

Abstract
It has been demonstrated that hypoxic preconditioning (HP) enhances the survival ability of the organism against the subsequent acute anoxia (AA). However, it is not yet clear whether necrosis induced by AA can be prevented by HP, and what are the underlying mechanisms. In this study, we examined the effect of HP (10% O2, 48 h) on necrosis induced by AA (0% O2, 24 h) in PC12 cells. We found that HP delayed the regulatory volume decrease and reduced cell swelling after 24 h of exposure to AA. Since aldose reductase (AR) is involved in cell volume regulation, we detected AR mRNA expression with reverse transcription-polymerase chain reaction (RT-PCR) techniques. The AR mRNA level was dramatically elevated by HP. Furthermore, an HP-induced decrease in cell injury was reversed by berberine chloride (BB), the inhibitor of AR. In addition, sorbitol synthesized from glucose catalyzed by AR is directly related to cell volume regulation. Subsequently, we tested sorbitol content in the cytoplasm. HP clearly elevated sorbitol content, while BB inhibited the elevation induced by HP. Further study showed that a strong inhibitor of sorbitol permease, quinidine, completely reversed the protection induced by HP after AA. These data provide evidence that HP prevents necrosis induced by AA and is mediated by AR and sorbitol pathway.
Keywords: Hypoxic preconditioning, Acute anoxia, Aldose reductase, Sorbitol
Introduction
Necrosis is a mode of cell death. It occurs more frequently in Alzheimer's disease (Bazan et al. 2002), acute anoxia (AA)/ischemia (Majno and Joris 1995; Northington et al. 2001), cerebrovascular disease (Fujikawa et al. 2000), and cerebral trauma resulting from acidosis (Ding et al. 2000).
Cell necrosis is triggered once cellular energy metabolism is at a lower level (Lipton 1999), which results in disturbance of ion homeostasis on membranes and an overloading of Na+ and Ca2+ (Carini et al. 1999; Lipton 1999), alteration of cytoskeleton function (Bellomo et al. 1990), degeneration of cellular membrane and, finally, cell disintegration. It is generally thought that cell necrosis is a non-specific, passive, and harmful form of cell death.
Anoxic necrosis is found predominantly in acute anoxic/ischemic injury. Prevention against necrosis after anoxic stress will contribute to the treatment of some diseases, such as stroke and cardiovascular disease. Our previous work demonstrated that after 3 days of hypertonic culture, PC12 cells developed some protection from necrosis following acute anoxic injury by increasing aldose reductase (AR) and sorbitol levels (Ma et al. 2002).
AR is the first enzyme of the polyol pathway. In this pathway, AR catalyzes conversion of glucose to sorbitol in the presence of nicotinamide adenine dinucleotide phosphate, while sorbitol dehydrogenase converts sorbitol to fructose in the presence of NAD+ (Hwang et al. 2005). Under osmotic stress, the abundance of AR is elevated by increasing the transcription of its gene. In turn, sorbitol synthesis is raised by increasing the amount and activity of AR (Burg 1995). Change of osmolarity causes sorbitol to leak rapidly to the external medium through a sorbitol permease transport pathway, which prevents excessive cell swelling. Efflux of sorbitol was the primary mechanism for regulatory volume decrease (RVD). RVD protects the cells by minimizing swelling (Garty et al. 1991). The roles of AR in oxidative damage induced by anoxia/ischemia are controversial. Some studies report that inhibition of AR increases peroxide, inflammation reactivity, and exacerbates ischemic injury (Kaiserova et al. 2008; Rittner et al. 1999; Spycher et al. 1997); other studies show that inhibition of AR protects the heart against myocardial ischemia and reperfusion injury (Hwang et al. 2002; Ramasamy et al. 1997, 1998).
In this study, we demonstrate for the first time that hypoxic preconditioning (HP) protects PC12 cells against necrosis after exposure to AA, and this protective role of HP is probably related to cell volume regulation by increasing AR and sorbitol levels.
