

Abstract
Objective
Misregulation of alternative splicing has become a molecular hallmark of myotonic dystrophy type 1 (DM1) in which neonatal splice variants are expressed in adult skeletal muscle. Splicing misregulation is induced by RNA containing expanded CUG repeats expressed from the expanded mutant allele by sequestration of Muscleblind-like 1 (MBNL1) protein within nuclear RNA foci and increased CUGBP, Elav-like family member 1 (CELF1) protein levels. Misregulated splicing has also been identified in other neuromuscular disorders suggesting either that diseases with different molecular causes share a common pathogenic mechanism or that misregulated splicing can also be a common secondary consequence of muscle degeneration and regeneration.
Methods
In this study we examined regulation of alternative splicing in four different mouse models of muscular dystrophy including DM1, limb-girdle muscular dystrophy, congenital merosin-deficient muscular dystrophy, Duchenne muscular dystrophy, and two myotoxin (cardiotoxin and notexin) muscle injury models.
Results
We show that DM1-like alternative splicing misregulation and altered expression of MBNL1 and CELF1 occurs in non-DM1 mouse models of muscular dystrophy and muscle injury, most likely due to recapitulation of neonatal splicing patterns in regenerating fibers. In contrast, CELF1 was elevated in nuclei of mature myofibers of the DM1 model consistent with a primary effect of pathogenic RNA expression.
Interpretation
Splicing misregulation in DM1 is a primary effect of RNA containing expanded CUG repeats. However, we conclude that splicing changes can also be observed secondary to muscle regeneration and this possibility must be taken into account when evaluating cause-effect relationships between misregulated splicing and disease processes.
Introduction
Myotonic dystrophy type 1 (DM1) is a multisystemic neuromuscular disease caused by a CTG trinucleotide repeat expansion in the 3’ untranslated region of the DM protein kinase (DMPK) gene.1 The expanded DMPK allele is transcribed and RNA containing expanded CUG repeats (CUGexp RNA) is retained in the nucleus within RNA foci.2 CUGexp RNA leads to DM1 pathogenesis, in part, through sequestration and nuclear depletion of the Muscleblind-like (MBNL) family, of which MBNL1 is the best characterized.3 Additionally, CUGexp RNA activates protein kinase C which stimulates hyperphosphorylation and stabilization of a member of the CUGBP, Elav-like family (CELF) called CELF1 (previously called CUGBP1).4 CELF and MBNL proteins coordinately regulate a subset of splicing transitions during postnatal heart and skeletal muscle development.5–8 MBNL1 loss-of-function and CELF1 gain-of-function in DM1 result in splicing misregulation of their respective targets with aberrant expression of neonatal splice isoforms in adult DM1 tissues.9
Several reports have shown that splicing misregulation occurs in other neuromuscular disorders. A mouse model for facioscapulohumeral muscular dystrophy (FSHD) exhibited splicing misregulation of Tnnt3 and Mtmr1, two transcripts aberrantly spliced in DM1.10 However, examination of FSHD muscle tissue has not shown these splicing changes.11 A mouse model for Kennedy disease, a CTG/CAG repeat expansion disorder, exhibits missplicing of Clcn1 exon 7a in skeletal muscle as well as increased CELF1 expression.12 Lastly, widespread alternative splicing defects occur in a mouse model for spinal muscular atrophy as a late event in motor neurons, perhaps secondary to cell injury.13, 14 Given that the misregulated splicing of DM1 is linked to a specific molecular mechanism unique to the disease-causing mutation, it was initially unexpected that similar splicing abnormalities would be found in different neuromuscular disorders. These results suggest that either different diseases converge on a common pathway of disrupted alternative splicing or that in addition to the mechanisms operative in DM1, splicing misregulation can also manifest as a secondary effect of muscle degeneration and regeneration.
The goal of this study was to determine the prevalence of alternative splicing misregulation in diverse causes of muscle damage. We examined six independent mouse models of muscular dystrophy and muscle injury. One was our previously described mouse model for DM1 (EpA960/HSA-Cre-ERT2) in which RNA containing 960 interrupted CUG repeats in the natural context of human DMPK 3' UTR is specifically induced in adult skeletal muscle.15 Muscle from induced animals contain intranuclear CUGexp RNA foci with MBNL1 colocalization, elevated CELF1 protein levels, misregulation of alternative splicing events found to be similarly misregulated in DM1 skeletal muscle tissue, myotonia, and muscle wasting.15 EpA960/HSA-Cre-ERT2 mice were compared to three other genetic models of muscular dystrophy (congenital merosin-deficient muscular dystrophy, limb-girdle muscular dystrophy, and Duchenne muscular dystrophy) and two myotoxin (cardiotoxin and notexin) muscle injury models. These six mouse models were examined histologically and analyzed for splicing misregulation as well as CELF1, CELF2, and MBNL1 protein expression. Our results indicate that altered splicing patterns appear both as a primary pathological event, as in DM1, and also as a consequence of skeletal muscle regeneration in which a progression of neonatal to adult splicing patterns is recapitulated.
Methods
Mouse models
Gastrocnemius muscle was collected at 3 months of age from DM1 (EpA960/HSA-Cre-ERT2) and control (HSA-Cre-ERT2) mice, one or four weeks post tamoxifen induction.15 Mice from both genotypes were the F1 progeny of EpA960xHSA-Cre-ERT2 matings. 129P1/ReJ-Lama2dy/J (stock #641) and B6.129-Sgcbtm1Kcam/2J (stock # 6833) were obtained from Jackson Laboratory (Bar Harbor, Maine). The gastrocnemius muscle was collected from 129P1/ReJ-Lama2dy/J mice at 1–2 months of age and B6.129-Sgcbtm1Kcam/2J mice at ~3 months of age. For both models, age matched wild type mice on the 129 background were used as controls. Mdx mice on the C57BL/6 background were obtained from Dr. Marta Fiorotto (Baylor College of Medicine, Houston, TX). Gastrocnemius muscle was collected at postnatal days 21, 23, and 26 and wild type C57BL/6 mice at postnatal day 21 were used as controls. Two myotoxin models were generated by intramuscular injection of cardiotoxin or notexin into the gastrocnemius muscle of C57BL/6 mice 2–4 months of age. Saline injected muscles were used as controls.
