

Abstract
Literature shows that Flaviviruses cause a variety of diseases, including fevers, encephalitis, and hemorrhagic fevers. NS3 is a multifunctional protein with an Nterminal protease domain (NS3pro) that is responsible for proteolytic processing of the viral polyprotein, and a C-terminal region that contains an RNA triphosphatase, RNA helicase and RNA-stimulated NTPase domain that are essential for RNA replication. Therefore, NS3 protein is the preferential choice for inhibition to stop the proteolytic processing. Hence, the 3D structure of NS3 protein was modeled using homology modeling by MODELLER 9v7. Evaluation of the constructed NS3 protein models were done by PROCHECK, VERYFY3D and through ProSA calculations. Ligands for the catalytic triad were designed using LIGBUILDER. The NS3 protein's catalytic triad was explored to find out the critical interactions pattern for inhibitor binding using molecular docking methodology using AUTODOCK Vina. It should be noted that these predicted data should be validated using suitable assays for further consideration.
Abbreviations
DOPE - Discrete optimized protein energy, WHO - World Health Organization, ADME/T - Absorption, Distribution, Metabolism, Excretion and Toxicity.
Keywords: NS3 protein, homology modeling, virtual screening, docking, ligand
Background
Flaviviruses are a group of more than 70 enveloped RNA viruses that cause serious diseases in humans and animals. Most of them are arthropod-borne viruses (arboviruses) and are transmitted to vertebrate hosts by either mosquitoes or ticks [1]. Flaviviruses cause a variety of diseases, including fevers, encephalitis, and hemorrhagic fevers [2]. It is believed that JE is responsible for more than 50,000 cases of encephalitis annually, with at least 10,000 deaths. Some 2.5 billion people – two fifths of the world's population – are now at risk for dengue, and despite 50 years of efforts, there is no dengue vaccine available on the market. WHO currently estimates that there may be 50 million dengue infections worldwide every year. In 2007 alone, there were more than 890,000 reported cases of dengue in the Americans, of which 26,000 cases were diagnosed as Dengue Hemorrhagic Fever (DHF). There are almost 100 asymptomatic infections for every reported flavivirus case, and the case fatality rates vary from 1 to 30% depending on the infecting flavivirus [WHO]. The family Flaviviridae consists of approximately 70 viruses, nearly 40 of which cause human disease [3]. Dengue virus (DENV), Japanese encephalitis virus (JEV), Murray Valley encephalitis virus (MVEV), Usutu virus (USUV), and West Nile virus (WNV) are a member of the genus Flavivirus and family Flaviviridae [4, 5].
Flaviviruses have a positive-sense single-stranded RNA (ssRNA) genome (approximately 11 kb) that encodes one large open reading frame containing a 5' type 1 cap and conserved RNA structures at both the 5' and 3' untranslated regions that are important for viral genome translation and replication. The genomic RNA is translated into a single polyprotein precursor consisting of three structural (C [capsid], prM [membrane], and E [envelope]) and seven nonstructural (NS1, NS2a, NS2b, NS3, NS4a, NS4b, and NS5) proteins arranged in the order C-prM-E-NS1-NS2a-NS2b-NS3-NS4a-NS4b-NS5 [6]. Only the structural proteins become part of the mature, infectious virion, whereas the nonstructural proteins are involved in polyprotein processing, viral RNA synthesis, and virus morphogenesis [7]. NS3 is a multifunctional protein with an N-terminal protease domain (NS3pro) that is responsible for proteolytic processing of the viral polyprotein, and a C-terminal region that contains an RNA triphosphatase, RNA helicase and RNA-stimulated NTPase domain that are essential for RNA replication. The serine protease domain of NS3 plays a central role in the replicative cycle of Flaviviruses [8] and is therefore a possible target for anti-flaviviral compounds [9].
In this in-silico study, we designed an inhibitor which showed inhibitory activity towards flavivirus (DENV, JEV, MVEV, USUV, and WNV) NS3 protein. The binding interactions between this inhibitor and NS3 protein were studied by docking methods using AutoDock vina software. The aim of this study was to get a better ligand that could inhibit polyprotein processing of Flavivirus (DENV, JEV, MVEV, USUV, and WNV), and to better understand the interactions between the inhibitor and the enzyme's binding sites via computational docking methods. We hope, this Drug will get success to clear out all the phases of clinical trial and it will be effective drug in the cure of encephalitis and hemorrhagic fevers.
