

Abstract
A bioinformatics analysis of sequences of enzymes of the glycoside hydrolase (GH) 13 family members such as α-amylase, cyclodextrin glycosyltransferase (CGTase), branching enzyme and cyclomaltodextrinase has been carried out in order to find out the sequence motifs that govern the reactions specificities of these enzymes by using hidden Markov model (HMM) profile. This analysis suggests the existence of such sequence motifs and residues of these motifs constituting the −1 to +3 catalytic subsites of the enzyme. Hence, by introducing mutations in the residues of these four subsites, one can change the reaction specificities of the enzymes. In general it has been observed that α -amylase sequence motif have low sequence conservation than rest of the motifs of the GH13 family members.
Keywords: α-Amylases, Glycoside hydrolase family 13, Sequence motif, Reaction specificity, Substrate specificity, HMM profile
Background
A large group of “Carbohydrate Active enzymes” that hydrolyse polysaccharides have been divided into the glycoside hydrolase (GH) families of the CAZy database [1] based on their amino acid sequence similarity. Members of the GH13, GH70 and GH77 families, being part of the same clan (GH-H), are believed to share a common ancestor and catalytic mechanism [2–5]. Further, these enzymes also share the A-, B- and C-domains. In addition to α-(1→4)- and α-(1→6)-linkages, even α-(1→1)-, α-(1→2)- and α-(1→3)- linkages are acted upon by members of the GH-H clan [6]. The enzymes of GH13 family have a wide range of substrate specificities and catalytic activities. This “polyspecificity” of the GH13 family has led to further division of this family into 35 subfamilies [7]. Most of the subfamilies of GH13 are apparently monospecific, however some also belong to polyspecific subfamilies. Amylolytic enzymes with different substrate specificities have poor overall sequence similarity in the domain A except for four regions of the conserved residues [8– 10]. An evolutionary tree constructed on the basis of these four conserved regions showed that α-(1→4)-hydrolases, α-(1→6)- hydrolases and transferases form distinct groups [9].
There are also regions wherein the sequence similarity is rather high; although strict conservation of the residues is not observed in these regions. These are region V (173LPDLD177), region VI (56GFTAIWITP64), and region VII (323GIPIIYAGQ331; through out the introduction residue number is given according to 6TAA (TAKA α-amylase), unless stated otherwise) [11]. Of these, region V is at the C-terminus of the domain B of these enzymes. A sequence analysis of 79 experimentally characterized proteins has suggested that the signature sequence QpDln and MPKln (single letter amino acid symbols are used; upper case letters indicate total conservation whereas lower case letters indicate partial conservation) define the oligo-1,6-glucosidase and neopullulanase subfamilies, respectively, in the region V. The signature sequence MPDLN characterized the intermediary group which includes enzymes with mixed specificities of α-amylase, cyclomaltodextrinase and neopullulanase [12]. Currently, the catalytic triad residues Asp206, Glu230 and Asp297 seem to be the only residues that are absolutely invariant among all the GH-H clan members [11]. In addition, a few residues such as Gly56 and Pro64 [13] (flanking the second β-strand), Tyr82 [13], His122 and His296 [11] are present in most of the members. However, Arg204 has been found to be conserved only in the amylolytic members [6, 14].