Toxin injection
Mice were anesthetized with 0.5 cc intraperitoneal injection of Avertin 20 mg/ml (2-2-2 Tribromoethanol and tert-amyl alcohol, Sigma T4840 and 240486 respectively). Either cardiotoxin 10 μM (Sigma C9759) or notexin 1 μg/μl (Sigma V0251) was injected (40 μl/injection) with a 28 gauge syringe (VWR BD-309305) in six sites along the left gastrocnemius muscle. For control samples an identical injection regime was used in the right gastrocnemius muscle with 0.85% NaCl.
RT-PCR
RNA was extracted from gastrocnemius muscle using TRIzol reagent (Invitrogen). cDNA was generated from 4 μg of RNA using oligo dT following standard procedures. PCR programs are as follows: [Clcn1 <3min 94°C (45sec 94°C, 45sec 58°C, 45sec 72°C) 27 cycles, 10min 72°C>; Serca1 <3min 95°C (45sec 95°C, 45sec 57°C, 1min 72°C) 20 cycles, 5min 72°C>. The Serca1 program was modified to include 22 cycles for Cypher, 27 cycles for Ank2, and 24 cycles for Capzb, Fxr1h, Mbnl1, H2afy, and Sorbs1]. For primer sequences see Supplementary Table 1.
Histology
Gastrocnemius muscle samples were fixed in 10% formalin, paraffin embedded, sectioned at 10 μm and then stained with hematoxylin and eosin according to standard procedures.
Western blot
Protein was extracted from the gastrocnemius muscle with a dounce homogenizer in 10 mM HEPES pH 7.5, 0.32 M sucrose, 5 μM MG132, 5 mM EDTA, protease inhibitor cocktail tablet (Roche), and 1% SDS. Protein extracts (50 μg per lane) were separated by SDS/PAGE and assayed by Western blot. The following antibodies were used for protein detection: CELF1 clone 3B1 (M. Swanson, 1:1,000) and CELF2 clone IH2 (M. Swanson, 1:500) followed by goat anti-mouse light chain (Jackson ImmunoResearch, 1:10,000); MBNL1 (C. Thornton, 1:1,000) followed by goat anti-rabbit (Calbiochem, 1:10,000); eMHC F1.652 (Developmental Studies Hybridoma Bank, 1:200) and GAPDH (Abcam, 1:5,000) followed by sheep anti-mouse (Jackson ImmunoResearch 1:10,000).
Fluorescence in situ hybridization and immunofluorescence
Fluorescence in situ hybridization was performed on paraffin-embedded sections (5 μm). Probe hybridization was performed as previously described16 using a (CTG)5-Cy3-labeled PNA probe (Applied Biosystems). Immunofluorescence was then performed as follows. Sections were incubated with primary antibodies [CELF1 clone 3B1 (1:100, mouse monoclonal, Millipore) and neonatal myosin heavy chain (1:350, rabbit polyclonal)17] overnight at 4°C, washed, and incubated with secondary antibodies [Alexa Fluor 488 goat anti-mouse IgG (1:500, Molecular Probes) and Alexa Fluor 594 donkey anti-rabbit IgG (1:500, Molecular Probes)] for 3 hours at room temperature.
Results
Histological examination in six models of muscle degeneration
A histological comparison was conducted between mouse models for DM1 (EpA960/HSA-Cre-ERT2),15 congenital merosin-deficient muscular dystrophy (129P1/ReJ-Lama2dy/J),18, 19 limb-girdle muscular dystrophy (B6.129-Sgcbtm1Kcam/2J),20 and Duchenne muscular dystrophy (mdx).21–23 To study a temporal progression of muscle degeneration and regeneration in a highly synchronized and reproducible manner, we performed myotoxin (cardiotoxin and notexin) induced muscle injury. Cardiotoxin, a peptide isolated from snake venom, causes depolarization and contraction of muscle cells leading to cell death via inhibition of protein kinase C.24 Notexin, a phospholipase A2 neurotoxin, hydrolyses lipids of the plasma membrane leading to depolarization and degradation of myofibers.25, 26
We examined histological sections of gastrocnemius muscle from each of the six models to compare the extent of degeneration/regeneration and to correlate tissue pathology with subsequent molecular analyses. Figure 1A shows wild type muscle histology as a reference in which the fibers are typically eosinophilic, polygonal in shape, uniform in size, and contain peripheral nuclei.
Figure 1. Histological analysis of six models of muscular dystrophy and muscle injury.

Hematoxylin and eosin stained cross-sections from paraffin embedded gastrocnemius muscle at 20X magnification. (A) Wild-type control, (B) EpA960/HSA-Cre-ERT2 (+ tam), (C) 129P1/ReJ-Lama2dy/J, (D) B6.129-Sgcbtm1Kcam/2J, (E) mdx at postnatal days 21, 23 and 26, (F) cardiotoxin and (G) notexin injured muscle at 1, 3, 5, 8 and 16 days post injection.