Methodology
Sequence alignment
The protein sequences of NS3 were obtained from GenBank (http://www.ncbi.nlm.nih.gov/genbank/) database (MVEV (NP_722535.1),JEV (NP_775670.1), USUV (YP_164814.1) and WNV (NP_776018.1)) and Swiss Prot (DENV (P27909)). Using the BLAST P (http://blast.ncbi.nlm.nih.gov/Blast.cgi) through NCBI, the homologous structure of MVEV NS3 was identified, which was used as template for the homology modeling. The sequence alignment was done using the online version of ClustalW (http://www.ebi.ac.uk/Tools/msa/clustalw2/) (Figure 1).
Figure 1.

The sequence alignment between the five NS3 proteins of MVEV, DENV, JEV, USUV and WNV. All three major amino acids forming the Catalytic Triad (H51, D75, and S135) have been highlighted.
Homology modeling
BLASTP was used to identify the most suitable template for homology modeling of NS3 protein. The available structure of NS3 protein from MVEV in the Protein Database (PDB entry 2WV9) was used as a template for DENV, JEV, USUV and WNV. The homology modeling was carried out using the Modeller (http://www.salilab.org/modeller/) 9v7 program. The target and the template sequences were aligned using Modeller 9v7, a comparative protein modeling program, was used for homology modeling to generate the 3-D structures of NS3 protein for MVEV, DENV, JEV, USUV and WNV. The energy computations were done with the GROMOS 96 by implementation of Swiss-PdbViewer (http://www.expasy.org/spdbv). Figure 2 shows the superimposed NS3 3D models of JEV, MVV, WNV, USUV and DENV in cartoons view with Pymol software.
Figure 2.
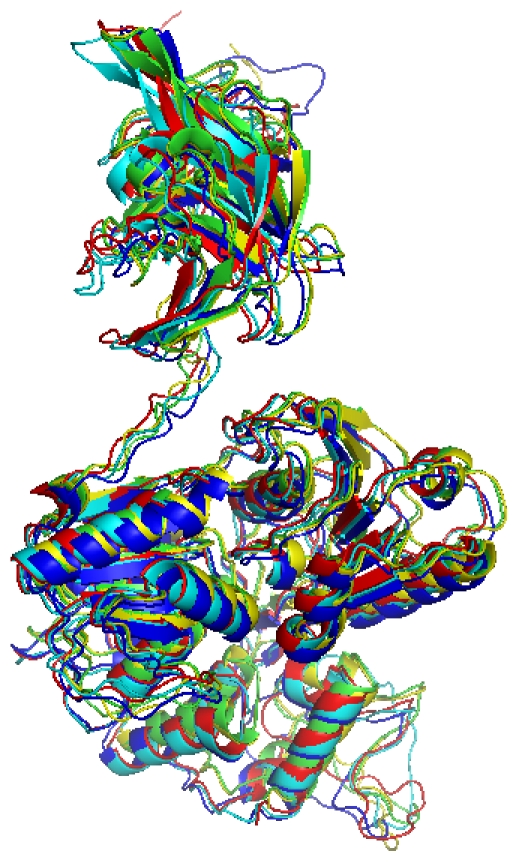
Structural model Superposition of NS3 proteins for JEV (Green), MVV (Red), WNV (Blue), USUV (Yellow) and DENV (Cyan).
Validation of the generated models
Different structure verification programs such as PROCHECK, VERIFY3D and ProSA (http://nihserver.mbi.ucla.edu/SAVES/) were used to evaluate the 3D-model of NS3 protein. The above mentioned validation programs validate the predicted structure by checking various parameters. While PROCHECK, a structure verification program relies on Ramachandran plot [10], determines the quality of the predicted structure by assessing various parameters such as lengths, angles and planarity of the peptide bonds, geometry of the hydrogen bonds, and side chain conformations of protein structures as a function of atomic resolution. The Verify3D determines the compatibility of an atomic model (3D) with its own amino acid sequence (1D) by assigning a structural class based on its location and environment (alpha, beta, loop, polar, nonpolar etc.) and comparing the results to valid structures [11].
