major psychotic, affective, neurodevelopmental, and substance use disorders.
Alternative Splicing: A Major Source of Functional Genomic and Phenotypic Diversity
High-throughput functional genomic (i.e., transcriptomic and proteomic) studies have revealed a great deal about the systems-level postmortem biological abnormalities that characterize a variety of neuropsychiatric conditions, including affective disorders such as bipolar disorder (BD) and major depressive disorder (MDD), and suicide (SUI) (Sequeira and Turecki, 2006; Sokolov, 2007), neurodevelopmental disorders such as autism (AUT) (Purcell et al., 2001), substance use disorders (SUDs) such as cocaine abuse/dependence and alcoholism (Sokolov, 2007), and psychotic disorders such as schizophrenia (SZ) (Iwamoto and Kato, 2006; Sokolov, 2007). Yet, most prior functional genomic studies of these illnesses have inadvertently overlooked one of the most potentially important sources of inter-individual (and perhaps disorder-related) phenotypic diversity: the expression of alternatively spliced variants (ASVs) or isoforms of these genes.
The primary transcripts (pre-mRNAs) of protein-coding genes include both introns and exons, requiring that the introns are removed and the exons are “spliced” or joined together before a mature, contiguous, translatable mRNA can be produced. This process is catalyzed by the “spliceosome”, which is usually comprised of five small nuclear ribonucleoproteins (snRNPs; the so-called “U proteins” U1, U2, U4, U5, and U6) and a large number of auxiliary factors (though a small number of pre-mRNAs are spliced by the cytosolic “minor spliceosome”, comprised of U5 along with U11, U12, U4atac, and U6atac). In its broadest sense, the term “splicing” refers to the joint and co-occurring processes of intron removal from pre-mRNAs and exon ligation into mRNAs that are ready to be translated into amino acids and, subsequently, proteins. Alternative splicing, therefore, refers to the variety of possible ways in which diverse mRNA species can be created from a single pre-mRNA through the skipping of exons, retention of introns, or use of alternative promoters, 5′ splice sites, 3′ splice sites, or poly-A tails (Figure 1).
Figure 1. Patterns of alternative splicing.
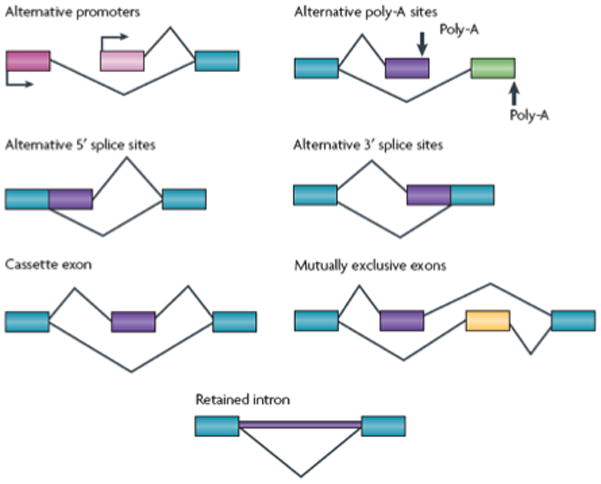
Transcripts from a gene can undergo many different patterns of alternative splicing. Transcriptional Initiation at different promoters (shown in two different shades of pink) generates alternative 5′-terminal exons that can be joined to a common 3′ exon (shown in blue) downstream. Similarly, alternative 3′ exons, with alternative polyadenylation sites, can be joined to a common upstream exon. Through the use of alternative 5′ or 3′ splice sites, exons can be extended or shortened in length. The most common pattern of alternative splicing is a cassette exon that can be included in the mRNA or skipped, inserting or deleting a portion of internal sequence. A special case of paired cassette exons show mutually exclusive splicing, where one exon or the other is included, but not both. Finally, the excision of an intron can be suppressed, to leave the retained intronic sequence in the mRNA that is exported to the cytoplasm. Many genes show multiple positions of alternative splicing, creating complex combinations of exons and alternative segments and a large family of encoded proteins.
Alternative splicing is one of the major mechanisms by which eukaryotes create enormous proteomic diversity from a smaller-than-expected number of genes; yet, it is but one of the many known mechanisms of RNA regulation and protein production, which include RNA editing (post-transcriptional changes to the base-sequence), post-transcriptional modification (by pseudouridylation, methylation, non-canonical polyadenlyation and RNA terminal polyuridylation), microRNA (miRNA) regulation (usually silencing) of translation, use of alternative translation start sites, and factors affecting RNA localization, stability, and turnover. (For a more thorough overview of splicing mechanics, see the comprehensive review by Li et al., 2007, and for a more thorough overview of other mechanisms of RNA and protein regulation, see the comprehensive reviews by Hartmann and Valcarcel, 2009, and Farajollahi and Maas, 2010). Collectively, these regulatory processes significantly complicate the widely perpetuated dogma of molecular biology in which transcribed mRNAs are simply and straightforwardly translated into amino acids and proteins. This complexity is further compounded by the spatial and temporal specificity of mRNA expression, and unfortunately, the rules that govern these processes are largely unknown. Yet, despite these uncertainties, we now have a much more accurate conceptualization than we did a decade ago of mRNA regulation and the important physiological consequences it can impart.
Biological Causes and Consequences of Alternative Splicing
The manner in which a gene’s exons are spliced together is a fundamental determinant of its protein’s functions. For example, the distinct ASVs of a gene may have diametrically opposed physiological functions (Clark et al., 2007). Thus, traditional discussions about a given gene’s function are moot unless a particular ASV of the gene is invoked. At the same time, modern neuropsychiatric geneticists must gain familiarity with alternative splicing in order to make biological sense of the large volumes of data being generated by genome-wide technologies, which may for example describe expression intensities of multiple discrete isoforms of a gene or implicate what superficially may appear to be innocuous intronic or synonymous variants as significant risk factors for neuropsychiatric disorders.
The human brain has unique patterns of alternative splicing, and expresses higher levels of splicing than most other tissues (Yeo et al., 2004). During neuronal development, many different splicing patterns are induced, probably via differential expression of splicing auxiliary factors or alternative splicing regulators (ASRs), which interact with the spliceosomal machinery that excises introns and ligates exons of pre-mRNAs to make functional genetic transcripts. These diverse splicing patterns dictate important regulatory decisions in many steps of neuronal development. By affecting proteins such as growth factors (e.g., FGF8) (Gemel et al., 1996; Ghosh et al., 1996; Li et al., 2007; MacArthur et al., 1995; Olsen et al., 2006) and cell-adhesion molecules (e.g., DSCAM, NLGNs, and NRXNs) (Nie et al., 2004; Ullrich et al., 1995), alternative splicing shapes processes such as cell-fate determination (via Numb proteins) (Dho et al., 1999; Dho et al., 2006; Reugels et al., 2006), axon guidance (via DSCAM) (Schmucker et al., 2000; Zipursky et al., 2006), and synaptogenesis (Li et al., 2007). In adult neurons, synaptic remodeling or strengthening is regulated by alternative splicing and the relative expression levels of ASVs. Proteins that are important for synaptic vesicle release (e.g., SNAP25) (Bark et al., 1995; Sorensen et al., 2003), post-synaptic density constitution (e.g., PSD95), and long-term potentiation (e.g., APOER2) (Beffert and others 2006; Beffert and others 2005) have been shown to have ASVs that each have a distinct functional role in relaying signals (Li et al., 2007). In turn, splicing events that alter neuronal activity are themselves differentially regulated by incoming stimuli, such as chronic excitatory depolarization (Hepp et al., 2001; Vallano et al., 1999; Zacharias and Strehler, 1996).
Other examples of alternatively spliced proteins include ion channels, neurotransmitter receptors, and proteins involved in calcium signaling (Li et al., 2007). For example, the N-type voltage-gated calcium channels located at presynaptic terminals control neurotransmitter release and are subject to alternative splicing (Bell et al., 2004). Two mutually exclusive isoforms (Cav2.2e37a and Cav2.2e37b) of this calcium channel differ in their intracellular domains (Lipscombe and Raingo, 2007). The inclusion of exon 37a creates a module that couples the N-type channel to a powerful form of G protein-dependent inhibition. Neurotransmitters such as GABA and enkephalins inhibit N-type channels in nociceptive neurons through their G-protein-coupled receptors and attenuate nociception. Critically, cells may control the sensitivity of their N-type channels to G-protein-mediated voltage-independent inhibition by adjusting the ratio of Cav2.2e37a to Cav2.2e37b isoforms (Lipscombe and Raingo, 2007; Raingo et al., 2007), illustrating the importance of ASV expression ratios in determining the physiological activity of cells.