EpA960/HSA-Cre-ERT2 (+ tam) mice have the most diffuse pattern of muscle pathology and show extensive fiber size variation including many small basophilic regenerating fibers with centrally located nuclei (Fig 1B).15 129P1/ReJ-Lama2dy/J mice exhibited significant hind limb weakness and muscle atrophy when sacrificed at 1–2 months of age. Despite an obvious impairment in muscle function, the muscle histology contains large areas of mature, healthy myofibers intermixed with patches of small regenerating fibers with centrally located nuclei (Fig 1C). Histological analysis of B6.129-Sgcbtm1Kcam/2J muscle from mice sacrificed at 3 months of age revealed fibers mostly uniform in size and a scattered presence of small basophilic regenerating fibers (Fig 1D).
Muscle histology of mdx mice was examined at postnatal days 21, 23, and 26 corresponding to the time of peak muscle degeneration.21 At day 21 the muscle histology was relatively normal with uniform large eosinophilic fibers and peripherally located nuclei (Fig 1E). At day 23 the muscle pathology was most severe with both necrotic fibers and small regenerating basophilic fibers containing centrally located nuclei. At day 26 there are an increased proportion of eosinophilic fibers with centrally located nuclei, due to progression of new myofiber maturation (Fig 1E). Consistent with previous studies in mdx mice,21 we find that muscle degeneration predominantly occurs at postnatal days 21 through 23 and stimulates the regeneration program at postnatal days 23 through 26.
The final two mouse models examined were myotoxin (cardiotoxin and notexin) injury models. The degree of necrosis and subsequent regenerative response in the myotoxin injury models is significantly more robust than the muscular dystrophy mouse models (Fig 1F to G). Several time points were examined to investigate the various stages of a synchronized degeneration/regeneration cycle, and the temporal progression was similar in both myotoxin injury models. Twenty-four hours post cardiotoxin or notexin injection (day 1, Fig 1F to G) there were large areas of muscle necrosis and infiltration of small basophilic inflammatory cells which progressed through day 3. By day 5 large areas of regenerating fibers were present with centrally located nuclei. By day 8, regenerating myofibers showed continued maturation, with increased size and eosinophilic staining. At day 16 the myofibers exhibited uniformity in size; however, they retained central nuclei distinguishing them as recently regenerated.
Overall, the extent of muscle necrosis and subsequent regeneration was milder in the muscular dystrophy mouse models than the toxin injury models with the exception of the EpA960/HSA-Cre-ERT2 mice.
Appearance of a neonatal splicing program temporally correlates with muscle regeneration
Misregulation of alternative splicing events are a characteristic feature of DM1 in human skeletal muscle tissue and in multiple DM1 mouse models in which neonatal isoforms are predominately expressed in place of the adult isoforms.9 The misregulated alternative splicing events tested thus far are regulated by either MBNL1 or CELF1, or jointly regulated by both splicing factors in an antagonistic manner.9, 27 We examined eight such events including Ank2, Capzb, Fxr1h, Mbnl1, Clcn1, H2afy, Cypher, and Serca1 in the six mouse models of skeletal muscle degeneration.
We previously defined splicing events that are regulated during heart and/or skeletal muscle development as responsive to expression of CELF1 alone, MBNL1 alone, or both CELF1 and MBNL1 using CELF1 transgenic and MBNL1 knock out mice.5, 28 In addition, splicing events that are regulated during skeletal muscle development and sensitive to MBNL1 knock out have been identified.8, 29 We first investigated three alternative splicing events that are responsive to CELF1 overexpression but not MBNL1 depletion (Ank2 exon 21, Capzb exon 8, and Fxr1h exon 15).5, 8, 28 We previously demonstrated that these CELF1 responsive splicing events are developmentally regulated in human and mouse skeletal muscle, and that their splicing patterns revert to a neonatal splice pattern in the EpA960/HSA-Cre-ERT2 (+ tam) mice and in individuals with DM1.15 Analysis across the six models of muscle degeneration demonstrated that there is a statistically significant reduction in the percent inclusion of Ank2 exon 21 in 129P1/ReJ-Lama2dy/J and B6.129-Sgcbtm1Kcam/2J mice, resembling a neonatal splicing pattern; however the misregulation is less severe than in the EpA960/HSA-Cre-ERT2 (+ tam) mice (Fig 2). Mdx mice at postnatal days 21, 23, and 26 do not show misregulation of Ank2 exon 21 when compared to their strain-matched controls. The cardiotoxin and notexin injury models show significant misregulation of Ank2 exon 21, similar in magnitude with the EpA960/HSA-Cre-ERT2 (+ tam) mice, as early as day 1 post myotoxin injection. All six muscular dystrophy and muscle injury models show statistically significant misregulation of Capzb exon 8 (Fig 2). Fxr1h exon 15 is not misregulated in the B6.129-Sgcbtm1Kcam/2J or mdx mice, but does show slight misregulation in 129P1/ReJ-Lama2dy/J mice (Fig 2). Misregulation in the myotoxin injured mice occurs at early time points and is similar to that seen in the EpA960/HSA-Cre-ERT2 (+ tam) mice.
Figure 2. Splicing misregulation in several muscular dystrophy and muscle injury models.

RT-PCR was performed on RNA extracted from gastrocnemius muscle using primers that anneal to constitutive exons flanking the alternative exon (Supp Table 1). The percentage inclusion of alternative exons from eight genes (Ank2, Capzb, Fxr1h, H2afy, Mbnl1, Cypher, Serca1 and Clcn1) is graphically represented. Lanes are as follows: newborn limb, postnatal day 1 wild-type limb; HSA-Cre, HSA-Cre- ERT2 (+ tam) age matched; EpA960/HSA-Cre, EpA960/HSA-Cre-ERT2 (+ tam); Lama2 control, 129P1 wild-type age matched; Lama2, 129P1/ReJ-Lama2dy/J; Sgcb control, B6 wild-type age matched; Scgb, B6.129-Sgcbtm1Kcam/2J; mdx control, C57BL/6 wild-type age matched; mdx postnatal day 21, 23, and 26; toxin control, C57BL/6 wild-type age matched; toxin post injection day 1, 3, 5, 8 and 16 (gray bars are cardiotoxin, white bars are notexin). Each bar represents the mean of three or four biological replicates with standard deviation, except for the newborn sample, which represents the mean of three technical replicates from a pool of 12 animals. Differences in percent exon inclusion between the six models and their controls marked with (*) were determined to be statistically significant by the Student’s t test, P < 0.05.