Materials and methods
Cell culture
PC12 cells are a neuronal cell line derived from a rat pheochromocytoma usually used as a model for neurons, in that they are derived from the neural crest and can be induced to differentiate to a neuronal phenotype. PC12 cells were obtained from American Type Culture Collection (Manassas, VA, USA) and cultured in 75-cm2 tissue culture flasks (Corning, Lowell, MA, USA) in Dulbecco's modified Eagle medium (DMEM; Gibco, Tulsa, OK, USA) as described previously (Greene and Tischler 1976). Briefly, cells were maintained in DMEM containing 5% newborn calf serum and 10% horse serum supplemented with 100 U/ml penicillin and 100 pg/ml streptomycin in a moist atmosphere of 5% CO2 at 37°C. Once a week, the PC12 cells were dissociated using 0.25% trypsin (Gibco, Tulsa, OK, USA) and plated at low density on dishes. Experiments were performed l–2 days after plating.
HP and AA treatment
For HP treatment, PC12 cells were placed in a 2,000-cm3 airtight chamber, and a gas mixture of 10% O2/5% CO2/85% N2 was continuously delivered into the chamber for 48 h as previous described (Wu et al. 2004). Subsequently, cells pretreated with HP and those cultured in normoxia were simultaneously exposed to a gas mixture of 5% CO2/95% N2 for 24 h. The former was designated as the HP + AA group, and the latter as the AA group.
Morphological analysis of necrotic cells
Cells were double-stained with propidium iodide (PI, 2.5 μg/ml; Sigma-Aldrich, Louis, MO, USA) and Hoechst 33258 (2.5 μg/ml; Sigma-Aldrich, Louis, MO, USA) and observed under a fluorescence microscope (Olympus IX71, Minneapolis, MN, USA) to distinguish necrotic cells from apoptotic cells according to the method described previously: intact blue nuclei, pink nuclei, and condensed blue nuclei were considered viable, necrotic, and apoptotic cells, respectively (Shimizu et al. 1996). Electron microscopy assay was carried out as previously described (Xiao et al. 2002). In brief, cells were fixed in 4% paraformaldehyde, pH 7.4, at 4°C for 1 h. After two washes in 0.1 M phosphate, cells were post-fixed with 2% OsO4 in the same buffer for 30 min. The cells were dehydrated in ethanol and then in 100% propylene oxide, followed by embedding overnight at 37°C for a further 3 days at 60°C. Ultrafine sections were cut and examined on an electron microscope (Philips EM120, Eindhoven, Netherlands).
Flow cytometry analysis of necrotic cells
Cells were harvested, permeabilized with 70% alcohol, and stained with 10 μg/ml of PI to determine necrotic cells by DNA content as previously described (Hetz et al. 2002). For DNA content analysis, samples containing roughly 1 × 104 cells were analyzed by FACS using the Cell Quest program (FACSalibur, Becton Dikinson, Franklin Lakes, NJ, USA).
Analysis of fragmented DNA by agarose gel electrophoresis
DNA was extracted from cultured PC12 cells according to the manufacturer's instructions (Trizol Reagent, Invitrozen, Carlsbad, CA, USA). DNA concentration was determined at 260 nm by spectrophotometry. DNA electrophoresis was carried out on 1% agarose gel containing 1 μg/ml ethidium bromide, and DNA bands were visualized under UV light (Alpha Innotech, San Leandro, CA, USA).
LDH measurement
Activity of lactate dehydrogenase (LDH) in the medium was measured by means of a test kit (Roche, Indianapolis, IN, USA). Total LDH activity was determined after thorough breakdown of cells caused by 0.1% Triton-X-100 in phosphate buffer solution. The percentage of LDH leakage from an index of cell injury was calculated following a formula: 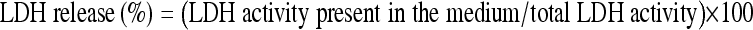 (Koh and Choi 1987; Wu et al. 2004).
(Koh and Choi 1987; Wu et al. 2004).
Cell volume assay
Cell volume was determined by the maximum cross-sectional area of a cell according to the method described previously (Leung et al. 1994). For volume measurements, cells were diluted to approximately 5 × 104 cell/ml. Measurement of cross-sectional areas was performed with the Imaging System (Panasonic, Osaka, Japan). Values of area were converted into the value of cell volume: absolute cell volumes were calculated from distribution curves of cell diameter using a standard curve generated by polystyrene latex beads of known diameter. Values were expressed as the relative value of cell volume change by dividing the normalized cell volume by that of the onset point of each group.