Alternative Splicing of Candidate Genes for Neuropsychiatric Disorders
Schizophrenia (SZ)
ASVs with discrete localization and competing functional profiles have been identified in the gene encoding the D2 dopamine receptor (DRD2), which is the main antagonized target of all effective antipsychotic medications. The two most common ASVs of DRD2 are the D2short and D2long isoforms, which differ in the exclusion or inclusion (respectively) of exon 6 that encodes 29 amino acids in the G-protein-binding third intracellular loop of the protein. Usiello et al. (2000) showed that the D2short receptor served as the prototypical inhibitory presynaptic autoreceptor, while D2long receptors were located primarily at postsynaptic sites. Further, Zhang et al. (2007) found that the T alleles of rs1076560 (which maps to intron 6) and rs2283265 (in intron 5) favored inclusion of exon 6 of DRD2, resulting in decreased expression of the D2short splice variant, greater fMRI activity of striatum and prefrontal cortex during working memory tests, and reduced working memory and attentional performance in healthy subjects. Bertolino et al. (2009) extended these molecular, neurobiological, and behavioral effects of the T/T haplotype to patients with schizophrenia, which is consistent with the association we have observed between these alleles and risk for the disorder (Glatt et al., Submitted; Glatt et al., 2009b). Although it is presently unknown how these polymorphisms influence the “spliceability” of DRD2 and the resultant behavioral, neurobiological, and clinical phenotypes, it is possible that these variants introduce, destroy, strengthen, or weaken binding sites for known ASRs. For example, the risk-associated T allele of rs2283265 strengthens a binding motif for hnRNP-I relative to the wild-type G allele, but it also creates a U2AF2 binding site while abolishing an SRp40 binding site. On the other hand, rs1076560 lengthens the binding motifs for two hnRNPs, including hnRNP-F and hnRNP-H. Thus, possessing the risk-associated alleles of these variants (or their haplotypic combination, which is often the case due to the high degree of linkage disequilibrium between them) might facilitate the inclusion of exon 6 leading to the observed over-expression of D2long receptors relative to D2short receptors in postmortem brain from both SZ and unaffected individuals. This possibility, coupled with the over-transmission of rs1076560 and rs2283265 risk alleles to patients with SZ, might provide a possible mechanism linking risk-associated polymorphisms with their molecular consequences. It should be noted that neither of these DRD2 polymorphisms, with odds ratios of approximately 1.1–1.2 for schizophrenia, has been identified as a major risk factor for the disorder, nor has either variant been found to be reliably associated with the disorder in any recent genome-wide association scans (GWAS); however, this does not preclude the possibility that these variants have a small but reliable influence on the overall susceptibility toward schizophrenia, which could be mediated via the effects of these polymorphisms on the ability or likelihood of alternative DRD2 splicing.
In addition to DRD2, a small handful of candidate genes for SZ have been found to exhibit abnormal splicing patterns in postmortem brain, including CTNNA2 (Mexal et al., 2008), DISC1 (Nakata et al., 2009), ERBB4 (Law et al., 2007; Silberberg et al., 2006), ESR1 (Weickert et al., 2008), GRIN1 (Kristiansen et al., 2006; Le Corre et al., 2000), GRM3 (Sartorius et al., 2008), NRG1 (Tan et al., 2007), and RGS4 (Ding and Hegde, 2009). Such results have further strengthened the support for this cadre of already strong candidate genes, but for most of these genes the functional difference between their alternate isoforms is not known, and thus the physiological consequence of dysregulation of a particular isoform is unclear. Yet, in some instances, the functional difference between isoforms is known, and this in turn sheds light on possible mechanisms by which these genes may influence susceptibility to the disorder. For example, the isoforms of ERBB4 that are reliably up-regulated in SZ include the alternatively spliced exon 16, which encodes a metalloprotease cleavable extracellular domain, and exon 26, which encodes a cytoplasmic domain with a phosphotidylinositol-3 kinase binding site that plays a role in chemotaxis and survival (Law et al., 2007; Silberberg et al., 2006). Although replication studies are needed to confirm these findings, they illustrate potential mechanisms whereby alternative splicing abnormalities might lead to disease.
Affective Disorders
Some candidate genes for SZ have been found to exhibit splicing irregularities in other psychiatric disorders as well. For example, ASVs of GABRB2 and PDE4B have been found to be similarly dysregulated in postmortem brain tissue samples from both SZ and BD patients (Fatemi et al., 2008; Huntsman et al., 1998; Zhao et al., 2009) while the β isoform of CAMKII was found to be up-regulated in both SZ and MDD but not BD (Novak et al., 2006). Such results highlight potentially common or unique molecular components of these disorders, or perhaps of their sometimes-shared phenotypic aspects (e.g., psychosis, or mood dysregulation).
Beyond GABRB2, PDE4B, and CAMKII, other genes have been found to be alternatively spliced in postmortem brain tissue samples from individuals with affective disorders. In fact, one of the earliest observations of disorder-associated alternative splicing in neuropsychiatry was provided by Vawter et al. (1998), who found increased NCAM isoform expression in postmortem hippocampus from BD patients compared to samples from SZ patients, SUI cases, and unaffected control subjects. Subsequently, Vawter’s group found under-expression of an isoform of NCAM1 containing a mini-exon “c” and the secreted “SEC” exon in postmortem cerebellum in BD but not SZ when compared to control tissue samples (Atz et al., 2007), again highlighting the potential regional (and phenotypic) specificity of splicing abnormalities in these disorders.
Suicide (SUI)
A number of psychiatric disorders (including affective disorders, SZ, and SUDs) are associated with an increased risk of SUI. In turn, diagnostic boundaries seem to have little bearing on the ability to detect alternative splicing events as they relate to SUI. For example, Dracheva et al. (2008) observed an increase in RNAsp2:RNAsp1 ratios of HTR2C in postmortem dorsolateral prefrontal cortex of SUI completers regardless of diagnosis, but no difference between SUI completers with BD and SUI completers with SZ. Interestingly, the ratio of these two HTR2C isoforms was correlated (but not perfectly) with RNA-editing efficiency at four of the five known editing sites in the mRNA. In other work, Ernst et al. (2009) found, in a subset of SUI completers, decreased expression of the T1 isoform of TRKB throughout the frontal lobes; however, this effect was not seen in the cerebellum, nor was it apparent in all SUI completers. Of note, the dysregulation of the T1 isoform was affected by the methylation status of various CpG dinucelotides in the gene, suggesting that disorder-relevant ASV expression (and ASV expression in general) may be governed by epigenetic processes.
Substance Use Disorders (SUDs)
Collectively, SUDs have not often been the object of alternative splicing analyses in human postmortem brain tissue. Yet, though not as often examined for alternative-splicing abnormalities as other neuropsychiatric disorders, SUDs have the advantage of more straightforward and face-valid modeling in animals. This is critical because research on postmortem brain tissue from individuals with SUDs (or any neuropsychiatric disorder, for that matter) will be unable to resolve whether dysregulation of particular splice variants is a consequence or a cause of the disorder. As such, there have been a number of investigations of the effects of either self- or investigator-administered substances of abuse (including alcohol, cocaine, and opiates) on the alternative splicing of candidate genes like BDNF, GNAO1, GRIN1, OPRK1, and OPRM1 in rat and mice models of human SUDs, or in animal neural tissue to which such substances are exogenously applied (Brunk et al., 2008; Kumari and Anji, 2005; Liu et al., 2006; Loftis and Janowsky, 2002; Pan et al., 2009; Saito et al., 2003; Winkler et al., 1999). These studies, by and large, have supported the hypothesis that exposure to addictive substances changes the expression of particular mRNA isoforms, which suggests a possible mechanism for the neuropsychiatric consequences of SUDs. These findings do not preclude the possibility that humans who inherit or acquire the capacity to over- or under-express such ASVs are at heightened susceptibility to develop an SUD once they are exposed. Future work in animal models could evaluate this hypothesis by artificially dysregulating particular implicated ASVs and monitoring differences in the acquisition, severity, and/or persistence of drug-seeking behavior in these animals. Unfortunately such linkages have yet to be made, but since SUDs constitute gene-environment disorders with at least one controllable environmental exposure (i.e., the ingestion of the drug), they are well-suited to further experimentation in animal models and present perhaps the best target among neuropsychiatric disorders for exhaustive clarification of the role of alternative splicing.