Two splicing events responsive to MBNL1 depletion but not CELF1 overexpression were examined (Mbnl1 exon 5 and Clcn1 exon 7a).5, 28, 29 MBNL1 nuclear depletion is expected in EpA960/HSA-Cre-ERT2 (+ tam) mice due to sequestration by large intranuclear CUGexp RNA foci; however, other models of muscle degeneration lack CUGexp RNA and thus a primary pathogenic effect on MBNL1 responsive splicing events is not expected.. Analysis across the muscular dystrophy and muscle injury models demonstrated a slight yet statistically significant misregulation of Mbnl1 exon 5 inclusion in B6.129-Sgcbtm1Kcam/2J, mdx, and myotoxin injured mice; however the degree of misregulation is considerably less than that seen in EpA960/HSA-Cre-ERT2 (+ tam) mice (Fig 2). Inclusion of Clcn1 exon 7a has been demonstrated to be responsible for the development of myotonia in mouse models of myotonic dystrophy.30–32 We show a statistically significant degree of Clcn1 exon 7a misregulation in EpA960/HSA-Cre-ERT2 (+ tam) (as shown previously),15 129P1/ReJ-Lama2dy/J, B6.129-Sgcbtm1Kcam/2J, mdx day 23, and myotoxin injured mice (Fig 2). It is unknown whether the degree of misregulation observed is sufficient to produce myotonia as in EpA960/HSA-Cre-ERT2 (+ tam) mice15.
We next examined three alternative splicing events that are regulated by both CELF1 and MBNL1 (H2afy mutually exclusive exon 6, Cypher exon 11, and Serca1 exon 22).8, 28, 29 H2afy is misregulated in EpA960/HSA-Cre-ERT2 (+ tam), B6.129-Sgcbtm1Kcam/2J, mdx at postnatal days 21 and 26, and in myotoxin injured mice at several time points (Fig 2). Misregulation of Cypher and Serca1 in EpA960/HSA-Cre-ERT2 (+ tam) mice leads to a statistically significant change in inclusion of exon 11 and exon 22, respectively (Fig 2). Myotoxin injured mice also demonstrate significant misregulation of Cypher exon 11 and Serca1 exon 22. There is also a small but statistically significant decrease in Serca1 splicing in the mdx mice.
We conclude that these alternative splicing events regulated by CELF1 overexpression and/or MBNL1 depletion, which have been shown to be misregulated in DM1 models, show strong splicing changes in response to myotoxin injury. While some alternative splicing events are misregulated in non-DM1 muscular dystrophy models, there is variability between each splicing event and the strength of misregulation is generally much weaker than the EpA960/HSA-Cre-ERT2 mouse model.
CELF1 and MBNL1 protein expression is altered in dystrophy models and during muscle regeneration
To determine if disruption of misregulated splicing events correlates with changes in CELF1 and MBNL1 protein expression, we performed Western blot analysis on muscle protein extracts from each of the muscular dystrophy and muscle injury mouse models. We previously demonstrated a four-fold increase in CELF1 protein steady state levels in skeletal muscle tissue from EpA960/HSA-Cre-ERT2 (+ tam) mice compared to control mice.15 CELF2, the only other CELF family member expressed in skeletal muscle, is 78% identical to CELF1, has a similar developmental down-regulation expression pattern, and has been shown to also affect alternative splicing of pre-mRNA targets regulated by CELF1.6, 7, 33 CELF2 shows a general but inconsistent trend towards up-regulation in EpA960/HSA-Cre-ERT2 (+ tam) mice (unpublished data). In contrast, in response to CUG repeat expression, MBNL1 colocalizes with CUGexp RNA nuclear foci.15
The 129P1/ReJ-Lama2dy/J and B6.129-Sgcbtm1Kcam/2J mice show about a two-fold up-regulation of CELF1, CELF2, and MBNL1 consistent with the subtle changes in alternative splicing (Fig 3 and Supp Fig 1). In agreement with the histology data that the regeneration response in the 129P1/ReJ-Lama2dy/J and B6.129-Sgcbtm1Kcam/2J mice is mild, the regeneration marker embryonic myosin heavy chain (eMHC) is not detectable by Western blot (Fig 3). Mdx mice show no consistent changes (less than 1.5-fold) in CELF1, CELF2, and MBNL1 expression during the periods of degeneration and regeneration. Two of the three mdx mice at postnatal day 26 show slight expression of eMHC, representing the progression of the regeneration program identified by histology.
Figure 3. Altered CELF and MBNL protein expression in muscle degeneration.

CELF1, CELF2, MBNL1, and eMHC protein levels were altered in multiple models of muscular dystrophy and muscle injury. Gastrocnemius skeletal muscle collected from 129P1/ReJ-Lama2dy/J, B6.129-Sgcbtm1Kcam/2J, mdx postnatal days 21, 23, and 26, cardiotoxin and notexin injured muscle at 1, 3, 5, 8, and 16 days post injection.