Inhibition site identification
The active sites were revealed on the basis of previous studies. The aminoterminal domain contains the serine protease catalytic triad consisting of amino acid residues H51, D75, and S135 and the substrate-binding pocket is contained within NS3 protein [12, 13, 14].
Generating novel ligands
The structure of the fragment, i.e. the “seed molecule” was revealed on the basis of previous studies of available inhibitors for NS3 protein [15]. The fragment “1 H-1, 2, 4-triazole” was identified on the basis of “Lipinski's Rule of Five” and may therefore represent suitable starting point for evolution of good quality lead compounds. The docking analysis of “1 H-1, 2, 4-triazole” compound with MVEV NS3 protein was carried out by HEX (http://hex.loria.fr/) docking software with default parameters. The conformation of the pre-placed “seed” ensuring the binding affinity decides the manner that ligands would be grown with Ligbuilder software. Novel ligands had been developed with Ligbuilder ( http://mdl.ipc.pku.edu.cn/drug_design/work/ligbuilder.html) v1.2 software. We developed 100 novel ligands for the inhibitory site in NS3 protein.
Virtual screening
Out of 100 novel ligands generated, 10 ligands were selected on the basis of maximum binding affinity measured in kcal/mol. The selected 10 ligands were then analyzed for drug- relevant properties based on “Lipinski's rule of five“ and other drug like properties of valid structures using OSIRIS Property Explorer (http://www.organic-chemistry.org/prog/peo/), Molsoft: Drug- Likeness and molecular property explorer (http://www.molsoft.com/mprop/), and Molinspiration property explorer (http://www.molinspiration.com/cgibin/properties). On the basis of binding affinity and drug like properties, one ligand (Figure 3F) was finally screened.
Figure 3.

The solvent-accessible surface of the protein is shown in color-coded atoms (white, carbon; red, oxygen; blue, nitrogen). Molecular docking of novel ligand onto NS3 protein of MVEV (A), JEV (B), USUV (C), WNV (D) and DENV (E). Figure 3(F) represents the structure of novel ligand.
Lead optimization
The goal of lead optimization is to improve the effectiveness of initial ‘hits’ from primary screening. There are many approaches to lead optimization, all of which have the same theme: reduce off-target effects, create an improved ADME/T profile and improve a compound's efficacy. The lead optimization was performed using Toxtree (http://toxtree.sourceforge.net/) v1.60 software.
Protein-ligand docking
The docking of ligands to the catalytic triad of NS3 protein for MVEV, DENV, JEV, USUV and WNV was performed using AutoDock Vina software. Docking was performed to obtain a population of possible conformations and orientations for the ligand at the binding site. Using the software, polar hydrogen atoms were added to the NS3 protein and its nonpolar hydrogen atoms were merged. All bonds of ligands were set to be rotatable. All calculations for protein-fixed ligand-flexible docking were done using the Lamarckian Genetic Algorithm (LGA) method. The grid box with a dimension of 20 × 20 × 20 points was used around the catalytic triad to cover the entire enzyme binding site and accommodate ligands to move freely. The best conformation was chosen with the lowest docked energy, after the docking search was completed. The interactions of complex NS3 protein-ligand conformations, including hydrogen bonds and the bond lengths were analyzed using Swiss-PdbViewer v4.0 and Pymol software.
Discussion
Sequence alignment of NS3 protein for MVEV, DENV, JEV, USUV and WNV from same family revealed MVEV to be the best template for homology modeling as the MVEV shared 61% identity with DENV, 86% identity with JEV, 88% identity with USUV and 81% identity with WNV. The crystal structure of the NS3 protease-helicase from Murray Valley Encephalitis Virus (PDB entry 2WV9) was used as a template to predict the structure of NS3 protein and the predicted 3D structure of NS3 protein was generated by Modeller and the structure with the lowest DOPE scores were selected. The qualities of the 3D models were evaluated using the PROCHECK program and assessed using the Ramachandran plot. It is evident from the Ramachandran plot that the predicted models have most favorable regions, the allowed regions, the generic regions and the disallowed regions. Such a percentage distribution of the protein residues determined by Ramachandran plot shows that the predicted models are of good quality. The models show all the main chain and side chain parameters to be in the ‘better’ region. The quality of the generated models of NS3 protein as evaluated by ProSA web server (https://prosa.services.came.sbg.ac.at/prosa.php) provided a z-score of -11.03 (MVEV), -11.09 (JEV), -11.63 (USUV),-11.18 (WNV) and -7.06 (DENV) which falls within the range of values observed for the experimentally determined structures of similar lengths. The validity of the predicted model of NS3 protein was also verified by employing the structure verification server Verify-3D. The DOPE scores of NS3 protein were 63437.13672, 52417.43359, 62345.25391, 63189.61719 and 62632.42969 for MVEV, DENV, JEV, USUV and WNV respectively.