RT-PCR analysis
PC12 cells were broken down with 40 μl water containing 1unit/μl RNase inhibitor (Promega, Madison, WI, USA), then frozen with liquid N2 for 10 min and thawed twice. The cell lysate was centrifuged at 10,000 × g for 10 min. Then, the supernatant was transferred into another tube to be mixed with the same volume of Trizol (Gibco, Tulsa, OK, USA) to extract total RNA. In reverse transcription-polymerase chain reaction (RT-PCR) procedure, cDNA synthesis was performed by using AMV transcriptase (Promega, Madison, WI, USA). Both AR primers and GAPDH primers (Promega, Madison, WI, USA) were added to the same tube to perform PCR amplification. The forward primer of AR was 5′-TGG ATC CAC TCT TGC GGG TCG TTG T, and the backward primer was 5′-TGA ATT CGC TTC AGA CTT CTG CGT G. The PCR was performed for 30 cycles defined as denaturing at 95°C for 1 min, annealing at 50°C for 1 min, and extending at 72°C for 1 min. The PCR product was run on a 1% agarose gel, and the gel was scanned under UV light in multi-image light cabinet (Alpha Innotech, San Leandro, CA, USA).
Measurement of sorbitol content
The content of sorbitol in cultured cytoplasm was enzymatically measured with sorbitol dehydrogenase (Roche, Indianapolis, IN, USA) according to the manufacturer's instructions. Briefly, cultures were centrifuged at 2,400 × g for 10 min at 4°C, and the supernatant was mixed with glycine buffer containing NAD+ and sorbitol dehydrogenase. The sorbitol contents were measured using spectrophotometric methods as described previously (Malone et al. 1980). To test the AR activity indirectly, 20 μg/ml berberine chloride (BB) (Sigma-Aldrich, Louis, MO, USA) was used as an inhibitor of AR (Liu et al. 2008).
Statistics
Data shown were expressed as mean ± SD from data obtained in at least three independent experiments. Student's t tests were used to compare the effects of all treatments. *P < 0.05 was considered statistically significant.
Results
The necrotic injury induced by AA in PC12 cells
After PC12 cells were subjected to different degrees of AA (4, 8, 12, 16, 20, and 24 h), the cellular DNA fragments were analyzed on 1% agarose electrophoresis. As shown in Fig. 1a, the pattern of DNA fragments isolated was smeared and dispersive at 20 and 24 h following exposure to AA. It suggested that 20 and 24 h of anoxia induced necrotic injury. Further investigation was analyzed by flow cytometry. Cell debris characteristic of necrosis (the bulk of which had been eliminated during acquisition with the flow cytometer by gate-off through eliminating the corresponding region) could be recognized by the smaller diameter and reduced DNA fluorescence compared to apoptotic cells. Figure 1b showed that there was no obvious hypo-diploid peak produced by apoptotic DNA fragments after 24 h of AA ahead of representation of the diploid peaks corresponding to normal nucleus DNA. Furthermore, the obvious cellular and organelle swelling, damage, and disintegration of the cytoplasmic membrane were seen under electron microscope (Fig. 1c). The above results demonstrated that necrosis rather than apoptosis was the main form of cell death induced by 24 h of AA. To assess the percentage of necrotic cells under these conditions, the number of necrotic cells and the total cells were counted by means of dyes with PI and Hoechst 33258, as shown in Fig. 1d. The result showed that the proportion of necrotic cells (PI-positive cells) in the total number of dead cells (necrotic and apoptotic cells) was 95% ± 5% (n = 12) after cells were exposed to AA for 24 h.
Fig. 1.

