

Abstract
Electrophysiological studies of wild-type and mutated forms of anthrax protective antigen (PA) suggest that the Phe clamp, a structure formed by the Phe427 residues within the lumen of the oligomeric PA pore, binds the unstructured N-terminus of the lethal factor and the edema factor during initiation of translocation. We now show by electrophysiological measurements and gel shift assays that a single Cys introduced into the Phe clamp can form a disulfide bond with a Cys placed at the N-terminus of the isolated N-terminal domain of LF. These results demonstrate direct contact of these Cys residues, supporting a model in which the interaction of the unstructured N-terminus of the translocated moieties with the Phe clamp initiates N- to C-terminal threading of these moieties through the pore.
Anthrax toxin is an ensemble of three proteins that cause symptoms of anthrax. One of the three proteins, protective antigen (PA; 83 kDa), serves to transport the other two, lethal factor (LF; 90 kDa) and edema factor (EF; 89 kDa), to the cytosol, where these factors enzymatically modify intracellular substrates, eliciting symptoms of the disease. PA binds to cellular receptors and, after being proteolytically converted to an active 63 kDa form (PA63), oligomerizes, generating heptameric and octameric pore precursors (prepores).1,2 The enzymatic “cargo proteins” LF and EF bind competitively to the prepores, generating complexes that are then endocytosed and trafficked to an acidic compartment. There, under the influence of acidic pH, the prepores transform into mushroom-shaped transmembrane pores (channels) capable of translocating bound cargo proteins to the cytosol.(3)
LF and EF bind to PA prepores via their homologous N-terminal domains (LFN and EFN, respectively).4,5 An unstructured segment, containing a high density of charged residues and with an overall positive charge, is present at the extreme N terminus of both LFN and EFN, and there is evidence that this segment initiates N- to C-terminal translocation through PA pores.6−8 A crystallographic model of LFN bound to the octameric prepore shows helix α1, the first secondary structure element of the domain, bound at the entrance to the lumen at a site (the α clamp) formed from two adjacent PA63 subunits.(9) The unstructured segment was not resolved in the crystallographic model, however.
Translocation of cargo proteins by the PA63 pore has been studied extensively in planar lipid bilayer systems.(10) PA63 forms ion-conducting pores in these bilayers, and the cargo proteins or their N-terminal domains (LFN is commonly used) bind to the cap of the pores. At symmetrical pH 5.5 and a low (δ20 mV) cis-positive potential, LFN binds to the pore and blocks ion conductance through it, an activity dependent upon the highly charged N-terminal segment of LFN. Blockage is released upon translocation, which may be initiated by either introducing a pH gradient or increasing the cis-positive potential to ≥50 mV.
Mutation of PA residue F427, which lies within the pore lumen, to Ala or many other amino acids causes severe defects in PA-mediated toxicity and in PA-dependent translocation across planar bilayers.(11) Results of site-directed spin-labeling studies indicate that the F427 side chains of PA oligomers move toward the axis of symmetry during prepore-to-pore conversion and come into close proximity with each other, forming a structure, called the Phe clamp, which plays a key role in translocation.(12) It has been hypothesized that the LFN N terminus interacts directly with the Phe clamp during translocation initiation,(13) but direct contact has not been demonstrated. To test for such contact, we conducted disulfide trapping experiments, in which we introduced a single Cys into the Phe clamp of PA and another on either the N-terminus or the C-terminus of LFN (Figure 1). We then tested the ability of these variants to form an interspecies disulfide bond in planar lipid bilayer measurements and gel-shift analyses.
Figure 1.