Autism (AUT)
Animal models also have been useful in identifying putative AUT-associated alternatively spliced isoforms of candidate genes. In a series of publications over the past five years, Sadakata and Furuichi et al. have mapped the tissue distribution (Sadakata et al., 2007c) and molecular physiological functions (Sadakata and Furuichi, 2009) of the AUT positional candidate gene CADPS2 in the mouse, and identified a plethora of cerebellar morphological and functional anomalies (Sadakata et al., 2007a). They also documented AUT-like behavioral abnormalities (Sadakata et al., 2007b) in mice lacking the gene, and discovered a novel CADPS2 ASV expressed in blood samples from several AUT patients (Sadakata et al., 2007b). The typically deleted exon (exon 3) encodes a dynactin 1-binding domain which, when absent, prevents axonal transport of CADPS2 protein. Further evaluation of the role of CADPS2 splicing in AUT-like behavior in both mouse and human studies seems strongly warranted.
Genetic Influences on Disorder-Associated Alternative Splicing
In many instances, the over- or under-expression of particular ASVs in a neuropsychiatric disorder has been tied to a particular genetic polymorphism, thus identifying these variants as splicing quantitative trait loci (sQTLs) (Pickrell et al., 2010; Ryten et al., 2009). DISC1 has long been recognized as one of the strongest risk factors for SZ, but the causal mechanism remains unknown; alternative splicing governed by an sQTL was recently identified as one potential contributor. Nakata et al. (2009) found Δ7, Δ8, and Δ3 ASVs, which encode truncated DISC1 proteins, were expressed more in the hippocampus of patients with SZ, and that this over-expression was associated with known SZ-risk single nucleotide polymorphisms (SNPs rs821616, rs6675281, and rs821597). Prefrontal cortical dysregulation of NCAM1 ASVs containing the mini-exon (c) and the secreted exon (SEC) in BD (not observed in SZ) has also been found to be under genomic control, in this case by rs2303377 (Atz et al., 2007).
In the realm of SUDs, Hishimoto et al. (2007) provided evidence for genetic association between polymorphisms in NRXN3 and alcohol dependence, identifying as the strongest susceptibility locus rs8019381, which is adjacent to the recognized splice site for exon 23 of the gene. This known alternatively spliced exon determines whether the NRXN3 protein is soluble or membrane-bound. In conjunction, these authors showed that the risk-conferring allele of this susceptibility polymorphism is associated with lower expression levels of the soluble and membrane-bound isoforms of the gene in postmortem samples of cerebral cortex from unaffected individuals; however, dysregulation of either ASV in the brains of alcohol-dependent subjects has yet to be demonstrated.
In contrast to these disorder-specific observations, splicing abnormalities in GABRB2 appear to be common to both SZ and BD. For example, Zhao et al. (2009) found increased expression of the β2S1 ASV and decreased expression of the β2S2 ASV in postmortem dorsolateral prefrontal cortex in both SZ and BD, both of which were correlated with GABRB2 SNPs (including rs187269 and rs2546620). Of note, all of the splicing-associated alleles of the sQTLs reviewed above are relatively common variants, occurring at more than 5% frequency in all evaluated populations; however, this does not preclude the possibility that these associations are merely signals (via linkage disequilibrium) for other undetected variants more closely and causally related to the observed effects on splicing, and such may comprise either common or possibly rare variants. Also, each of the sQTLs described previously has been cis-acting (i.e., influences the splicing of the gene in which it is situated), but it is also probable that there are many trans-acting sQTLs that influence the splicing of genes elsewhere in the genome, and that the splicing of some susceptibility genes for neuropsychiatric disorders will be influenced by these as well.
Additional evidence for the possible existence of psychiatric disorder-associated sQTLs in the genome comes from the recent wave of GWASs in psychiatry, which have identified at least 19 SNPs meeting or surpassing the most commonly accepted threshold for asserting genome-wide significant association (p<5×10−8) with one or more disorders (Hindorff et al., 2011). At first blush, it has been difficult to understand the biological significance of most of these results, since the SNPs typically included in GWAS platforms have not been chosen based on their functionality (especially with regard to splicing), but rather in regard to their genomic positions and their abilities to tag blocks of linkage disequilibrium within which the actual functional SNPs may reside. However, we note here the possibility that, in several instances, these disorder-associated tagging SNPs may themselves have biological significance mediated via their effects on splicing. For example, 10 of the 19 GWAS-significant SNPs associated with one or more psychiatric disorders are within genes (the remaining nine are intergenic) and, as determined by the Splicing Rainbow software tool (Morais and Valcarel, 2010), each one of these 10 SNPs has the capacity to influence splicing via the introduction, abolition, strengthening, or weakening of one or more ASR-binding motifs (Table 1). Each of these 10 polymorphisms is in linkage disequilibrium with many other potentially functional polymorphisms (including some that affect splicing and some that do not), so it is premature to assert that the detected variants have a direct impact on risk for these disorders, and that these effects are mediated via alterations in splicing, but these are clearly strong possibilities that should be followed up.
Table 1.
Genetic Polymorphisms Surpassing Criterion for Genome-Wide Significant Association (p≤5×10−8) with Psychiatric Disorders
| Disorder | P-Value | SNP | Gene | Location | Effect on ASR-Binding Motif(s)* |
|---|---|---|---|---|---|
| Attention-deficit/hyperactivity disorder | 1 × 10−8 | rs7995215 | GPC6 | Intron | (>)ASF/SF2; (+)hnRNPU; (>)SC35 |
| Attention-deficit/hyperactivity disorder | 1 × 10−8 | rs864643 | MOBP | Intron | (>)ASF/SF2 |
| Alcohol dependence | 1 × 10−8 | rs7590720 | --- | Intergenic | |
| Autism | 4 × 10−8 | rs4141463 | MACROD2 | Intron | (−)hnRNPC1/C2; (<)hnRNPI; (>)HuR; (−)SRp55; (>)Sxl; (>)U2AF65 |
| Autism | 2 × 10−10 | rs4307059 | --- | Intergenic | |
| Bipolar disorder | 9 × 10−9 | rs10994336 | --- | Intergenic | |
| Bipolar disorder | 2 × 10−8 | rs1012053 | DGKH | Intron | (+)ASF/SF2; (−)hnRNPK; (>)SRp40; (>)SRp55 |
| Bipolar disorder and major depressive disorder | 3 × 10−8 | rs1006737 | CACNA1C | Intron | (+)hnRNPF; (+)hnRNPH; (>)hnRNPU; (>)SRp40; (>)SRp55; (+) Tra2Beta |
| Bipolar disorder and major depressive disorder | 2 × 10−9 | rs2251219 | PBRM1 | Exon | (+)hnRNPF; (+)hnRNPH; (−)Tra2Beta |
| Bipolar disorder and schizophrenia | 6 × 10−9 | rs11789399 | --- | Intergenic | |
| Bipolar disorder and schizophrenia | 4 × 10−8 | rs12201676 | --- | Intergenic | |
| Narcolepsy | 3 × 10−8 | rs2858884 | --- | Intergenic | |
| Narcolepsy | 3 × 10−22 | rs1154155 | TRAA | Exon | (>)ASF/SF2; (−)hnRNPG; (<)SR9GB; (>)SRp55; (−)Tra2Beta |
| Nicotine dependence | 6 × 10−20 | rs1051730 | CHRNA3 | Exon | (−)SRp20; (<)SRp40; (<)SRp55 |
| Schizophrenia | 1 × 10−12 | rs6932590 | --- | Intergenic | |
| Schizophrenia | 2 × 10−10 | rs3131296 | NOTCH4 | Intron | (+)ASF/SF2; (<)SRp40; (<)SRp55 |
| Schizophrenia | 2 × 10−9 | rs12807809 | --- | Intergenic | |
| Schizophrenia | 4 × 10−9 | rs9960767 | TCF4 | Exon | (+)SRp40; (>)SRp55 |
| Schizophrenia | 1 × 10−8 | rs13194053 | --- | Intergenic |
(+) introduces; (−) abolishes; (>) strengthens; (<) weakens
The Next Dimension: Whole-Exome and Whole-Spliceome Profiling
The average number of exons in each of the approximately 23,000 protein-coding human genes is approximately nine (Sakharkar et al., 2004), and therefore the human “exome” (i.e., the full compendium of expressed exons) can be estimated to include at least 207,000 exons, though probably many more. In turn, as many as 95% of human genes are alternatively spliced (i.e., have at least two different isoforms) (Barash et al.), and the average number of ASVs per gene is approximately three (Banks et al., 2000), so the human “spliceome” (i.e., the full compendium of alternatively spliced protein-coding variants, not to be confused with the above-mentioned and similarly spelled “spliceosome”) can be estimated to include at least 65,500 ASVs (though, again, likely more). This degree of biological variability (especially such physiologically important variability) should not be ignored.