The necrotic occurrence of PC12 cells under acute anoxia (AA) exposure. a 1% agarose electrophoresis graph of 4, 12, 16, 20, and 24 h after AA. Smear band characteristic of necrosis appears at 20 and 24 h after AA. b Flow cytometric analysis of cells treated with propidium iodide (PI) shows that there are no typical apoptotic sub-diploid peaks, but there are some peaks characteristic of necrosis after 24 h of AA exposure. c The morphology under electron microscopy. The upper image shows a cultured PC12 cell under normoxia, and the lower image shows a cultured PC12 cell exposed to AA. Disintegration of cytoplasmic membrane and cellular and organellar swelling are clearly seen in the cell exposed to AA. d PI and Hoechst 33258 double staining under fluorescent microscope. Intact blue nuclei, pink nuclei, and condensed blue nuclei are considered viable, necrotic, and apoptotic cells, respectively
The anti-necrotic protection of HP against AA
To investigate the effect of HP on necrotic injury induced by AA in PC12 cells, cultures were divided into five groups: (1) cells were cultured in normoxia (control); (2) cells were exposed to AA for 24 h (AA); (3) cells were treated with HP for 48 h followed by AA exposure for 24 h (HP + AA); (4) cells were treated with HP and 20 μg/ml of BB for 48 h followed by AA exposure for 24 h (HP + BB).
The effect of HP on LDH release
The necrotic injury was evaluated by LDH release. As shown in Fig. 2, the data indicated that the ratio of LDH release in the control group was 17.46 ± 8.66%, and it rose to 48.21 ± 6.46% in the AA group, while in the HP + AA group, it was lowered to 29.28 ± 3.42% (P < 0.01 compared with AA group, n = 10), and in HP + BB group, the ratio of the LDH release rose again to 42.89 ± 4.97% (P < 0.05 compared with HP + AA group, n = 10). These results indicate that HP induces a protective role against anoxic injury, while inhibition of AR activity with BB destroys the protection induced by HP.
Fig. 2.

Lactate dehydrogenase (LDH) leakage of PC12 cells. Activity of LDH in the medium was measured according to the manufacturer's instructions. Control: cells were cultured in normoxia. AA: cells were exposed to anoxia for 24 h. HP + AA: cells were treated with hypoxic preconditioning (HP) for 48 h followed by 24 h of anoxia. HP + BB: cells were treated with HP for 48 h in the medium containing 20 μg/ml of berberine chloride followed by AA exposure for 24 h. **P < 0.01 compared with AA group; #P < 0.05 compared with HP + AA group. Data represent the mean ± SD for four experiments
The effect of HP on changes of cell volume
The change process of cell volume is shown in Fig. 3a. At 4 h of anoxia exposure, PC12 cells showed clear RVD behavior. Subsequently, cell volume increased gradually to 4.9-fold of the initial level 24 h after anoxia in the AA group; in contrast, the RVD behavior appeared at 8 h of anoxia exposure, and the cell volume was 2.6-fold of the initial volume after 24 h of anoxia in HP + AA group. The statistical data demonstrated that the increase of cell volume in the HP + AA group at 24 h of exposure to anoxia was significantly less than that in the AA group (Fig. 3b). It is clear that HP stabilizes the cell volume when cells are exposed to AA.
Fig. 3.

Cell volume regulation under acute anoxia (AA). a The process of cell volume regulation during 24 h of AA exposure. AA: cells cultured in normoxia were directly exposed to AA for 0, 4, 8, 16, and 24 h. HP + AA: cells treated with hypoxic preconditioning (HP) were followed by exposure to AA for 0, 4, 8, 16, and 24 h. A regulatory volume decrease was delayed in the HP group compared with the AA group. Cell swelling occurred after 16 h of AA exposure. b Analysis of the increase of cell volume 24 h after anoxia. After that, the cell volume increased approximately fivefold compared to the initial volume in the AA group, while there was less than a threefold increase in the HP + AA group. *P < 0.05 compared with AA group. Data are expressed as the mean ± SD from three independent experiments
Elevation of AR mRNA expression by HP
Cell volume regulation is related to AR (Burg 1995; Czekay et al. 1994). Additionally, in our present study, inhibition of AR activity attenuated the protection induced by HP (Fig. 2). We investigated whether HP alone affected the expression of AR. To analyze the effect of HP on the mRNA expression of AR, RT-PCR technique was used to test the mRNA levels. Figure 4 shows that AR expression was significantly up-regulated in the HP group compared with the control. This suggests that the protection of HP may be related to elevated AR expression.
Fig. 4.