A larger number of residues are conserved within subgroups of enzymes such as neopullulanases and oligo- α-1,6-glucosidases [12]. For example, Lys or Arg are conserved at position 209 in 91“ of the α-(1→4)-linkage specific members of the GH13 and GH77 family [06]. Similarly, Gly207 and His210 are present in many α-(1→4)-linkage-specific enzymes. In some of the enzymes, Gly207 is replaced by an aromatic residue and mimics the interactions of His210. However, in case of archaeal and plant α-amylases His210 is replaced with a Gly. Trp and Tyr/Phe are found at positions 231 and 232 in CGTases and one maltogenic amylase but not in other GH13 family members [06]. Enzymes which act on α-(1→6)-linkages (e.g., pullulanases, isoamylases, glycogen debranching enzymes) have a conserved aromatic residue in the loop that links the second β-strand and second helix. On the other hand, the enzymes that act on α-(1→4)-linkages, have a conserved aliphatic residue in this position (Ala120) suggesting that such regions provide the enzyme with a distinct “activity” and/or “substrate” specificities. Certain fungal proteins of the GH13 family, some of which are involved in cell wall synthesis, share a few conserved residues that are absent in α-amylases from other phyla (plant, animals and bacteria) [15]. These residues are His (Thr41 in Taka-amylase), Arg (Gly44), Cys (Thr66), Leu (Ala120), Tyr (Val231), Trp (Leu232), Cys (Ile326) and Leu (Glu332). The sequence motifs that are responsible for the reaction specificity of the enzymes of the GH13 family are not very well understood. So, the identification of the sequence motif of α-amylase, CGTase, branching enzymes and CDase subfamily of the GH13 family was performed.
All these motifs are in continuation with the conserved region III and are in almost same position with respect to each other. These sequence motifs belong to the region that has not been explored so far, and include residue number 225 to 264. This analysis identifies sequence motif that is responsible for reaction specificity of the GH13 family. The newly discovered sequence motif along with the previous analyses of the structures of these enzymes [16, 17] will not only help in the understanding of structure-function relationship of these enzymes but also in the identification of the GH13 family members.
Materials and Methodology
Databases and software
The experimentally characterized sequences of the GH13 family were taken from the UniProt database (http://www.uniprot.org/) [18]. This analysis was carried out on sequences retrieved from UniProt database in November 2008 and hence, the results of this analysis correspond to the database status of that period. Multiple sequence alignment was performed by using the locally installed T coffee (version 6.92) [19]. Multiple aligned sequences were visualized by using BioEdit [20]. The conserved part of the aligned sequences was used as a seed to generate the hidden Markov model (HMM) profile by using hmmbuild module of the HMMER (version 2.3.2) [21] and hmmcalibrate module used to calibrate E-value scores. The hmmsearch module was used to search the Swiss-Prot database (4,00,771 sequences; version 56.4; 4 Nov. 2008) at E value cutoff of 0.1. Sequences having highest sequence similarity with the profile were given a score and ‘Expect value’ (E) by HMMER program. The E-value of a sequence with a score z indicates the number of sequences that are expected to score z by chance, when searching a sequence database with the given size. Sequence logos were created using WebLogo (version 2.8.2) [22]. All parameters of different softwares had default values unless specified.
Generation of dataset and analysis strategy
The analysis was performed on those members of the GH13 family that use the α‐glucan as a substrate and produce disaccharides to polysaccharides as a final product. 90% sequence identity cutoff option present on the UniProt database (http://www.uniprot.org/) was used to retrieve the sequences and only reviewed Swiss-Prot sequences for α‐amylase, branching enzyme and cyclodextrin glycosyltransferase (CGTase) were selected. For CDase enzymes all the reviewed Swiss-Prot entries were chosen, as the number of the sequences was very low. Analysis was performed on the experimentally characterized sequences rather than computationally annotated sequences (having larger size of data set), because; despite of having high overall sequence similarity, changes in key residues may confer different activity or no activity at all. All the peptides and exceptionally large sequences were ignored to ensure the proper alignment. This selection criteria lead to generation of the dataset consisting of 59 α‐amylases, 12 CGTases, 166 branching enzymes, 3 maltogenic α‐amylases, 3 neopullulanases and 2 cyclomaltodextrinases (CDase) (Supplementary Table 1 - available with author). The CDase, neopullulanase and maltogenic α‐amylase enzymes are considered together in CDase subfamily as these enzymes have similar enzymatic activities [23].