Following the more severe muscle damage in both cardiotoxin and notexin injured tissue, CELF1 and CELF2 protein levels are strongly increased greater than ten-fold by day 3 post myotoxin injection and peak by days 5 through 8 (Fig 3 and Supp Fig 1). CELF1 and CELF2 expression returns to control levels by day 16 when the overall muscle morphology has been restored. MBNL1 is up-regulated three- to ten-fold by day 3 post myotoxin injection and peaks at day 5. The up-regulation of these splicing regulators following muscle injury is striking and likely contributes to the strong splicing misregulation in these muscle tissues. The myotoxin injury models show robust expression of eMHC days 5 and 8 post injection, correlating with the times of peak CELF1, CELF2, and MBNL1 protein expression. This data clearly demonstrates that altered expression of these splicing regulators occurs not only in DM1, but also in response to different causes of muscle damage.
CELF1 up-regulation in EpA960/HSA-Cre-ERT2 (+ tam) mice is observed in mature myofibers
The results above indicate that analysis of skeletal muscle tissues for misregulation of alternative splicing and alterations in expression of splicing regulators must distinguish between primary pathological processes and secondary effects due to muscle regeneration. The combination of unaffected and regenerating myofibers within a muscle tissue prevent accurate discrimination of primary and secondary effects on protein expression by Western blotting since it is unclear whether an elevation in protein expression is within the mature muscle fibers or from regenerating fibers. For example, the EpA960/HSA-Cre-ERT2 (+ tam) mice exhibit elevated CELF1 and misregulated splicing of demonstrated CELF1 targets in patterns that are consistent with increased CELF1 activity.15 However, the histology of EpA960/HSA-Cre-ERT2 (+ tam) muscle demonstrates the presence of regenerating skeletal muscle fibers four weeks following induction of CUGexp RNA that are coincident with maximal changes in alternative splicing.
To determine whether CELF1 was increased in mature muscle fibers, we performed immunofluorescent staining of EpA960/HSA-Cre-ERT2 (+ tam) muscle at one and four weeks following induction of CUGexp RNA. We also examined HSA-Cre-ERT2 (+ tam) muscle one week following tamoxifen administration as a control for muscle that expresses Cre-ERT2 but not CUGexp RNA. The results demonstrated that one week and more so at four weeks following induction of CUGexp RNA, nuclear staining for CELF1 was substantially increased within peripheral nuclei of myofibers lacking expression of a regeneration marker, neonatal myosin heavy chain (nMHC), consistent with being nuclei of mature myofibers (Fig 4A and Supp Fig 2). In addition, in situ hybridization using Cy3-labeled CAG PNA probes indicate that a majority of nuclei exhibiting increased CELF1 also contain nuclear foci of CUGexp RNA (Fig 4A). The expression of CUGexp RNA is restricted to the myofibers by cell-specific expression of Cre-ERT2 by the skeletal alpha-actin promoter.34
Figure 4. CELF1 up-regulation in mature and regenerating myofibers.
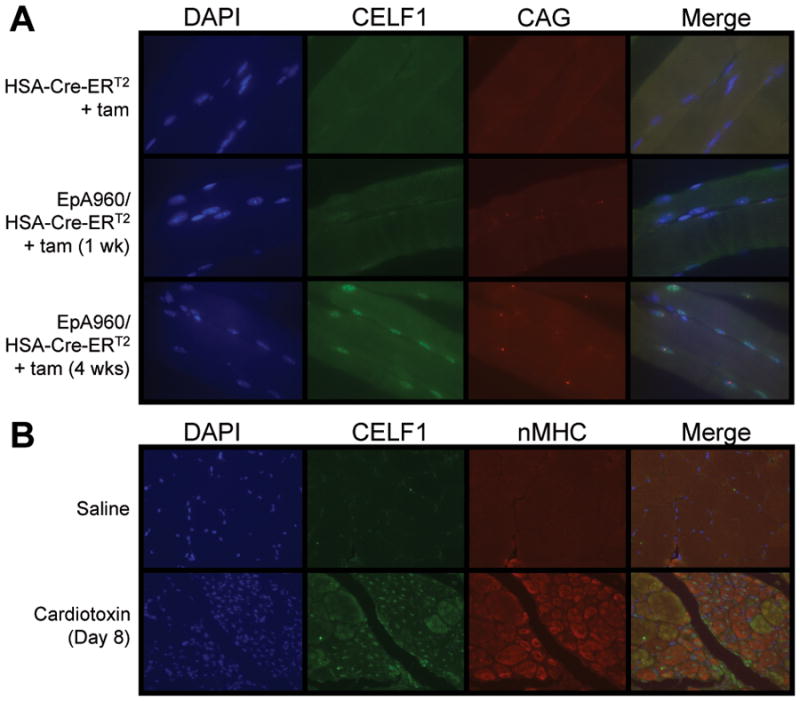
(A) CELF1 immunofluorescence and in situ hybridization to detect CUGexp RNA foci in EpA960/HSA-Cre-ERT2 (+ tam) gastrocnemius muscle longitudinal sections. 63X magnification. (B) Co- immunofluorescence of CELF1 and nMHC on cardiotoxin injured gastrocnemius muscle cross-sections. 20X magnification.