The reverse transcription-polymerase chain reaction (RT-PCR) analysis of aldose reductase (AR) mRNA expression. RT-PCR products, separated and visualized by agarose gel electrophoresis, represent AR mRNA levels. Marker: DNA ladder molecular size marker; control: cells were cultured in normoxia; HP: cells were treated with hypoxic preconditioning (HP) for 48 h; GAPDH: loading control. The mRNA level of AR was elevated after HP. This graph is typical of two separate experiments
The role of sorbitol in HP-induced protection against AA
To further verify whether the protection of HP is mediated by AR, its enzymatic product sorbitol was measured using spectrophotometric methods. As shown in Fig. 5a, there was a significant increase in sorbitol levels in the HP group compared with the control, while the inhibitor of AR and BB attenuated the increase of sorbitol content induced by HP to less than half that in the HP group. In order to test the role of sorbitol in HP-induced protection, a strong inhibitor of sorbitol permease, quinidine (Garty et al. 1991), was added in the HP group and then exposed to AA. Cellular injury was assessed by LDH assay. The result showed that the LDH leakage decreased in the HP group versus the control; in contrast, the LDH leakage in the quinidine-treated HP (HP + Qui) group significantly increased compared with the HP group (Fig. 5b). These results suggest that the increase of sorbitol content by HP might contribute to the protective effect of HP against AA.
Fig. 5.

The role of sorbitol in hypoxic preconditioning (HP)-induced protection against acute anoxia (AA). a The percentage of sorbitol content compared with the control value. The value of the sorbitol content in the control group was taken as 100%. Control: cells were cultured in normoxia. HP: cells were treated with HP for 48 h. HP + BB: 20 μg/ml berberine chloride was added in the medium of HP. **P < 0.01 compared with the control group; #P < 0.05 compared with HP group. Data represent the mean ± SD for three experiments. b The lactate dehydrogenase leakage. Control: cells were cultured in normoxia. AA: cells cultured in normoxia were directly exposed to AA for 24 h. HP + AA: cells pretreated with HP for 48 h were subsequently exposed to AA for 24 h. HP + Qui: 0.5 mmol/L quinidine was added to the medium of HP cells for 48 h before cells were exposed to AA. *P < 0.05 compared with AA group; ##P < 0.01 compared with HP + AA group. Data are expressed as the mean ± SD from three independent experiments
Discussion
Under anoxia exposure, whether cells die via the apoptosis or necrosis pathway is determined by many factors. Tissue and cell type specificity, and the extent or nature of injury and death signals should all be taken into consideration (Barros et al. 2001; Rosser and Gores 1995; Strauss and Morton 2003). It was found that after anoxia or 5–12 mM NaCN exposure, PC12 cells or NGF-induced differentiated PC12 cells were equally injured leading to death by necrosis. Although necrosis cannot be reversed, we found that necrotic death induced by AA could be prevented by HP.
After AA exposure, cells presented the characteristics of necrosis: for example, a smear band in DNA electrophoresis and obvious cellular and organellar swelling and disintegration of cytoplasmic membrane. Many papers have reported that apoptosis induced by severe hypoxia or ischemia could be blocked by HP (Rybnikova et al. 2006; Wu et al. 2005; Zhang et al. 2007). However, there are few reports that HP can protect cells from necrosis induced by AA. Moreover, the underlying mechanism is still not clear. In this study, we show that HP protects PC12 cells from necrotic death after exposure to AA. We also demonstrate for the first time that the protection is related to increased AR and sorbitol levels by HP.
AA caused a sharp rise in LDH leakage indicative of cell injury, while HP clearly inhibited it. In addition, BB, an inhibitor of AR, completely reversed the protection of HP. These results indicated that AR was involved in the protection produced by HP.
AA furthermore caused the increase in cell volume, which would result in swelling and eventually lead cells to necrosis. By observing the change of cell volume at different time points, we found that HP not only delayed the appearance of RVD but also inhibited the increase of cell volume during 24 h of AA exposure. This suggested that cell volume regulation could be a potential mechanism in the protection exerted by HP against AA.
AR is involved in cell volume regulation (Burg 1995). In this study, we found HP significantly elevated the abundance of AR gene products by increasing its transcription. Therefore, it is necessary to test further whether its function as a protease is affected by HP.