Technological advances in the last half-decade (e.g., the Affymetrix Human Gene and Exon 1.0 ST Arrays) now allow for improved sensitivity in measuring the transcriptome while simultaneously querying the exome and spliceome. Recently our group and others have employed this technology to study BD and SZ, and the results of our initial work support the notion that exomic and spliceomic abnormalities are common in these disorders. For example, we recently published work in which we used the Exon array to measure exome and spliceome expression in peripheral blood mononuclear cells (PBMCs) from 13 SZ patients, 9 BD patients, and 8 non-mentally ill control subjects (Glatt et al., 2009a). Each diagnostic group was compared to each other, and the combined group of BD and SZ patients was also compared to the control group. Furthermore, we compared subjects with a history of psychosis (PSYCH+) to subjects without such history (PSYCH−), regardless of diagnostic boundaries (the former included 6 of the 9 BD patients, while the latter included the remaining 3 BD patients). Even after applying very stringent Bonferroni corrections for the 21,866 full-length gene transcripts analyzed, and statistically controlling for the potential confounding effects of exposure to antipsychotic medication and mood stabilizers, we found significant interactions between diagnosis and exon identity, consistent with group differences in rates or types of alternative splicing of one or more exons. By far, the greatest disparity in patterns of expression of alternatively spliced genes was seen when comparing groups with a different history of psychosis. Of the 16,555 nominally significantly dysregulated exons in PSYCH+, only 64 were down-regulated relative to the PSYCH- group, while the remaining 16,491 exons were up-regulated; this represented a highly significant departure from the ratio expected by chance (p<1.00×10−10).
These results suggested to us, for the first time, that there might be a systematic abnormality in the splicing machinery causing widespread exonic over-inclusion in psychotic disorders such as SZ and BD. Indeed, there are known examples of “spliceopathies” involving disruption of the basal splicing machinery, such as spinal muscular atrophy (caused by a loss-of-function mutation in SMN1, which is an assembler of snRNP complexes) and retinitis pigmentosa (caused by mutations in HPRP3, which is a U4- and U6-associated protein, and PRPF31, which activates the spliceosome). It seems unlikely however that SZ and BD are also examples of true spliceopathies, as disruption of any of the core spliceosomal snRNPs would result in abnormal splicing of most genes in the genome, which is not what has been observed by our group or others. Thus, although the single most significantly dysregulated transcript in PBMCs from the PSYCH+ group was SNRPC, which encodes one of the specific protein components of the U1 spliceosomal snRNP (p=0.0001), this dysregulation did not lead to universally faulty splicing of other genes. Disruption of splicing auxiliary factors or ASRs, each of which regulates the splicing of only a select structurally conserved portion of genes in the transcriptome, was also observed. The detected dysregulation of PUF60 (poly-U-binding splicing factor), SF3A2 (splicing factor 3A subunit 2), and SFRS12 (splicing factor, arginine/serine-rich 12), for example, may provide more attractive theoretical mechanisms for the widespread (but not necessarily global) and directionally uniform (i.e., consistent exon up-regulation) splicing abnormalities we have seen in psychosis. Alternatively, neuropsychiatric disorders such as SZ and BD may not have any reliable link to alterations in the spliceosome, its auxiliary factors, or ASRs, but still be characterized by the simultaneous dysregulation of particular physiologically important ASVs of a number of different genes.
Beyond our blood-based work, we recently completed a pilot study of alternative splicing in postmortem tissue samples of two brain regions (Brodmann area 10 [BA10] and caudate) from SZ and unaffected control subjects in the Harvard Brain Tissue Resource Center, again using an exon-profiling array from Affymetrix. The results of this study are useful for the present discussion for three reasons. First, we detected a large number of genes that show evidence of dysregulation of particular ASVs in SZ in one or both brain regions in SZ, and these are targets worthy of further validation efforts. Second, the study provides further support for the notion of abnormal or dysregulated ASRs in the disorder. For example, we found significant dysregulation of several splicing factors, including HNRNPH1, HNRNPH3, HNRNPC, and SFRS16, in BA10 in SZ. Third, this work serves as a functional genomic validation of our prior blood-based biomarker study of ASVs in SZ and BD. For example, 44 (28%) of the 156 genes that we previously found to have highly reliable Bonferroni-corrected significant abnormal expression of an ASV in blood in psychotic subjects were also found to have at least nominally significant abnormal ASV expression in either caudate or BA10 in postmortem brains from SZ patients. Further, eight of these genes (ADAR, ARHGAP26, BIRC6, MAPK14, STXBP2, SYNE2, UTRN, and ZDHHC17) had ASVs that were significantly dysregulated in the same direction in blood (with Bonferroni-corrected significance) and in both brain regions (with at least nominal significance). After performing a Bonferroni correction in this pilot study of postmortem brain tissue from just four cases and four control subjects, we lacked inferential power to declare statistically significant replication of the results that attained Bonferroni-corrected significance in the study of peripheral blood; however, the convergence of results across tissue types and studies reveals a degree of overlap that is quite compelling, even at this early stage of investigation. Sullivan et al. (2006) previously documented that gene expression levels in various regions of the brain were reasonably well correlated with the expression levels of those same genes in peripheral blood, and our work suggests that potentially disorder-related changes in ASV expression may have some degree of correspondence across tissues as well. If the magnitude of the effects observed in postmortem brain persists as more subjects are studied and sample size (and resultant power) increases, then the genes identified as dysregulated in peripheral blood and both brain regions in SZ will highlight and validate two areas of further work which we are vigorously pursuing, including the detection of factors (especially sQTLs) that may be capable of disrupting the expression of particular ASVs regardless of tissue type, and the validation of blood-based biomarkers for these mental disorders. Lastly, this pilot study lends support to hypotheses generated by prior work. In particular, it was gratifying to observe significantly differential expression of alternatively constituted CHI3L1 transcripts in the peripheral blood in SZ as well as comparable dysregulation of this gene’s ASVs in both caudate and BA10 in SZ. These results are consistent with results from Zhao et al. (2007) who first discovered that SZ-associated risk haplotypes of CHI3L1 were associated with lower transcriptional activity and lower expression of the gene. Subsequent multi-center case-control studies, as well as meta-analysis, have provided further evidence of association between this gene and risk for schizophrenia in Asian samples (Ohi et al., 2010), but further collaborative work will be required to determine if the risk-associated variants of CHI3L1 also influence the expression of particular ASVs of the gene, rather than simply influencing overall transcriptional activity. In observing dysregulation of ERBB4 ASVs in both BA10 and caudate, we have also confirmed prior postmortem work linking particular splice variants of this NRG1 cofactor to risk for the disorder (Law et al., 2007; Silberberg et al., 2006). Lastly, several genes for which functionally distinct and neurodevelopmentally important ASVs are known (including NUMBL, DSCAM, FGF, SNAP25, and NLGN2 and NLGN4X, all of which were described above) were also found to have SZ-associated dysregulation of one or more ASVs in postmortem BA10, but not caudate, reinforcing the importance of pursuing both ubiquitous and regionally specific alterations in ASV expression.