The conserved region of α‐amylase, CGTase, branching enzyme and CDase subfamily was obtained by multiple sequence alignment and was further used for generation of sequence logos. The conserved region was selected by visualization in BioEdit. While selecting the conserved region, the length and region were kept same as far as possible. A sequence logo shows the relative frequencies of the various residues at a given position. This is indicated by proportionally varying the size of the symbol. The order of predominance of the residues at a given position are indicated by showing the most frequently occurring residue at the top of the heap and least frequently occurring residue at the bottom of the heap. The height of the logo at a given position is proportional to the degree of conservation at that position.
Sensitivity and specificity
Sensitivity is a parameter that reflects the ability of a profile to detect true positive sequences, while specificity reflects their ability to reject false positive sequences. Sensitivity = TP/(TP+FN), where TP is true positive, FN is false negative. Specificity = TP/(TP+FP), where FP is false positive.
Results and Discussion
The conserved region of α‐amylase, CGTase, branching enzyme and CDase are present in equivalent position in the multiple sequence alignment (Supplementary Figure 1 - available with author) and also includes conserved region III. Some of the residues of these motifs constitute the −1 to +3 catalytic subsites in 3D structure of the enzymes (that is present within the 4.5Å from the ligand, data not shown).
α‐amylase
Searching by the HMM profile of α-amylase sequence motif against Swiss-Prot database, the α-amylase enzymes with sensitivity of 95% and specificity of 99% were identified (Figure 1a). The four false positive hits included two CGTases and two uncharacterized glycosyl hydrolases. The E-values of CGTases are 0.0023 and 0.014; while that of uncharacterized glycosyl hydrolases are 4.5e-05 and 0.0085. The results suggest that the similar sequences present in both the α-amylase motif and false positive CGTases may be responsible for the α-(1→4) hydrolytic activity. However, the role of these residues needs to be experimentally investigated. The low E-value for the uncharacterized glycosyl hydrolases hit indicates these enzymes to be α- amylase. The sequence logo of this motif suggests that, besides catalytic glutamate, this motif also contains three highly conserved residues (Glu, Arg and Tyr and Trp as aromatic residue) that are absent in other enzymes at equivalent positions. It has been seen that mutations in the α-amylase of Bacillus stearothermophillus (A271Y, A271F) [25] and Bacillus licheniformis (V271F) [26] (Figure 1a; P04745 residue number, position 59 in sequence logo) caused an increase in transglycosylation reaction as compared to the wild-type enzymes. Interestingly, the mutant A271Y performed transglycosylation reaction more efficiently than A271F. In case of human salivary α-amylase the introduction of bulkier tryptophan residue in place of Phe271 (Figure 1a; P04745 residue number, position 59 in sequence logo) caused a disruption of the water chain involved in hydrolysis leading to reduction in hydrolytic activity by 70 folds [27]. Thus mutational analyses suggest that residues of this motif may contribute to the hydrolytic activity in α-amylases (Figure 1a).
Figure 1.
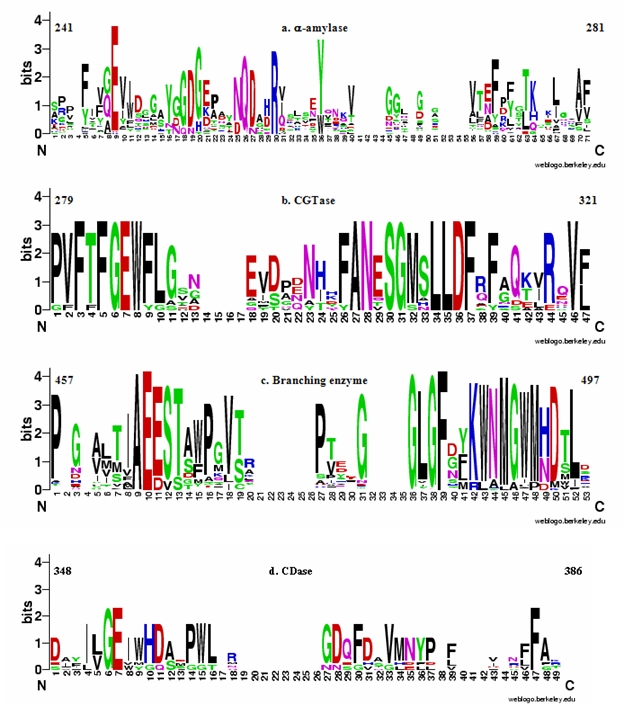
Sequence logo of (a) α‐amylase, (b) CGTase, (c) branching enzyme and (d) CDase. These sequence logos were generated from the conserved region of multiple sequence alignment of experimentally characterized enzymes (Supplementary Table 1 - available with author). The number above the sequence logo is according to P04745, P26827, A0PUI6, Q08751 and Q08751 Uniprot Id for α‐amylase, CGTase, branching enzyme, amylase subfamily and CDase, respectively. The numbers along the abscissa indicate the position of residues within the conserved region. The ordinates are in units of bits and are indicative of the information content at each position (Schneider & Stephens 1990) [24].