Sorbitol synthesized from glucose catalyzed by AR is directly related to cell volume regulation (Czekay et al. 1994; Schuttert et al. 2002; Siebens and Spring 1989). After synthesis, sorbitol is packed into the secretory vesicles to carry and fuse to cytoplasmic membrane, and finally sorbitol is released from it, which is a procedure involving the help of cytoskeleton (Czekay et al. 1994). Its biological significance is related to cell volume regulation (Burg 1995; Siebens and Spring 1989). In this study, we showed for the first time that HP significantly increased sorbitol levels, while the inhibitor of AR, BB, attenuated the increase in sorbitol content induced by HP. According to the above results, we hypothesized that sorbitol might be correlated with increased AR by HP. The fact that quinidine, a stronger inhibitor of sorbitol, reversed the protection afforded by HP indicates that sorbitol contributes to the protection of HP.
Taken together, HP can prevent PC12 cells from necrosis after AA, and the protection of HP is mediated by elevating AR expression. AR increases sorbitol synthesis, which inhibits the swelling of cell volume induced by AA. Ultimately, cell necrosis was prevented.
Acknowledgements
We are grateful to Dr. Lawrence Hightower for editorial help and Dr. Helen Neumann for language corrections. We also thank Dr. Ping Liang, the executive editor of Progress in Natural Science, for her critical reading of this manuscript. This work was supported by the Chinese National Key Basic Research Project (2006CB504100) and the Key Grant of the National Nature Science Foundation of China (30393130).
Contributor Information
Ling-Ling Zhu, Phone: +86-10-68210077, FAX: +86-10-68213039, Email: linglingzhu@hotmail.com.
Ming Fan, Phone: +86-10-68214026, FAX: +86-10-68213039, Email: fanming@nic.bmi.ac.cn.
References
- Barros LF, Hermosilla T, Castro J. Necrotic volume increase and the early physiology of necrosis. Comp Biochem Physiol Part A Mol Integr Physiol. 2001;130:401–409. doi: 10.1016/S1095-6433(01)00438-X. [DOI] [PubMed] [Google Scholar]
- Bazan NG, Palacios-Pelaez R, Lukiw WJ. Hypoxia signaling to genes: significance in Alzheimer's disease. Mol Neurobiol. 2002;26:283–298. doi: 10.1385/MN:26:2-3:283. [DOI] [PubMed] [Google Scholar]
- Bellomo G, Mirabelli F, Vairetti M, Iosi F, Malorni W. Cytoskeleton as a target in menadione-induced oxidative stress in cultured mammalian cells. I. Biochemical and immunocytochemical features. J Cell Physiol. 1990;143:118–128. doi: 10.1002/jcp.1041430116. [DOI] [PubMed] [Google Scholar]
- Burg MB. Molecular basis of osmotic regulation. Am J Physiol. 1995;268:F983–F996. doi: 10.1152/ajprenal.1995.268.6.F983. [DOI] [PubMed] [Google Scholar]
- Carini R, Autelli R, Bellomo G, Albano E. Alterations of cell volume regulation in the development of hepatocyte necrosis. Exp Cell Res. 1999;248:280–293. doi: 10.1006/excr.1999.4408. [DOI] [PubMed] [Google Scholar]
- Czekay RP, Kinne-Saffran E, Kinne RK. Membrane traffic and sorbitol release during osmo- and volume regulation in isolated rat renal inner medullary collecting duct cells. Eur J Cell Biol. 1994;63:20–31. [PubMed] [Google Scholar]
- Ding D, Moskowitz SI, Li R, Lee SB, Esteban M, Tomaselli K, Chan J, Bergold PJ. Acidosis induces necrosis and apoptosis of cultured hippocampal neurons. Exp Neurol. 2000;162:1–12. doi: 10.1006/exnr.2000.7226. [DOI] [PubMed] [Google Scholar]