Abnormalities in the expression of individual ASVs might help account for the failure to observe reliable functional genomic (i.e., transcriptomic) signatures of these disorders, as only the most robust differences in important ASV expression abnormalities would have been detected previously on platforms that assess “full-length” transcript expression, i.e., effects of genes with no known ASVs or genes for which all ASVs were similarly dysregulated. In appreciation of the prevalence of alternatively spliced genes and the physiological capabilities and differences between ASVs of the same gene, we expect such scenarios will be few and far between relative to the number of reliable differences awaiting discovery at the level of individual exons and ASVs. In fact, we already know of many instances in which ASVs of the same gene perform very different (even antagonistic) functions (Clark et al., 2007). Thus it is simply no longer productive to attempt to analyze “whole-gene” expression —or even discuss “gene function”—without reference to individual ASVs. Beyond the realm of functional genomics, we hope and anticipate that an appreciation of the importance of alternative splicing will filter through to other areas of psychiatric genetic investigation, thus allowing for a more precise depiction of causal molecular mechanisms by epidemiologists and neurobiologists alike.
In our own initial studies of alternative splicing in peripheral blood and various postmortem brain regions in SZ and BD, we have utilized the Affymetrix platform of microarrays, which provide a relatively sensitive, specific, reliable, and affordable method for assaying exonic mRNA expression intensities. This approach has limitations too; chiefly, the detection of abnormally expressed exons cannot unequivocally be ascribed to the dysregulation of a given ASV, only inferred based on expected patterns of expression given the configuration of known isoforms. Furthermore, even the latest tiling microarrays lack the ability to detect novel isoforms for which no probe is present on the pre-fabricated microarray. A relatively novel alternative approach is the generalization of so-called “next-generation” sequencing to the identification and quantification of mRNAs, a process known as RNA-seq (Marguerat and Bahler, 2010). This technique overcomes many of the major hurdles of microarray-based approaches. For example, RNA-seq has a much broader dynamic range than microarray-based assays (perhaps spanning five orders of magnitude) and, with enough sequencing depth, can detect very low-expressed transcripts. RNA-seq is also more sensitive than microarray analysis, and thus typically detects more transcripts per sample that are expressed significantly above background noise levels. RNA-seq is much better suited than microarray techniques to detect post-transcriptional modifications such as alternative splicing, but also RNA editing and other modifications that drive dissimilarity of expressed transcripts and their predicted probe sequences. These advantages come at a cost however. Specifically, whole-transcriptome RNA-seq is much more expensive than microarray analysis on a per-sample basis. In addition, RNA-seq currently requires larger investments of time in preparation stages (e.g., in the creation of libraries) and at data-analytic stages due to the volumes of sequence to be interrogated and data to be generated. The actual costs of RNA-seq are projected to come down sharply as the technology improves and sees wider adoption by more end-users; so too will the amounts of time and effort required of bioinformaticians and biostatisticians working with RNA-seq projects as protocols for library generation and data analysis become more refined and readily accessible as a consequence of stronger market demand. The imminent widespread conversion from microarray-based hybridization techniques to RNA-seq should have an enormous impact on our ability to generate accurate profiles of aberrant ASV expression in various tissues and neuropsychiatric disorders, but for the time being there is still a place for both microarray-based and RNA-seq approaches in the arsenal of neuropsychiatric geneticists and neurobiologists.
Future Directions
Aside from building a theoretical foundation and conducting preliminary studies, due to a lack of empirical data we cannot yet definitively describe the contributions of alternative splicing to BD, SZ, and other neuropsychiatric disorders. This void immediately indicates areas of high priority for future research into the nature, extent, causes, and consequences of alternative splicing anomalies in these major mental illnesses. Today the field is presented with an enormous opportunity, as the methods of microarray analysis of gene expression are already well worked out from prior transcriptomic studies while, concurrently, new exon-specific profiling technologies are ready to be deployed to generate exon- and ASV-level data. Thus, first and foremost, it seems most logical to utilize whole-exome and whole-spliceome technologies, such as the Affymetrix Exon array or RNA-seq, to determine which ASVs of all genes in the human transcriptome are dysregulated in a variety of postmortem brain regions—and peripheral blood samples (Rollins et al., 2010)—from individuals with neuropsychiatric disorders relative to unaffected individuals.
By identifying ASV expression abnormalities that are distinct to (or shared between) various neuropsychiatric disorders, we will be able to formulate new hypotheses regarding the fundamental nature of the biological relatedness/independence of these conditions. By identifying which abnormalities occur throughout the brain and which exist only in specific brain regions, we may also be able to shed light on the existence of sQTLs for some ASVs while illuminating epigenetic influences on the expression of others. Consistent with a pseudo-spliceopathic basis for these disorders, it should also be possible to identify particular protein domains that are commonly dysregulated in one or more conditions, indicating possible targeting by the same ASR(s). Aside from identifying specific dysregulated ASVs or groups of functionally, structurally, or ontologically related ASVs that are dysregulated in neuropsychiatric disorders, it will be useful to survey known ASRs and splicing auxiliary factors for structural, functional, or regulatory genetic variations as a possible means of identifying a heritable basis for splicing anomalies that may be found to characterize these disorders.
Neuropsychiatric disorders are common, chronic, and disabling conditions undoubtedly caused by the interactions of genes and environmental factors. The limited number of reliable risk factors identified to date collectively account for very little of the variance in who becomes affected and who is spared from one or more of these illnesses. A complete accounting of ASV-expression abnormalities in these disorders will undoubtedly allow the identification of candidate genes for further study as causal factors. At the same time it will allow the identification of dysregulated ASVs that are not governed by genetic polymorphisms, thus identifying these as putative environmentally regulated transcripts. By highlighting potential lines of investigation into ASVs in neuropsychiatry, we hope to inspire work aimed at generating a more sophisticated understanding of the functional genomic correlates (if not causes) of these disorders. Longitudinal studies of at-risk individuals, as well as the examination of unaffected (and thus, untreated) first-degree relatives of affected individuals and the continued use of animal models, should allow the advancement of stronger inferences linking ASV abnormalities to these disorders in a causal manner.
Although palliative treatments for most neuropsychiatric disorders exist, none are curative and most have serious side effects. Current treatments largely were discovered serendipitously or through incremental expansion of the prevailing disease models, while none are based on a strict understanding of the mechanisms that cause the disorders. Functional molecular genetic studies (including surveys of dysregulated exons and ASVs) hold open the possibility of discovering new causal pathways and dramatically improving our knowledge of causal mechanisms. If such abnormalities can be reliably detected in neuropsychiatric disorders, this ultimately may foster medicinal chemistry applications and the development of novel therapeutics that more precisely target mis-spliced mRNAs or dysregulated exons to more efficiently reduce suffering. If these disorders are shown to result from abnormalities in the splicing machinery (i.e., involving one or more ASR or splicing auxiliary factor that target groups of structurally related genes, rather than to the dysregulation of any particular ASV or exon), then that would suggest a more efficient alternate avenue for therapeutic intervention. For example, rather than targeting the numerous ASVs dysregulated by an underlying abnormality in the splicing machinery, the most efficient medication might be one developed to modulate overall functionality of individual ASRs or auxiliary proteins (Buratti et al., 2006; Garcia-Blanco et al., 2004). Any of these achievements would constitute a major public health success, translating into marked improvements in the lives of individuals with neuropsychiatric disorders as well as their families and society in general.
Acknowledgments
This work was supported in part by U.S. National Institutes of Health grants R21MH075027 (M.T.T.) and P50MH081755-0003 (S.J.G.), and a Katowitz/Radin Young Investigator Award and the Sidney R. Baer, Jr. Prize for Schizophrenia Research from NARSAD (S.J.G.). The authors wish to thank Dr. Francine Benes and George Tejada of the Harvard Brain Tissue Resource Center for providing postmortem brain tissue samples, and Elizabeth Sergison, Sean Bialosuknia, Justin Zelenka, Cheryl A. Roe, M.S., and Sharon D. Chandler, Ph.D., for their critical appraisal of—and additions to—the manuscript.