To determine whether CELF1 was up-regulated in regenerating fibers, cardiotoxin injured skeletal muscle was co-stained for CELF1 and nMHC. Eight days following cardiotoxin administration, when skeletal muscle is undergoing substantial regeneration, a large number of small fibers containing centrally located nuclei could be identified. These fibers fit the morphological criteria of regenerating fibers.24 Consistent with their morphology, these fibers stained for nMHC and the majority of fibers staining for nMHC contained centrally located nuclei that stained strongly for CELF1 (Fig 4B). In contrast, larger fibers do not contain nuclei exhibiting increased CELF1 staining. Interestingly, some small and intermediate sized fibers exhibit staining for CELF1 in the sarcoplasm. While it is possible that this signal is artifactual and results from cross-reactivity to a transiently expressed unrelated protein, there are also well established cytoplasmic roles for CELF1 as well as demonstration that members of the CELF family shuttle between the nucleus and the cytoplasm.35, 36 Based on these results we conclude that CELF1 is specifically up-regulated within nuclei of regenerating muscle fibers. Furthermore, analysis of EpA960/HSA-Cre-ERT2 (+ tam) muscle indicates up-regulation of CELF1 in mature skeletal muscle fibers in which CUGexp RNA is expressed. These results demonstrate the coexistence of two independent processes: induced expression of CELF1 as a primary effect of CUGexp RNA and up-regulation of CELF1 expression in regenerating fibers.
Discussion
Expression of neonatal splice isoforms in adult skeletal muscle has become a classic molecular feature of DM1: more than 21 alternative splicing events have been shown to be misregulated in adult DM1 heart, skeletal muscle, or brain tissues.9, 15 The mechanisms of splicing misregulation in DM1 are directly linked to the effects of the CUGexp RNA on the functions of MBNL and CELF protein families, RNA binding proteins that normally regulate splicing during development. The expression of CUGexp RNA causes a loss of MBNL1 function by sequestration and a gain of CELF1 function by stabilizing the protein via protein kinase C-mediated hyperphosphorylation.37 The identification of some of the same splicing abnormalities in mouse models of neuromuscular disorders generated by different mutations prompted us to examine the relationship between degenerating/regenerating skeletal muscle and altered splicing patterns.
In this study, we examined eight alternative splicing events in six independent mouse models of muscular dystrophy and muscle injury including EpA960/HSA-Cre-ERT2 (+ tam), 129P1/ReJ-Lama2dy/J, B6.129-Sgcbtm1Kcam/2J, mdx, cardiotoxin injury, and notexin injury. The results demonstrated that all of the alternative splicing events that are misregulated in skeletal muscle tissue from one or more DM1 mouse model(s) and human DM1 patients5, 15, 38 are also misregulated in non-DM1 mouse models of skeletal muscle damage and degeneration.
Each of the six mouse models displayed histological evidence of degeneration and regeneration with varying levels of severity. Importantly, the degree of misregulation of the alternative splicing events correlated with the degree of degeneration/regeneration observed histologically. In addition, the severity of the histopathology was consistent with the eMHC expression level by Western blot analyses. These results indicate that while splicing misregulation can reflect a primary disease process, analysis of splicing is complicated by overlapping splicing abnormalities resulting from muscle regeneration. Histological evaluation of the extent of regeneration provides an important parameter when evaluating the basis for abnormalities in splicing regulation or express of splicing regulators.
Regeneration of skeletal muscle requires rapid activation and proliferation of satellite stem cells followed by differentiation to myoblasts and fusion to form myotubes.24 This process resembles that of normal skeletal muscle development24 and it is not surprising that regenerating muscle recapitulates neonatal alternative splicing patterns. Western blot data showed increased protein levels of CELF1, CELF2, and MBNL1 in the non-DM1 mouse models of muscle degeneration. This result is also consistent with reversion to neonatal expression patterns because all three proteins decrease during postnatal skeletal muscle development7, 8 (and data not shown). It is important to note, however, that an increase in nuclear MBNL1 due to nuclear-cytoplasmic shuttling is proposed to be the main mechanism of MBNL1-mediated splicing transitions during postnatal skeletal muscle development.8 Reversion of MBNL1-regulated splicing events to early postnatal patterns is not consistent with increased MBNL1 protein levels observed in the dystrophy models based on Western blot analysis. However, the lack of complete correlation between protein levels and misregulated alternative splicing is likely to reflect an absence of information regarding the nuclear concentration of MBNL1 that would be obtained by immunolocalization studies.
Several results support the contention that CELF1 up-regulation and altered splicing in DM1 is a primary response to the CUGexp RNA rather than secondary to regeneration. First, the splicing misregulation and increased CELF1 protein levels observed in skeletal muscle tissue from individuals with DM1 are not associated with a robust regenerative response.1, 39–41 This observation strongly suggests that aberrant expression of neonatal splice isoforms in adult DM1 skeletal muscle likely occurs in mature myofibers. Second, misregulated splicing and up-regulation of CELF1 is observed in DM1 cardiac tissue, which does not undergo regeneration.5, 42 Third, a heart-specific and inducible transgenic mouse model for DM1 has shown that misregulated splicing and elevated CELF1 is observed within 6 hours following induced expression of CUGexp RNA demonstrating these as primary responses to the pathogenic RNA.42 Finally, in the present study, we used immunofluorescent staining for CELF1 combined with in situ hybridization to detect CUGexp RNA foci and detected elevated CELF1 in nuclei of non-regenerating muscle fibers and that were coincident with RNA foci.
We conclude that expression of neonatal alternative splicing isoforms in adult skeletal muscle occurs in several diverse models of muscle degeneration and muscle damage as a result of a robust regenerative process. Therefore, it is important to distinguish whether changes in alternative splicing patterns and expression of splicing regulators are primary effects of the pathogenic mechanism or secondary to muscle regeneration. It is important to note that these processes are not mutually exclusive. The EpA960/HSA-Cre-ERT2 (+ tam) DM1 model in skeletal muscle exhibits misregulated splicing, up-regulated CELF1, and diffuse degeneration/regeneration. Our results indicate that assays for splicing misregulation and altered expression of CELF1 are likely to reflect the combined effects of CUGexp RNA in mature skeletal muscle fibers as well as effects secondary to regeneration. The results described here for skeletal muscle tissue from mouse models are likely to be applicable to tissue samples from individuals with degenerative muscle disease, and stress the importance of evaluating cause-effect relationships between disease processes and misregulated splicing.