CGTase
CGTase specific motif was identified from 12 experimentally characterized sequences. Searching by the HMM profile of CGTase sequence motif against Swiss-Prot database, CGTase enzymes with sensitivity of 100% and specificity of 89% were identified (Figure 1b). The lower specificity of this motif is due to the two false positive hits (α‐amylase and maltogenic α‐amylase). Both of these false positive hits have a very low E-value with 4.7e-28 for α‐amylase and 3.2e-06 for maltogenic α‐amylase. However, the bit score for maltogenic α‐amylase is low and aligns with the short stretch of the query motif. As CGTase can perform both hydrolysis and transglycosylation reactions of α‐ (1→4) linked polysaccharide, it might have lead to picking of these false positive hits. Maltogenic α‐amylase can efficiently catalyze both hydrolysis and transglycosylation reactions, suggesting that the similar sequences in CGTase motif and false positive maltogenic α‐amylase may be responsible for hydrolytic and/or transglycosylation activity. The role of these residues needs to be experimentally validated. The sequence logo of this motif shows the presence of highly conserved sequence in different positions. Mutation, W286V, in Bacillus stearothermophillus CGTase (Figure 1b; P26827 residue number, position 8 in sequence logo) leads to a decrease in the cyclization and amylase activity [28] of CGTase. On the other hand, mutations in CGTase of Bacillus stearothermophillus (F287I) [28– 30], Bacillus sp.1011 (F287L) [31], Bacillus circulans strain 251 (F287N) [32] and Thermoanaerobacterium thermosulfurigenes (F287/N/L/I/E) [33, 34] (Figure 1b; P26827 residue number, position 9 in sequence logo) caused a decrease in cyclization and disproportionation reactions along with increase in hydrolytic activity. These mutational analyses suggest that these residues are central for cyclization reaction [35]. The replacement of E292A (Figure 1b; P26827 residue number, position 18 in sequence logo) in Bacillus circulans strain 251 implies that this residue may be involved in disproportionation reaction [32]. Thus, the above mutational analyses suggest that this CGTase specific motif may be conferring the reaction specificity in this enzyme.
Branching enzyme
The sequence logo of 166 multiple aligned sequences show the presence of a highly conserved sequence. On searching the HMM profile of branching enzyme sequence motif against Swiss-Prot database, the branching enzymes with a very high sensitivity and specificity of 100% were identified (Figure 1c). There are many residues like Ala, which are highly conserved and might be responsible for the reaction specificity of the branching enzymes. Unlike other enzymes where an aromatic residue or a hydrophobic residue is present next to the catalytic Glu, the branching enzymes have an acidic residue like Glu or Asp present. Thus, the conserved residues of this motif may be responsible for the reaction specificity of the enzyme.