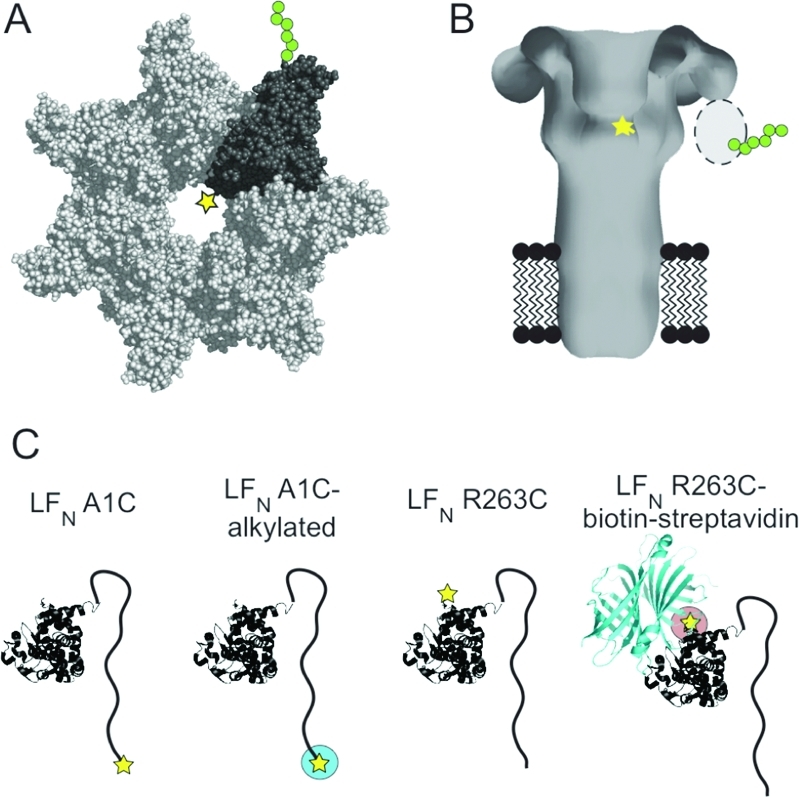
Schematic of proteins used in this study. (A) Space-filling model of heteroheptameric PA prepore, based on the crystal structure of [WT]7 prepore (PDB 1TZN(21)), showing a single subunit (dark gray) containing F427C (★) and a C-terminal hexa-His tag (green circles). (B) Membrane-inserted model of heteroheptameric PA pore based on the 3D reconstruction of negatively stained EM particles(22) showing a single mutated F427C (yellow star) at the predicted location of the Phe-clamp. Domain 4 was not resolved in the EM reconstruction, and a dotted oval is shown where one domain 4, with its C-terminal hexa-His tag (green circles) attached, is expected to be located. (C) Schematic of the four mutated LFN constructs used in the study. The cysteine substitution is shown as a yellow star, alkylation of the cysteine is represented by a green circle surrounding the yellow star, and biotin−streptavidin linkage is shown as a pink circle and a cyan ribbon diagram, respectively. The LFN models are based on the crystal structure of LF (PDB 1J7N(5)), and the streptavidin model is based on its crystal structure (PDB 1MEP(23)).
Experimental Procedures
Materials
Biochemical reagents were purchased from Sigma unless indicated otherwise. Oligonucleotides for mutagenesis were synthesized by Integrated DNA Technologies (Coralville, IA). E. coli BL21 (DE3) used for expression of proteins was grown in ECPM1 medium.(14)
Expression and Purification of Proteins
Recombinant WT PA and PA F427C were overexpressed in the periplasm of E. coli BL21 (DE3) and purified by anion-exchange chromatography.(15) The PA F427C monomer contained a C-terminal hexa-His affinity tag and was stored in 5 mM tris(2-carboxyethyl)phosphine (TCEP) in order to maintain the thiol in the reduced state. WT LFN, LFN A1C, and LFN R263C were overexpressed in E. coli BL21 (DE3) as N-terminal Hexa-His tagged SUMO-fusion proteins and purified by Ni-NTA chromatography followed by SUMO cleavage, resulting in untagged proteins.(6)
Formation and Purification of Homoheptameric and Heteroheptameric PA63 Prepore
WT PA63 prepore ([WT]7) was formed by limited trypsin digestion followed by anion-exchange chromatography.(16) Heteroheptamers with predominantly one mutant subunit containing F427C (Figure 1A,B) were prepared by a similar method as previously reported.17,18 Briefly, WT PA monomers were mixed with PA F427C (containing a hexa-His affinity tag) monomers in a 40:1 molar ratio. The mixture was then nicked with trypsin, and the resulting homoheptamer/heteroheptamer mixture was purified by anion-exchange chromatography. The preparation was then passed over Ni-NTA resin, and bound heptamers were eluted with a gradient of 40−500 mM imidazole. The product was desalted to remove imidazole and passed over a second Ni-NTA column to ensure the removal of [WT]7.