- Fujikawa DG, Shinmei SS, Cai B. Seizure-induced neuronal necrosis: implications for programmed cell death mechanisms. Epilepsia. 2000;41(Suppl 6):S9–S13. doi: 10.1111/j.1528-1157.2000.tb01549.x. [DOI] [PubMed] [Google Scholar]
- Garty H, Furlong TJ, Ellis DE, Spring KR. Sorbitol permease: an apical membrane transporter in cultured renal papillary epithelial cells. Am J Physiol. 1991;260:F650–F656. doi: 10.1152/ajprenal.1991.260.5.F650. [DOI] [PubMed] [Google Scholar]
- Greene LA, Tischler AS. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci U S A. 1976;73:2424–2428. doi: 10.1073/pnas.73.7.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz CA, Hunn M, Rojas P, Torres V, Leyton L, Quest AF. Caspase-dependent initiation of apoptosis and necrosis by the Fas receptor in lymphoid cells: onset of necrosis is associated with delayed ceramide increase. J Cell Sci. 2002;115:4671–4683. doi: 10.1242/jcs.00153. [DOI] [PubMed] [Google Scholar]
- Hwang YC, Sato S, Tsai JY, Yan S, Bakr S, Zhang H, Oates PJ, Ramasamy R. Aldose reductase activation is a key component of myocardial response to ischemia. FASEB J. 2002;16:243–245. doi: 10.1096/fj.01-0732com. [DOI] [PubMed] [Google Scholar]
- Hwang YC, Shaw S, Kaneko M, Redd H, Marrero MB, Ramasamy R. Aldose reductase pathway mediates JAK-STAT signaling: a novel axis in myocardial ischemic injury. FASEB J. 2005;19:795–797. doi: 10.1096/fj.04-2780fje. [DOI] [PubMed] [Google Scholar]
- Kaiserova K, Tang XL, Srivastava S, Bhatnagar A. Role of nitric oxide in regulating aldose reductase activation in the ischemic heart. J Biol Chem. 2008;283:9101–9112. doi: 10.1074/jbc.M709671200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh JY, Choi DW. Quantitative determination of glutamate mediated cortical neuronal injury in cell culture by lactate dehydrogenase efflux assay. J Neurosci Methods. 1987;20:83–90. doi: 10.1016/0165-0270(87)90041-0. [DOI] [PubMed] [Google Scholar]
- Leung S, O'Donnell ME, Martinez A, Palfrey HC. Regulation by nerve growth factor and protein phosphorylation of Na/K/2Cl cotransport and cell volume in PC12 cells. J Biol Chem. 1994;269:10581–10589. [PubMed] [Google Scholar]
- Lipton P. Ischemic cell death in brain neurons. Physiol Rev. 1999;79:1431–1568. doi: 10.1152/physrev.1999.79.4.1431. [DOI] [PubMed] [Google Scholar]
- Liu W, Liu P, Tao S, Deng Y, Li X, Lan T, Zhang X, Guo F, Huang W, Chen F, Huang H, Zhou SF. Berberine inhibits aldose reductase and oxidative stress in rat mesangial cells cultured under high glucose. Arch Biochem Biophys. 2008;475:128–134. doi: 10.1016/j.abb.2008.04.022. [DOI] [PubMed] [Google Scholar]
- Ma ZM, Xie YZ, Ding AS, Wu LY, Zhao T, Wang YX, FZ FM. The mechanism of cell volume regulation on acute hypoxic tolerance in PC12 cells. Prog Nat Sci. 2002;12:994–996. [Google Scholar]
- Majno G, Joris I. Apoptosis, oncosis, and necrosis. An overview of cell death. Am J Pathol. 1995;146:3–15. [PMC free article] [PubMed] [Google Scholar]
- Malone JI, Knox G, Benford S, Tedesco TA. Red cell sorbitol: an indicator of diabetic control. Diabetes. 1980;29:861–864. doi: 10.2337/diabetes.29.11.861. [DOI] [PubMed] [Google Scholar]
- Northington FJ, Ferriero DM, Graham EM, Traystman RJ, Martin LJ. Early neurodegeneration after hypoxia-ischemia in neonatal rat is necrosis while delayed neuronal death is apoptosis. Neurobiol Dis. 2001;8:207–219. doi: 10.1006/nbdi.2000.0371. [DOI] [PubMed] [Google Scholar]