References
- Anthony K, Gallo JM. Aberrant RNA processing events in neurological disorders. Brain Res. 2010;1338:67–77. doi: 10.1016/j.brainres.2010.03.008. [DOI] [PubMed] [Google Scholar]
- Atz ME, Rollins B, Vawter MP. NCAM1 association study of bipolar disorder and schizophrenia: polymorphisms and alternatively spliced isoforms lead to similarities and differences. Psychiatric Genetics. 2007;17(2):55–67. doi: 10.1097/YPG.0b013e328012d850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks RE, Dunn MJ, Hochstrasser DF, Sanchez JC, Blackstock W, Pappin DJ, Selby PJ. Proteomics: new perspectives, new biomedical opportunities. Lancet. 2000;356(9243):1749–1756. doi: 10.1016/S0140-6736(00)03214-1. [DOI] [PubMed] [Google Scholar]
- Barash Y, Calarco JA, Gao W, Pan Q, Wang X, Shai O, Blencowe BJ, Frey BJ. Deciphering the splicing code. Nature. 465(7294):53–59. doi: 10.1038/nature09000. [DOI] [PubMed] [Google Scholar]
- Bark IC, Hahn KM, Ryabinin AE, Wilson MC. Differential expression of SNAP-25 protein isoforms during divergent vesicle fusion events of neural development. Proceedings of the National Academy of Science of the United States of America. 1995;92(5):1510–1514. doi: 10.1073/pnas.92.5.1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell TJ, Thaler C, Castiglioni AJ, Helton TD, Lipscombe D. Cell-specific alternative splicing increases calcium channel current density in the pain pathway. Neuron. 2004;41(1):127–138. doi: 10.1016/s0896-6273(03)00801-8. [DOI] [PubMed] [Google Scholar]
- Bertolino A, Fazio L, Caforio G, Blasi G, Rampino A, Romano R, Di Giorgio A, Taurisano P, Papp A, Pinsonneault J, et al. Functional variants of the dopamine receptor D2 gene modulate prefronto-striatal phenotypes in schizophrenia. Brain. 2009;132(Pt 2):417–425. doi: 10.1093/brain/awn248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunk I, Blex C, Sanchis-Segura C, Sternberg J, Perreau-Lenz S, Bilbao A, Hortnagl H, Baron J, Juranek J, Laube G, et al. Deletion of Go2alpha abolishes cocaine-induced behavioral sensitization by disturbing the striatal dopamine system. FASEB Journal. 2008;22(10):3736–3746. doi: 10.1096/fj.08-111245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buratti E, Baralle M, Baralle FE. Defective splicing, disease and therapy: searching for master checkpoints in exon definition. Nucleic Acids Research. 2006;34(12):3494–3510. doi: 10.1093/nar/gkl498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark TA, Schweitzer AC, Chen TX, Staples MK, Lu G, Wang H, Williams A, Blume JE. Discovery of tissue-specific exons using comprehensive human exon microarrays. Genome Biology. 2007;8(4):R64. doi: 10.1186/gb-2007-8-4-r64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dho SE, French MB, Woods SA, McGlade CJ. Characterization of four mammalian numb protein isoforms. Identification of cytoplasmic and membrane-associated variants of the phosphotyrosine binding domain. Journal of Biological Chemistry. 1999;274(46):33097–33104. doi: 10.1074/jbc.274.46.33097. [DOI] [PubMed] [Google Scholar]
- Dho SE, Trejo J, Siderovski DP, McGlade CJ. Dynamic regulation of mammalian numb by G protein-coupled receptors and protein kinase C activation: Structural determinants of numb association with the cortical membrane. Molecular Biology of the Cell. 2006;17(9):4142–4155. doi: 10.1091/mbc.E06-02-0097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L, Hegde AN. Expression of RGS4 splice variants in dorsolateral prefrontal cortex of schizophrenic and bipolar disorder patients. Biological Psychiatry. 2009;65(6):541–545. doi: 10.1016/j.biopsych.2008.10.026. [DOI] [PubMed] [Google Scholar]
- Dracheva S, Chin B, Haroutunian V. Altered serotonin 2C receptor RNA splicing in suicide: association with editing. Neuroreport. 2008;19(3):379–382. doi: 10.1097/WNR.0b013e3282f556d2. [DOI] [PubMed] [Google Scholar]
- Ernst C, Deleva V, Deng X, Sequeira A, Pomarenski A, Klempan T, Ernst N, Quirion R, Gratton A, Szyf M, et al. Alternative splicing, methylation state, and expression profile of tropomyosin-related kinase B in the frontal cortex of suicide completers. Archives of General Psychiatry. 2009;66(1):22–32. doi: 10.1001/archpsyc.66.1.22. [DOI] [PubMed] [Google Scholar]
- Farajollahi S, Maas S. Molecular diversity through RNA editing: a balancing act. Trends in Genetics. 2010;26(5):221–230. doi: 10.1016/j.tig.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatemi SH, King DP, Reutiman TJ, Folsom TD, Laurence JA, Lee S, Fan YT, Paciga SA, Conti M, Menniti FS. PDE4B polymorphisms and decreased PDE4B expression are associated with schizophrenia. Schizophrenia Research. 2008;101(1–3):36–49. doi: 10.1016/j.schres.2008.01.029. [DOI] [PubMed] [Google Scholar]
- Garcia-Blanco MA, Baraniak AP, Lasda EL. Alternative splicing in disease and therapy. Nature Biotechnology. 2004;22(5):535–546. doi: 10.1038/nbt964. [DOI] [PubMed] [Google Scholar]
- Gemel J, Gorry M, Ehrlich GD, MacArthur CA. Structure and sequence of human FGF8. Genomics. 1996;35(1):253–257. doi: 10.1006/geno.1996.0349. [DOI] [PubMed] [Google Scholar]
- Ghosh AK, Shankar DB, Shackleford GM, Wu K, T’Ang A, Miller GJ, Zheng J, Roy-Burman P. Molecular cloning and characterization of human FGF8 alternative messenger RNA forms. Cell Growth and Differentiation. 1996;7(10):1425–1434. [PubMed] [Google Scholar]
- Glatt SJ, Chandler SD, Bousman CA, Chana G, Lucero GR, Tatro E, May T, Lohr JB, Kremen WS, Everall IE, et al. Alternatively spliced genes as biomarkers for schizophrenia, bipolar disorder and psychosis: A blood-based spliceome-profiling exploratory study. Current Pharmacogenomics and Personalized Medicine. 2009a;7(3):164–188. doi: 10.2174/1875692110907030164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glatt SJ, Faraone SV, Cohen OS, Xu Q, Shen Y, Zhang D, Yue W, Yan J, Wang L, Lu T, et al. Collective evidence from six independent samples implicates rs1076560 of DRD2 as a risk factor for schizophrenia in Han Chinese. Submitted. [Google Scholar]
- Glatt SJ, Faraone SV, Lasky-Su JA, Kanazawa T, Hwu H-G, Tsuang MT. Family-based association testing strongly implicates DRD2 as a risk gene for schizophrenia in Han Chinese from Taiwan. Molecular Psychiatry. 2009b;14:885–893. doi: 10.1038/mp.2008.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabowski PJ, Black DL. Alternative RNA splicing in the nervous system. Progress in Neurobiology. 2001;65(3):289–308. doi: 10.1016/s0301-0082(01)00007-7. [DOI] [PubMed] [Google Scholar]
- Hartmann B, Valcarcel J. Decrypting the genome’s alternative messages. Current Opinion in Cell Biology. 2009;21(3):377–386. doi: 10.1016/j.ceb.2009.02.006. [DOI] [PubMed] [Google Scholar]
- Hepp R, Dupont JL, Aunis D, Langley K, Grant NJ. NGF enhances depolarization effects on SNAP-25 expression: induction of SNAP-25b isoform. Neuroreport. 2001;12(4):673–677. doi: 10.1097/00001756-200103260-00011. [DOI] [PubMed] [Google Scholar]
- Hindorff LA, Junkins HA, Hall PN, Mehta JP, Manolio TA. A Catalog of Published Genome-Wide Association Studies. 2011 Available at: www.genome.gov/gwastudies.