Modification of Mutant Proteins
LFN A1C was alkylated by bromoacetamide.(6) Briefly, 500 μM LFN A1C was fully reduced by incubation with 10 mM dithiolthreitol (DTT) for 15 min at room temperature (RT) and, after desalting to remove the DTT, was reacted with 50 mM 2-bromoacetamide for 15 min (RT). Sodium 2-mercaptoethanesulfonate (100 mM) was added to stop the reaction, and the proteins were again desalted to remove free bromoacetamide and sodium 2-mercaptoethanesulfonate. The resulting protein is referred to as LFN-AlC-alkylated (Figure 1C). As a negative control for translocation, LFN R263C was biotinylated and bound to streptavidin, a nontranslocatable protein, as described(17) (Figure 1C).
Electrophysiology
Planar phospholipid bilayer experiments were performed in a Warner Instruments Planar Lipid Bilayer Workstation (BC 525D, Hamden, CT). Planar bilayers were painted(19) onto a 200 μm aperture of a Delrin cup in a Lucite chamber, with 3% 1,2-diphytanoyl-sn-glycerol-3-phosphocholine (DPhPC) in n-decane (Avanti Polar Lipids, Alabaster, AL). 1 mL aliquots of buffer were added to the cup and the chamber, and both compartments were stirred continuously. Cis (side to which PA prepore and LFN were added) and trans compartments contained 100 mM KCl, 1 mM ethylenediaminetetraacetic acid (EDTA), and 10 mM each of sodium oxalate, potassium phosphate, and 2-(N-morpholino)ethanesulfonic acid (MES), pH 5.5.
Once a membrane was formed in the planar lipid bilayer system, PA prepore (25 pM) was added to the cis compartment, which was held at a Vm = +20 mV with respect to the trans compartment. After appropriate current increase, the cis compartment was perfused with ∼10 mL of non-PA-containing buffer at a flow rate of ∼3 mL/min to remove any free PA. Once the current was constant, LFN was added to the cis compartment (1 mg/mL), and binding to PA channels was monitored by the decrease in conductance. The cis compartment was perfused again after LFN addition to eliminate free ligand. The total time between LFN addition and initiation of translocation was kept constant at 10 min, unless otherwise noted. Translocation was initiated by raising the pH of the trans compartment to pH 7.2 with 2 M KOH, while maintaining the cis compartment at pH 5.5. Experiments for each PA protein tested were normalized to control experiments where KOH was added to the trans compartment in the absence of LFN in order to adjust for current changes due to salt addition alone. Translocation was monitored at Vm = +20 mV by the rise in current. In parallel experiments to translocation, PA channels blocked by LFN or an LFN mutant construct were unblocked by reversing the membrane potential from Vm = +20 mV to Vm = −20 mV. The fraction of LFN that was reversed was monitored over 1 min.
Gel-Shift Analysis
WT or [WT]6[F427C]1 PA heptamer (100 μg) was mixed with an equimolar amount (7.3 μg) of LFN WT, LFN A1C, LFN A1C-alkylated, or LFN R263C in a volume of 50 μL. The mixtures were incubated for 30 min at pH 8.5 with 10 mM DTT to limit nonspecific disulfide bond formation. Samples were then desalted using Zeba (Thermo Scientific) desalting spin columns into buffer (20 mM Tris, pH 8.5, 150 mM NaCl) with or without 15 mM DTT. The pH was dropped to pH 5.5 with the addition of 6.8 μL of 100 mM HCl (sample volume = 60 μL), and the samples were incubated for 1 h at RT to allow for oxidation. 2-Bromoacetamide was then added to a final concentration of 5 mM to quench any unreacted cysteines, and the reaction was allowed to proceed for 10 min at RT. Trichloroacetic acid (TCA) was then added to precipitate the proteins, and the samples were incubated on ice for 10 min. Protein pellet was collected by centrifugation and washed twice with acetone to remove TCA. Following the final acetone wash step, samples were incubated for 5−10 min in a 95 °C heat block to evaporate any remaining acetone. The protein pellets were then resuspended in SDS sample buffer and boiled for 10 min before being loaded onto an SDS-PAGE gel. Results were visualized by Coomassie staining of the resulting gel.