CDase
CDase subfamily includes cylcomaltodextrinase (CDase), maltogenic α‐ amylase and neopullulanase. Despite of having different EC number, these enzymes have similar enzymatic activities [23], and hence are treated together in the present analysis. Searching by the HMM profile of CDase subfamily sequence motif against Swiss-Prot database, the enzymes of CDase subfamily with sensitivity of 100% and specificity of 53% were identified (Figure 1d). The false positive hits include four amylopullulanase and one maltodextrin glucosidase. The sequences similar in CDase motif and false positive amylopullulanase may be responsible for the hydrolytic activity. I355W (Figure 1d; Q08751residue number, position 8 in sequence logo) mutation in the CDase of Bacillus stearothermophillus reduced the affinity of this enzyme for α- (1→6) glycosidic linked substrate. It also lead to reaction specificity similar to that of typical starch-saccharifying α-amylase [36]. However, I355V mutant have high affinity for α- (1→6) glycosidic linked substrate. A mutation, W356A, of Thermoactinomyces vulgaris neopullulanase II suggests that W356 (Figure 1d; Q08751residue number, position 9 in sequence logo) is crucial for the binding of different substrate and it does so by making stacking interaction [37]. However, to make this stacking interaction possible, Y374 residue is required. The replacement of Y374A (Figure 1d; Q08751residue number, position 36 in sequence logo) results in a decrease in Km value for the pullulan as a substrate [38]. Y374 residue also helps in the hydrolysis of the different substrates by providing catalytic water near the catalytic site. It has been observed that on replacing Y374 with hydrophilic residue (D/S) in Bacillus stearothermophillus neopullulanase, there was a decrease in transglycosylation. Further, M372L and Y374F (Figure 1d; Q08751residue number, position 34 and 36 in sequence logo) mutants have been observed to have higher transglycosylation activity than the wild-type enzyme [36]. Thus, above mutational analyses clearly indicates that the residues of CDase motif may govern the reaction specificities in enzymes of this subfamily.
Conclusion
Sequence variation in the α‐amylase enzyme is higher as compared to rest of the enzyme of GH13 family members. This may be due to the presence of α‐ amylase in diverse variety of organisms and it may have evolved earlier than rest of the enzymes of the GH13 family. Thereby, during the evolution more mutations may have occurred in α‐amylase to perform its activity in diverse variety of biological systems or environment. As suggested by a number of mutational studies the replacement of residues belonging to one motif with sequence of another motif at equivalent positions may have changed the reaction-specificities of the enzyme. Hence, these motifs can be used as a guide for the inter-conversion of the GH13 family. Residues of these motifs constitute the −1 to +3 catalytic subsites of the GH13 family members, suggesting that these four subsites are mainly responsible for the reaction specificities of the enzymes of the some of the GH13 family members.
Acknowledgments
The author is grateful to University Grants Commission (UGC), India, for providing the senior research fellowship and Indian Institute of Technology Bombay, India, for providing the computing facility.
Footnotes
Citation:Kumar. Bioinformation 6(2): 61-63 (2011)
References
- 1.BL Cantarel, et al. Nucleic Acids Res. 2008;37:D233. [Google Scholar]
- 2.G Davies, B Henrissat. Structure. 1995;3(9):853. doi: 10.1016/S0969-2126(01)00220-9. [DOI] [PubMed] [Google Scholar]
- 3.B Henrissat, A Bairoch. Biochem J. 1996;316:695. doi: 10.1042/bj3160695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.T Kuriki, T Imanaka. J Biosci Bioeng. 1999;87(557) doi: 10.1016/s1389-1723(99)80114-5. [DOI] [PubMed] [Google Scholar]