The Protein-ligand interaction plays a significant role in structure based drug designing. Overall, the best confirmation shows that the free energy of binding (ΔGbind kcal/mol) for the designed ligand were -5.9 kcal/mol, -6.0 kcal/mol, - 6.7 kcal/mol, -6.6 kcal/mol and -6.6 kcal/mol for MVEV, JEV, USUV, WNV and DENV respectively (Figure 3). The negative and low value of ΔGbind indicates strong favorable bonds between NS3 protein and the novel ligand indicating that the ligand was in its most favourable conformations. The information about the number of hydrogen bonds formed and catalytic site residues involved in protein-ligand complex are shown in Table 1 (see Table 1).
Conclusion
In this study, we designed a novel ligand against NS3 protein of MVEV, DENV, JEV, USUV and WNV. The molecular docking was applied to explore the binding mechanism and studies on the novel ligand against the NS3 protein showed that the free binding energy for the inhibitor was small, indicating that the ligand binds favorably to the binding site. The ligand was observed as the best inhibitor candidate, which may be considered as a potential ligand for treatment of diseases caused by flavivirus. The ligand thus developed is likely to inhibit viral infections, which share high sequence similarity with the five NS3 proteins of MVEV, DENV, JEV, USUV and WNV from same family. We plan to calculate ADME/T (Absorption, Distribution, Metabolism, Excretion / Toxicity) properties of the designed ligand using the available commercial ADME/T tools in future.
Supplementary material
Footnotes
Citation:Jitendra & Vinay, Bioinformation 6(2): 57-60 (2011)
References
- 1.MD Fernandez-Garcia, et al. Cell Host Microbe. 2009;5:318. doi: 10.1016/j.chom.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 2.Gould EA, T Solomon. Lancet. 2008;371:500. doi: 10.1016/S0140-6736(08)60238-X. [DOI] [PubMed] [Google Scholar]
- 3.I Kurane, et al. Emerg Infect Dis. 2000;6:569. doi: 10.3201/eid0606.000603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.H Weissenböck, et al. Emerg Infect Dis. 8:652. [Google Scholar]
- 5.D Vlachakis, et al. Theor Biol Med Model. 2009;6:9. doi: 10.1186/1742-4682-6-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.R Assenberg, et al. J Virol. 2009;83:12895. doi: 10.1128/JVI.00942-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.CL Murray, et al. Nat Rev Microbiol. 2008;6:699. doi: 10.1038/nrmicro1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.S Natarajan. Genet Mol Biol. 2010;33(2):214. doi: 10.1590/S1415-47572010000200002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.EJ Mancini, et al. Protein Sci. 2007;16:2294. doi: 10.1110/ps.072843107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.GN Ramachandran, V Sasisekharan. Adv Protein Chem. 1968;23:283. doi: 10.1016/s0065-3233(08)60402-7. [DOI] [PubMed] [Google Scholar]
- 11.G Chhabra, et al. Bioinformation. 2010;4(7):278. doi: 10.6026/97320630004278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.NH Mueller, et al. Antimicrob Agents Chemother. 2008;52(9):3385. doi: 10.1128/AAC.01508-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.LR Jan, et al. J Gen Virol. 1995;76:573. doi: 10.1099/0022-1317-76-3-573. [DOI] [PubMed] [Google Scholar]
- 14.Tambunan US, et al. Bioinformation. 2010;5(6):250. doi: 10.6026/97320630005250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.P Borowski, et al. Acta Biochim Pol. 2002;49(3):597. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.