- Hishimoto A, Liu QR, Drgon T, Pletnikova O, Walther D, Zhu XG, Troncoso JC, Uhl GR. Neurexin 3 polymorphisms are associated with alcohol dependence and altered expression of specific isoforms. Human Molecular Genetics. 2007;16(23):2880–2891. doi: 10.1093/hmg/ddm247. [DOI] [PubMed] [Google Scholar]
- Huntsman MM, Tran BV, Potkin SG, Bunney WE, Jr, Jones EG. Altered ratios of alternatively spliced long and short gamma2 subunit mRNAs of the gamma-amino butyrate type A receptor in prefrontal cortex of schizophrenics. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(25):15066–15071. doi: 10.1073/pnas.95.25.15066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamoto K, Kato T. Gene expression profiling in schizophrenia and related mental disorders. Neuroscientist. 2006;12(4):349–361. doi: 10.1177/1073858406287536. [DOI] [PubMed] [Google Scholar]
- Kristiansen LV, Beneyto M, Haroutunian V, Meador-Woodruff JH. Changes in NMDA receptor subunits and interacting PSD proteins in dorsolateral prefrontal and anterior cingulate cortex indicate abnormal regional expression in schizophrenia. Molecular Psychiatry. 2006;11(8):737–747. 705. doi: 10.1038/sj.mp.4001844. [DOI] [PubMed] [Google Scholar]
- Kumari M, Anji A. An old story with a new twist: do NMDAR1 mRNA binding proteins regulate expression of the NMDAR1 receptor in the presence of alcohol? Annals of the New York Academy of Science. 2005;1053:311–318. doi: 10.1196/annals.1344.027. [DOI] [PubMed] [Google Scholar]
- Law AJ, Kleinman JE, Weinberger DR, Weickert CS. Disease-associated intronic variants in the ErbB4 gene are related to altered ErbB4 splice-variant expression in the brain in schizophrenia. Human Molecular Genetics. 2007;16(2):129–141. doi: 10.1093/hmg/ddl449. [DOI] [PubMed] [Google Scholar]
- Le Corre S, Harper CG, Lopez P, Ward P, Catts S. Increased levels of expression of an NMDARI splice variant in the superior temporal gyrus in schizophrenia. Neuroreport. 2000;11(5):983–986. doi: 10.1097/00001756-200004070-00017. [DOI] [PubMed] [Google Scholar]
- Li Q, Lee JA, Black DL. Neuronal regulation of alternative pre-mRNA splicing. Nature Reviews Neuroscience. 2007;8(11):819–831. doi: 10.1038/nrn2237. [DOI] [PubMed] [Google Scholar]
- Lipscombe D, Raingo J. Alternative splicing matters: N-type calcium channels in nociceptors. Channels (Austin) 2007;1(4):225–227. doi: 10.4161/chan.4809. [DOI] [PubMed] [Google Scholar]
- Liu QR, Lu L, Zhu XG, Gong JP, Shaham Y, Uhl GR. Rodent BDNF genes, novel promoters, novel splice variants, and regulation by cocaine. Brain Res. 2006;1067(1):1–12. doi: 10.1016/j.brainres.2005.10.004. [DOI] [PubMed] [Google Scholar]
- Loftis JM, Janowsky A. Cocaine treatment- and withdrawal-induced alterations in the expression and serine phosphorylation of the NR1 NMDA receptor subunit. Psychopharmacology (Berl) 2002;164(4):349–359. doi: 10.1007/s00213-002-1209-9. [DOI] [PubMed] [Google Scholar]
- MacArthur CA, Lawshe A, Xu J, Santos-Ocampo S, Heikinheimo M, Chellaiah AT, Ornitz DM. FGF-8 isoforms activate receptor splice forms that are expressed in mesenchymal regions of mouse development. Development. 1995;121(11):3603–3613. doi: 10.1242/dev.121.11.3603. [DOI] [PubMed] [Google Scholar]
- Marguerat S, Bahler J. RNA-seq: from technology to biology. Cellular and Molecular Life Sciences. 2010;67(4):569–579. doi: 10.1007/s00018-009-0180-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mexal S, Berger R, Pearce L, Barton A, Logel J, Adams CE, Ross RG, Freedman R, Leonard S. Regulation of a novel alphaN-catenin splice variant in schizophrenic smokers. American Journal of Medical Genetics B Neuropsychiatric Genetics. 2008;147B(6):759–768. doi: 10.1002/ajmg.b.30679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morais NL, Valcarel J. Splicing Rainbow. 2010. [Google Scholar]
- Nakata K, Lipska BK, Hyde TM, Ye T, Newburn EN, Morita Y, Vakkalanka R, Barenboim M, Sei Y, Weinberger DR, et al. DISC1 splice variants are upregulated in schizophrenia and associated with risk polymorphisms. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(37):15873–15878. doi: 10.1073/pnas.0903413106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie J, Li SS, McGlade CJ. A novel PTB-PDZ domain interaction mediates isoform-specific ubiquitylation of mammalian Numb. Journal of Biological Chemistry. 2004;279(20):20807–20815. doi: 10.1074/jbc.M311396200. [DOI] [PubMed] [Google Scholar]
- Novak G, Seeman P, Tallerico T. Increased expression of calcium/calmodulin-dependent protein kinase IIbeta in frontal cortex in schizophrenia and depression. Synapse. 2006;59(1):61–68. doi: 10.1002/syn.20211. [DOI] [PubMed] [Google Scholar]
- Ohi K, Hashimoto R, Yasuda Y, Yoshida T, Takahashi H, Iike N, Iwase M, Kamino K, Ishii R, Kazui H, et al. The chitinase 3-like 1 gene and schizophrenia: evidence from a multi-center case-control study and meta-analysis. Schizophrenia Research. 2010;116(2–3):126–132. doi: 10.1016/j.schres.2009.12.002. [DOI] [PubMed] [Google Scholar]
- Olsen SK, Li JY, Bromleigh C, Eliseenkova AV, Ibrahimi OA, Lao Z, Zhang F, Linhardt RJ, Joyner AL, Mohammadi M. Structural basis by which alternative splicing modulates the organizer activity of FGF8 in the brain. Genes and Development. 2006;20(2):185–198. doi: 10.1101/gad.1365406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan YX, Xu J, Xu M, Rossi GC, Matulonis JE, Pasternak GW. Involvement of exon 11-associated variants of the mu opioid receptor MOR-1 in heroin, but not morphine, actions. Proceedings of the National Academy of Science of the United States of America. 2009;106(12):4917–4922. doi: 10.1073/pnas.0811586106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell JK, Marioni JC, Pai AA, Degner JF, Engelhardt BE, Nkadori E, Veyrieras JB, Stephens M, Gilad Y, Pritchard JK. Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature. 2010;464(7289):768–772. doi: 10.1038/nature08872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell AE, Jeon OH, Zimmerman AW, Blue ME, Pevsner J. Postmortem brain abnormalities of the glutamate neurotransmitter system in autism. Neurology. 2001;57(9):1618–28. doi: 10.1212/wnl.57.9.1618. [DOI] [PubMed] [Google Scholar]
- Raingo J, Castiglioni AJ, Lipscombe D. Alternative splicing controls G protein-dependent inhibition of N-type calcium channels in nociceptors. Nature Neuroscience. 2007;10(3):285–292. doi: 10.1038/nn1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reugels AM, Boggetti B, Scheer N, Campos-Ortega JA. Asymmetric localization of Numb:EGFP in dividing neuroepithelial cells during neurulation in Danio rerio. Developmental Dynamics. 2006;235(4):934–948. doi: 10.1002/dvdy.20699. [DOI] [PubMed] [Google Scholar]
- Rollins B, Martin MV, Morgan L, Vawter MP. Analysis of whole genome biomarker expression in blood and brain. American Journal of Medical Genetics B Neuropsychiatric Genetics. 2010;153B(4):919–936. doi: 10.1002/ajmg.b.31062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryten M, Trabzuni D, Hardy J. Genotypic analysis of gene expression in the dissection of the aetiology of complex neurological and psychiatric diseases. Briefings in Functinal Genomics and Proteomics. 2009;8(3):194–198. doi: 10.1093/bfgp/elp028. [DOI] [PubMed] [Google Scholar]