Supplementary Material
Acknowledgments
All histological samples were processed by the Center for Comparative Medicine pathology core facility at Baylor College of Medicine, special thanks to Bilqees Bhatti for her assistance. Thanks to Marta Fiorotto, Ph.D. (Baylor College of Medicine, Houston, TX) for providing mdx mice as well as general advice. We thank Onnik Agbulut, Ph.D. (Paris Diderot University, Paris, France) for providing neonatal myosin heavy chain polyclonal antibody. Support for this work was provided by National Institutes of Health Grants R01AR45653 (T.A.C), F30NS057884 (J.P.O) and F31NS067740 (A.J.W), and by the Muscular Dystrophy Association (T.A.C).
References
- 1.Harper PS, Brook JD, Newman E. Myotonic dystrophy. 3. ix. London; New York: W. B. Saunders; 2001. p. 436. [Google Scholar]
- 2.Davis BM, McCurrach ME, Taneja KL, et al. Expansion of a CUG trinucleotide repeat in the 3' untranslated region of myotonic dystrophy protein kinase transcripts results in nuclear retention of transcripts. Proc Natl Acad Sci U S A. 1997;94:7388–7393. doi: 10.1073/pnas.94.14.7388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miller JW, Urbinati CR, Teng-Umnuay P, et al. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J. 2000;19:4439–4448. doi: 10.1093/emboj/19.17.4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kuyumcu-Martinez NM, Wang GS, Cooper TA. Increased steady-state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation. Mol Cell. 2007;28:68–78. doi: 10.1016/j.molcel.2007.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kalsotra A, Xiao X, Ward AJ, et al. A postnatal switch of CELF and MBNL proteins reprograms alternative splicing in the developing heart. Proc Natl Acad Sci U S A. 2008;105:20333–20338. doi: 10.1073/pnas.0809045105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ladd AN, Charlet N, Cooper TA. The CELF family of RNA binding proteins is implicated in cell-specific and developmentally regulated alternative splicing. Mol Cell Biol. 2001;21:1285–1296. doi: 10.1128/MCB.21.4.1285-1296.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ladd AN, Stenberg MG, Swanson MS, Cooper TA. Dynamic balance between activation and repression regulates pre-mRNA alternative splicing during heart development. Dev Dyn. 2005;233:783–793. doi: 10.1002/dvdy.20382. [DOI] [PubMed] [Google Scholar]
- 8.Lin X, Miller JW, Mankodi A, et al. Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Hum Mol Genet. 2006;15:2087–2097. doi: 10.1093/hmg/ddl132. [DOI] [PubMed] [Google Scholar]
- 9.Osborne RJ, Thornton CA. RNA-dominant diseases. Hum Mol Genet. 2006;15(Spec No 2):R162–169. doi: 10.1093/hmg/ddl181. [DOI] [PubMed] [Google Scholar]
- 10.Gabellini D, D'Antona G, Moggio M, et al. Facioscapulohumeral muscular dystrophy in mice overexpressing FRG1. Nature. 2006;439:973–977. doi: 10.1038/nature04422. [DOI] [PubMed] [Google Scholar]
- 11.Osborne RJ, Welle S, Venance SL, et al. Expression profile of FSHD supports a link between retinal vasculopathy and muscular dystrophy. Neurology. 2007;68:569–577. doi: 10.1212/01.wnl.0000251269.31442.d9. [DOI] [PubMed] [Google Scholar]
- 12.Yu Z, Wang AM, Robins DM, Lieberman AP. Altered RNA splicing contributes to skeletal muscle pathology in Kennedy disease knock-in mice. Dis Model Mech. 2009;2:500–507. doi: 10.1242/dmm.003301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baumer D, Lee S, Nicholson G, et al. Alternative splicing events are a late feature of pathology in a mouse model of spinal muscular atrophy. PLoS Genet. 2009;5:e1000773. doi: 10.1371/journal.pgen.1000773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Z, Lotti F, Dittmar K, et al. SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell. 2008;133:585–600. doi: 10.1016/j.cell.2008.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Orengo JP, Chambon P, Metzger D, et al. Expanded CTG repeats within the DMPK 3' UTR causes severe skeletal muscle wasting in an inducible mouse model for myotonic dystrophy. Proc Natl Acad Sci U S A. 2008;105:2646–2651. doi: 10.1073/pnas.0708519105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mankodi A, Urbinati CR, Yuan QP, et al. Muscleblind localizes to nuclear foci of aberrant RNA in myotonic dystrophy types 1 and 2. Hum Mol Genet. 2001;10:2165–2170. doi: 10.1093/hmg/10.19.2165. [DOI] [PubMed] [Google Scholar]
- 17.Launay T, Noirez P, Butler-Browne G, Agbulut O. Expression of slow myosin heavy chain during muscle regeneration is not always dependent on muscle innervation and calcineurin phosphatase activity. Am J Physiol Regul Integr Comp Physiol. 2006;290:R1508–1514. doi: 10.1152/ajpregu.00486.2005. [DOI] [PubMed] [Google Scholar]
- 18.Sunada Y, Bernier SM, Utani A, et al. Identification of a novel mutant transcript of laminin alpha 2 chain gene responsible for muscular dystrophy and dysmyelination in dy2J mice. Hum Mol Genet. 1995;4:1055–1061. doi: 10.1093/hmg/4.6.1055. [DOI] [PubMed] [Google Scholar]