- Ramasamy R, Oates PJ, Schaefer S. Aldose reductase inhibition protects diabetic and nondiabetic rat hearts from ischemic injury. Diabetes. 1997;46:292–300. doi: 10.2337/diabetes.46.2.292. [DOI] [PubMed] [Google Scholar]
- Ramasamy R, Trueblood N, Schaefer S. Metabolic effects of aldose reductase inhibition during low-flow ischemia and reperfusion. Am J Physiol. 1998;275:H195–H203. doi: 10.1152/ajpheart.1998.275.1.H195. [DOI] [PubMed] [Google Scholar]
- Rittner HL, Hafner V, Klimiuk PA, Szweda LI, Goronzy JJ, Weyand CM. Aldose reductase functions as a detoxification system for lipid peroxidation products in vasculitis. J Clin Invest. 1999;103:1007–1013. doi: 10.1172/JCI4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosser BG, Gores GJ. Liver cell necrosis: cellular mechanisms and clinical implications. Gastroenterology. 1995;108:252–275. doi: 10.1016/0016-5085(95)90032-2. [DOI] [PubMed] [Google Scholar]
- Rybnikova E, Sitnik N, Gluschenko T, Tjulkova E, Samoilov MO. The preconditioning modified neuronal expression of apoptosis-related proteins of Bcl-2 superfamily following severe hypobaric hypoxia in rats. Brain Res. 2006;1089:195–202. doi: 10.1016/j.brainres.2006.03.053. [DOI] [PubMed] [Google Scholar]
- Schuttert JB, Fiedler GM, Grupp C, Blaschke S, Grunewald RW. Sorbitol transport in rat renal inner medullary interstitial cells. Kidney Int. 2002;61:1407–1415. doi: 10.1046/j.1523-1755.2002.00285.x. [DOI] [PubMed] [Google Scholar]
- Shimizu S, Eguchi Y, Kamiike W, Itoh Y, Hasegawa J, Yamabe K, Otsuki Y, Matsuda H, Tsujimoto Y. Induction of apoptosis as well as necrosis by hypoxia and predominant prevention of apoptosis by Bcl-2 and Bcl-XL. Cancer Res. 1996;56:2161–2166. [PubMed] [Google Scholar]
- Siebens AW, Spring KR. A novel sorbitol transport mechanism in cultured renal papillary epithelial cells. Am J Physiol. 1989;257:F937–F946. doi: 10.1152/ajprenal.1989.257.6.F937. [DOI] [PubMed] [Google Scholar]
- Spycher SE, Tabataba-Vakili S, O'Donnell VB, Palomba L, Azzi A. Aldose reductase induction: a novel response to oxidative stress of smooth muscle cells. FASEB J. 1997;11:181–188. doi: 10.1096/fasebj.11.2.9039961. [DOI] [PubMed] [Google Scholar]
- Strauss KA, Morton DH. Type I glutaric aciduria, part 2: a model of acute striatal necrosis. Am J Med Genet C Semin Med Genet. 2003;121C:53–70. doi: 10.1002/ajmg.c.20008. [DOI] [PubMed] [Google Scholar]
- Wu LY, Ding AS, Zhao T, Ma ZM, Wang FZ, Fan M. Involvement of increased stability of mitochondrial membrane potential and overexpression of Bcl-2 in enhanced anoxic tolerance induced by hypoxic preconditioning in cultured hypothalamic neurons. Brain Res. 2004;999:149–154. doi: 10.1016/j.brainres.2003.09.081. [DOI] [PubMed] [Google Scholar]
- Wu LY, Ding AS, Zhao T, Ma ZM, Wang FZ, Fan M. Underlying mechanism of hypoxic preconditioning decreasing apoptosis induced by anoxia in cultured hippocampal neurons. NeuroSignals. 2005;14:109–116. doi: 10.1159/000086293. [DOI] [PubMed] [Google Scholar]
- Xiao AY, Wei L, Xia S, Rothman S, Yu SP. Ionic mechanism of ouabain-induced concurrent apoptosis and necrosis in individual cultured cortical neurons. J Neurosci. 2002;22:1350–1362. doi: 10.1523/JNEUROSCI.22-04-01350.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Park TS, Gidday JM. Hypoxic preconditioning protects human brain endothelium from ischemic apoptosis by Akt-dependent survivin activation. Am J Physiol Heart Circ Physiol. 2007;292:H2573–H2581. doi: 10.1152/ajpheart.01098.2006. [DOI] [PubMed] [Google Scholar]