- Sadakata T, Furuichi T. Developmentally regulated Ca2+-dependent activator protein for secretion 2 (CAPS2) is involved in BDNF secretion and is associated with autism susceptibility. Cerebellum. 2009;8(3):312–322. doi: 10.1007/s12311-009-0097-5. [DOI] [PubMed] [Google Scholar]
- Sadakata T, Kakegawa W, Mizoguchi A, Washida M, Katoh-Semba R, Shutoh F, Okamoto T, Nakashima H, Kimura K, Tanaka M, et al. Impaired cerebellar development and function in mice lacking CAPS2, a protein involved in neurotrophin release. Journal of Neuroscience. 2007a;27(10):2472–2482. doi: 10.1523/JNEUROSCI.2279-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadakata T, Washida M, Iwayama Y, Shoji S, Sato Y, Ohkura T, Katoh-Semba R, Nakajima M, Sekine Y, Tanaka M, et al. Autistic-like phenotypes in Cadps2-knockout mice and aberrant CADPS2 splicing in autistic patients. Journal of Clinical Investigation. 2007b;117(4):931–943. doi: 10.1172/JCI29031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadakata T, Washida M, Morita N, Furuichi T. Tissue distribution of Ca2+-dependent activator protein for secretion family members CAPS1 and CAPS2 in mice. Journal of Histochemistry and Cytochemistry. 2007c;55(3):301–311. doi: 10.1369/jhc.6A7033.2006. [DOI] [PubMed] [Google Scholar]
- Saito M, Ehringer MA, Toth R, Oros M, Szakall I, Sikela JM, Vadasz C. Variants of kappa-opioid receptor gene and mRNA in alcohol-preferring and alcohol-avoiding mice. Alcohol. 2003;29(1):39–49. doi: 10.1016/s0741-8329(02)00322-1. [DOI] [PubMed] [Google Scholar]
- Sakharkar MK, Chow VT, Kangueane P. Distributions of exons and introns in the human genome. In Silico Biology. 2004;4(4):387–393. [PubMed] [Google Scholar]
- Sartorius LJ, Weinberger DR, Hyde TM, Harrison PJ, Kleinman JE, Lipska BK. Expression of a GRM3 splice variant is increased in the dorsolateral prefrontal cortex of individuals carrying a schizophrenia risk SNP. Neuropsychopharmacology. 2008;33(11):2626–2634. doi: 10.1038/sj.npp.1301669. [DOI] [PubMed] [Google Scholar]
- Schmucker D, Clemens JC, Shu H, Worby CA, Xiao J, Muda M, Dixon JE, Zipursky SL. Drosophila Dscam is an axon guidance receptor exhibiting extraordinary molecular diversity. Cell. 2000;101(6):671–684. doi: 10.1016/s0092-8674(00)80878-8. [DOI] [PubMed] [Google Scholar]
- Sequeira A, Turecki G. Genome wide gene expression studies in mood disorders. OMICS. 2006;10(4):444–54. doi: 10.1089/omi.2006.10.444. [DOI] [PubMed] [Google Scholar]
- Silberberg G, Darvasi A, Pinkas-Kramarski R, Navon R. The involvement of ErbB4 with schizophrenia: association and expression studies. American Journal of Medical Genetics B Neuropsychiatric Genetics. 2006;141B(2):142–148. doi: 10.1002/ajmg.b.30275. [DOI] [PubMed] [Google Scholar]
- Sokolov BP. Oligodendroglial abnormalities in schizophrenia, mood disorders and substance abuse. Comorbidity, shared traits, or molecular phenocopies? Int J Neuropsychopharmacol. 2007;10(4):547–55. doi: 10.1017/S1461145706007322. [DOI] [PubMed] [Google Scholar]
- Sorensen JB, Nagy G, Varoqueaux F, Nehring RB, Brose N, Wilson MC, Neher E. Differential control of the releasable vesicle pools by SNAP-25 splice variants and SNAP-23. Cell. 2003;114(1):75–86. doi: 10.1016/s0092-8674(03)00477-x. [DOI] [PubMed] [Google Scholar]
- Sullivan PF, Fan C, Perou CM. Evaluating the comparability of gene expression in blood and brain. American Journal of Medical Genetics B Neuropsychiatric Genetics. 2006;141B(3):261–268. doi: 10.1002/ajmg.b.30272. [DOI] [PubMed] [Google Scholar]
- Tan W, Wang Y, Gold B, Chen J, Dean M, Harrison PJ, Weinberger DR, Law AJ. Molecular cloning of a brain-specific, developmentally regulated neuregulin 1 (NRG1) isoform and identification of a functional promoter variant associated with schizophrenia. Journal of Biological Chemistry. 2007;282(33):24343–24351. doi: 10.1074/jbc.M702953200. [DOI] [PubMed] [Google Scholar]
- Ullrich B, Ushkaryov YA, Sudhof TC. Cartography of neurexins: more than 1000 isoforms generated by alternative splicing and expressed in distinct subsets of neurons. Neuron. 1995;14(3):497–507. doi: 10.1016/0896-6273(95)90306-2. [DOI] [PubMed] [Google Scholar]
- Usiello A, Baik JH, Rouge-Pont F, Picetti R, Dierich A, LeMeur M, Piazza PV, Borrelli E. Distinct functions of the two isoforms of dopamine D2 receptors. Nature. 2000;408(6809):199–203. doi: 10.1038/35041572. [DOI] [PubMed] [Google Scholar]
- Vallano ML, Beaman-Hall CM, Benmansour S. Ca2+ and pH modulate alternative splicing of exon 5 in NMDA receptor subunit 1. Neuroreport. 1999;10(17):3659–3664. doi: 10.1097/00001756-199911260-00036. [DOI] [PubMed] [Google Scholar]
- Vawter MP, Hemperly JJ, Hyde TM, Bachus SE, VanderPutten DM, Howard AL, Cannon-Spoor HE, McCoy MT, Webster MJ, Kleinman JE, et al. VASE-containing N-CAM isoforms are increased in the hippocampus in bipolar disorder but not schizophrenia. Experimental Neurology. 1998;154(1):1–11. doi: 10.1006/exnr.1998.6889. [DOI] [PubMed] [Google Scholar]
- Weickert CS, Miranda-Angulo AL, Wong J, Perlman WR, Ward SE, Radhakrishna V, Straub RE, Weinberger DR, Kleinman JE. Variants in the estrogen receptor alpha gene and its mRNA contribute to risk for schizophrenia. Human Molecular Genetics. 2008;17(15):2293–2309. doi: 10.1093/hmg/ddn130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler A, Mahal B, Kiianmaa K, Zieglgansberger W, Spanagel R. Effects of chronic alcohol consumption on the expression of different NR1 splice variants in the brain of AA and ANA lines of rats. Brain Research Molecular Brain Research. 1999;72(2):166–175. doi: 10.1016/s0169-328x(99)00218-1. [DOI] [PubMed] [Google Scholar]
- Yeo G, Holste D, Kreiman G, Burge CB. Variation in alternative splicing across human tissues. Genome Biology. 2004;5(10):R74. doi: 10.1186/gb-2004-5-10-r74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacharias DA, Strehler EE. Change in plasma membrane Ca2(+)-ATPase splice-variant expression in response to a rise in intracellular Ca2+ Current Biology. 1996;6(12):1642–1652. doi: 10.1016/s0960-9822(02)70788-4. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Bertolino A, Fazio L, Blasi G, Rampino A, Romano R, Lee ML, Xiao T, Papp A, Wang D, et al. Polymorphisms in human dopamine D2 receptor gene affect gene expression, splicing, and neuronal activity during working memory. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(51):20552–20557. doi: 10.1073/pnas.0707106104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Xu Z, Wang F, Chen J, Ng SK, Wong PW, Yu Z, Pun FW, Ren L, Lo WS, et al. Alternative-splicing in the exon-10 region of GABA(A) receptor beta(2) subunit gene: relationships between novel isoforms and psychotic disorders. PLoS One. 2009;4(9):e6977. doi: 10.1371/journal.pone.0006977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Tang R, Gao B, Shi Y, Zhou J, Guo S, Zhang J, Wang Y, Tang W, Meng J, et al. Functional variants in the promoter region of Chitinase 3-like 1 (CHI3L1) and susceptibility to schizophrenia. American Journal of Human Genetics. 2007;80(1):12–18. doi: 10.1086/510438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zipursky SL, Wojtowicz WM, Hattori D. Got diversity? Wiring the fly brain with Dscam. Trends in Biochemical Science. 2006;31(10):581–588. doi: 10.1016/j.tibs.2006.08.003. [DOI] [PubMed] [Google Scholar]