- 19.Xu H, Wu XR, Wewer UM, Engvall E. Murine muscular dystrophy caused by a mutation in the laminin alpha 2 (Lama2) gene. Nat Genet. 1994;8:297–302. doi: 10.1038/ng1194-297. [DOI] [PubMed] [Google Scholar]
- 20.Durbeej M, Cohn RD, Hrstka RF, et al. Disruption of the beta-sarcoglycan gene reveals pathogenetic complexity of limb-girdle muscular dystrophy type 2E. Mol Cell. 2000;5:141–151. doi: 10.1016/s1097-2765(00)80410-4. [DOI] [PubMed] [Google Scholar]
- 21.Bulfield G, Siller WG, Wight PA, Moore KJ. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc Natl Acad Sci U S A. 1984;81:1189–1192. doi: 10.1073/pnas.81.4.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hoffman EP, Brown RH, Jr, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- 23.Sicinski P, Geng Y, Ryder-Cook AS, et al. The molecular basis of muscular dystrophy in the mdx mouse: a point mutation. Science. 1989;244:1578–1580. doi: 10.1126/science.2662404. [DOI] [PubMed] [Google Scholar]
- 24.Charge SB, Rudnicki MA. Cellular and molecular regulation of muscle regeneration. Physiol Rev. 2004;84:209–238. doi: 10.1152/physrev.00019.2003. [DOI] [PubMed] [Google Scholar]
- 25.Harris JB. Myotoxic phospholipases A2 and the regeneration of skeletal muscles. Toxicon. 2003;42:933–945. doi: 10.1016/j.toxicon.2003.11.011. [DOI] [PubMed] [Google Scholar]
- 26.Plant DR, Colarossi FE, Lynch GS. Notexin causes greater myotoxic damage and slower functional repair in mouse skeletal muscles than bupivacaine. Muscle Nerve. 2006;34:577–585. doi: 10.1002/mus.20616. [DOI] [PubMed] [Google Scholar]
- 27.Orengo JP, Cooper TA. Alternative splicing in disease. Adv Exp Med Biol. 2007;623:212–223. doi: 10.1007/978-0-387-77374-2_13. [DOI] [PubMed] [Google Scholar]
- 28.Ward AJ, Rimer M, Killian JM, et al. CUGBP1 overexpression in mouse skeletal muscle reproduces features of myotonic dystrophy type 1. Hum Mol Genet. 2010 doi: 10.1093/hmg/ddq277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kanadia RN, Johnstone KA, Mankodi A, et al. A muscleblind knockout model for myotonic dystrophy. Science. 2003;302:1978–1980. doi: 10.1126/science.1088583. [DOI] [PubMed] [Google Scholar]
- 30.Charlet BN, Savkur RS, Singh G, et al. Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing. Mol Cell. 2002;10:45–53. doi: 10.1016/s1097-2765(02)00572-5. [DOI] [PubMed] [Google Scholar]
- 31.Mankodi A, Takahashi MP, Jiang H, et al. Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol Cell. 2002;10:35–44. doi: 10.1016/s1097-2765(02)00563-4. [DOI] [PubMed] [Google Scholar]
- 32.Wheeler TM, Lueck JD, Swanson MS, et al. Correction of ClC-1 splicing eliminates chloride channelopathy and myotonia in mouse models of myotonic dystrophy. J Clin Invest. 2007;117:3952–3957. doi: 10.1172/JCI33355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Faustino NA, Cooper TA. Identification of putative new splicing targets for ETR-3 using sequences identified by systematic evolution of ligands by exponential enrichment. Mol Cell Biol. 2005;25:879–887. doi: 10.1128/MCB.25.3.879-887.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schuler M, Ali F, Metzger E, et al. Temporally controlled targeted somatic mutagenesis in skeletal muscles of the mouse. Genesis. 2005;41:165–170. doi: 10.1002/gene.20107. [DOI] [PubMed] [Google Scholar]
- 35.Barreau C, Paillard L, Mereau A, Osborne HB. Mammalian CELF/Bruno-like RNA-binding proteins: molecular characteristics and biological functions. Biochimie. 2006;88:515–525. doi: 10.1016/j.biochi.2005.10.011. [DOI] [PubMed] [Google Scholar]
- 36.Ladd AN, Cooper TA. Multiple domains control the subcellular localization and activity of ETR-3, a regulator of nuclear and cytoplasmic RNA processing events. J Cell Sci. 2004;117:3519–3529. doi: 10.1242/jcs.01194. [DOI] [PubMed] [Google Scholar]
- 37.Lee JE, Cooper TA. Pathogenic mechanisms of myotonic dystrophy. Biochem Soc Trans. 2009;37:1281–1286. doi: 10.1042/BST0371281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mankodi A, Logigian E, Callahan L, et al. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science. 2000;289:1769–1773. doi: 10.1126/science.289.5485.1769. [DOI] [PubMed] [Google Scholar]
- 39.Furling D, Coiffier L, Mouly V, et al. Defective satellite cells in congenital myotonic dystrophy. Hum Mol Genet. 2001;10:2079–2087. doi: 10.1093/hmg/10.19.2079. [DOI] [PubMed] [Google Scholar]
- 40.Savkur RS, Philips AV, Cooper TA. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat Genet. 2001;29:40–47. doi: 10.1038/ng704. [DOI] [PubMed] [Google Scholar]
- 41.Thornell LE, Lindstom M, Renault V, et al. Satellite cell dysfunction contributes to the progressive muscle atrophy in myotonic dystrophy type 1. Neuropathol Appl Neurobiol. 2009;35:603–613. doi: 10.1111/j.1365-2990.2009.01014.x. [DOI] [PubMed] [Google Scholar]
- 42.Wang GS, Kearney DL, De Biasi M, et al. Elevation of RNA-binding protein CUGBP1 is an early event in an inducible heart-specific mouse model of myotonic dystrophy. J Clin Invest. 2007;117:2802–2811. doi: 10.1172/JCI32308. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.