- 5.MR Stam, et al. Carbohydr Res. 2005;340:2728. doi: 10.1016/j.carres.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 6.EA MacGregor, et al. Biochim Biophys Acta. 2001;1546:1. doi: 10.1016/s0167-4838(00)00302-2. [DOI] [PubMed] [Google Scholar]
- 7.MR Stam, et al. Protein Eng Des Sel. 2006;19:555. doi: 10.1093/protein/gzl044. [DOI] [PubMed] [Google Scholar]
- 8.S Janecek, et al. Biochem J. 1992;288:1069. doi: 10.1042/bj2881069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.HM Jespersen, et al. J Protein Chem. 1993;12:791. doi: 10.1007/BF01024938. [DOI] [PubMed] [Google Scholar]
- 10.G Pujadas, J Palau. Mol Biol Evol. 2001;18:38. [Google Scholar]
- 11.Janecek S, et al. Biologia Bratislava. 2002;11:29. [Google Scholar]
- 12.A Oslancova, S Janecek. Cell Mol Life Sci. 2002;59:1945. doi: 10.1007/PL00012517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.EA MacGregor, et al. FEBS Lett. 1996;378:263. doi: 10.1016/0014-5793(95)01428-4. [DOI] [PubMed] [Google Scholar]
- 14.Machovik M, S Janecek. Biologia Bratislava. 2003;58:1127. [Google Scholar]
- 15.RM Van der Kaaij, et al. Microbiology. 2007;153:4003. doi: 10.1099/mic.0.2007/008607-0. [DOI] [PubMed] [Google Scholar]
- 16.V Kumar, et al. Carbohydr Res. 2010;345:893. doi: 10.1016/j.carres.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 17.V Kumar, et al. Carbohydr Res. 2010;345:1564. doi: 10.1016/j.carres.2010.04.027. [DOI] [PubMed] [Google Scholar]
- 18.A Bairoch, et al. Nucleic Acids Res. 2005;33:D154. doi: 10.1093/nar/gki070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.C Notredame, et al. J Mol Biol. 2000;302:205. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- 20. http://www.mbio.ncsu.edu/BioEdit/bioedit.html.
- 21.SR Eddy, et al. Bioinformatics. 1998;14:755. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- 22.GE Crooks, et al. Genome Res. 2004;14:1188. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.HS Lee, et al. J Biol Chem. 2002;277:21891. doi: 10.1074/jbc.M201623200. [DOI] [PubMed] [Google Scholar]
- 24.TD Schneider, RM Stephens. Nucleic Acids Res. 1990;18:6097. doi: 10.1093/nar/18.20.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.G Saab-Rincón, et al. FEBS Lett. 1999;453:100. [Google Scholar]
- 26.MH Rivera, et al. Protein Eng. 2003;16:505. doi: 10.1093/protein/gzg060. [DOI] [PubMed] [Google Scholar]
- 27.N Ramasubbu, et al. Biologia Bratislava. 2005;16:47. [Google Scholar]
- 28.S Fujiwara, et al. J Bacteriol. 1992;174:7478. doi: 10.1128/jb.174.22.7478-7481.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.SH Lee, et al. J Agric Food Chem. 2002;50:1411. doi: 10.1021/jf010928q. [DOI] [PubMed] [Google Scholar]
- 30.BA van der Veen, et al. Biochim Biophys Acta. 2000;5543:336. [Google Scholar]
- 31.A Nakamura, et al. Biochemistry. 1994;33:9929. doi: 10.1021/bi00199a015. [DOI] [PubMed] [Google Scholar]
- 32.BA van der Veen, et al. J Biol Chem. 2001;276:44557. doi: 10.1074/jbc.M107533200. [DOI] [PubMed] [Google Scholar]
- 33.H Leemhuis, et al. FEBS Lett. 2002;514:189. [Google Scholar]
- 34.RM Kelly, et al. Biochemistry. 2007;46:11216. doi: 10.1021/bi701160h. [DOI] [PubMed] [Google Scholar]
- 35.RM Kelly, et al. Appl Microbiol Biotechnol. 2009;84:119. doi: 10.1007/s00253-009-1988-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.T Kuriki, et al. J Biol Chem. 1996;271:17321. doi: 10.1074/jbc.271.29.17321. [DOI] [PubMed] [Google Scholar]
- 37.A Ohtaki, et al. Carbohydr Res. 2006;341:1041. doi: 10.1016/j.carres.2006.01.029. [DOI] [PubMed] [Google Scholar]
- 38.M Mizuno, et al. Eur J Biochem. 2004;271:2530. doi: 10.1111/j.1432-1033.2004.04183.x. [DOI] [PubMed] [Google Scholar]