To confirm the identity of the bands observed in the gel-shift analysis, we performed a parallel analysis in which we probed the bands with an antibody specific to LFN. Specifically, we mixed either WT or [WT]6[F427C]1 PA heptamer (50 μg) with an equimolar amount (3.8 μg) of either WT LFN or LFN A1C, in the presence of 10 mM DTT, in a total volume of 100 μL. After allowing time for the formation of the PA prepore−LFN complex, the samples were desalted against buffer containing 20 mM Tris, pH 8.5, 150 mM NaCl, with or without 15 mM DTT. After buffer exchange, the pH of the solutions was dropped to pH 5.5 by addition of 100 mM HCl to mimic the conditions of the endosome, and the samples were incubated for 1 h at RT to promote oxidation. After oxidation, a 20 μL aliquot of each sample was applied to a 4−20% acrylamide SDS-PAGE gel. Following electrophoresis, the proteins were transferred to a nitrocellulose membrane using a 25 V electrophoresis for 90 min. The membranes were then probed with a polyclonal antibody raised in goats against the N-terminal domain of LF.
Results and Discussion
To test for direct contact between the Phe clamp of the PA63 pore and the LFN N terminus, we first created a heteroheptameric form of the pore containing the F427C mutation in a single subunit ([WT]6[F427C]1, Figure 1A,B). We then tested whether this variant could form a disulfide bond with either LFN A1C or LFN R263C, residue 263 representing the C terminus (Figure 1C). Earlier reports suggested that the LFN N-terminus enters the pore lumen during translocation initiation, while the LFN C-terminus remains outside.(8) As controls, we constructed an LFN A1C variant in which the Cys thiol was alkylated to prevent disulfide bond formation (LFN A1C-alkylated) and an LFN R263C variant in which a biotin was attached to the Cys. Binding of streptavidin to the biotinylated Cys yielded the complex, LFN R263C-biotin:streptavidin, which is known not to translocate8,17 (Figure 1C).
To test for disulfide bond formation between PA [WT]6[F427C]1 and the LFN variants, we first used electrophysiological assays in planar lipid bilayers. Briefly, channels were formed with PA [WT]7 or PA [WT]6[F427C]1 at pH 5.5 and Vm = cis-positive 20 mV, and an LFN variant was then added to the cis compartment, causing a decrease in ion conductance. Oxidation was allowed to proceed for 10 min at RT before introducing a pH gradient to initiate translocation, detected by restoration of current. With channels formed from WT PA63, all of the LFN variants translocated at essentially the same rate and efficiency as WT LFN, except for the LFN R263C−biotin−streptavidin complex, a negative control (Figure 2A). However, with channels formed from [WT]6[F427C]1, translocation of LFN A1C was virtually nil, whereas WT LFN and LFN R263C retained the ability to translocate (Figure 2B). LFN A1C in which the thiol had been alkylated also translocated efficiently through F427C-containing channels. These findings supported the hypothesis that the loss of translocation resulted from the formation of a disulfide bond between the cysteine of LFN A1C and that of the Phe clamp of [WT]6[F427C]1 (Figure 2B). Curiously, addition of DTT did not alleviate the blockage of translocation, suggesting that the disulfide is protected from reduction, perhaps by shielding within the hydrophobic environment of the Phe clamp.
Figure 2.

Translocation of various LFN constructs through either (A) WT PA channels or (B) heteroheptamer F427C channels. Macroscopic conductance was measured at symmetrical pH of 5.5 and Vm = +20 mV. At time 0, translocation was initiated by adding 2 M KOH to the trans compartment to raise the pH to 7.2; there was an ∼20 s mixing delay in this system with both compartments continuously stirred. Representative data are shown from n ≥ 3 trials. The five LFN constructs were as follows: WT LFN (black), LFN A1C (blue), LFN A1C-alkylated (green), LFN R263C (red), and LFN R263C−biotin−streptavidin (orange).
While cross-linked dimers of LFN are known not to translocate through PA channels,(20) the decreased translocation rate seen with [WT]6[F427C]1 and LFN A1C was almost certainly not caused by LFN dimerization because both LFN A1C and LFN R263C translocated efficiently through WT channels (Figure 2A). Presumably, disulfide formation did not occur with LFN R263C during translocation because contact of this residue with F427C was transient and brief.
As shown in Figure 3, LFN A1C lost the ability to translocate under the influence of a pH gradient within seconds to a few minutes after the protein bound to [WT]6[F427C]1 channels. The ability to relieve channel blockage by reversing the polarity of the transmembrane potential was also lost with approximately the same kinetics. The regain of conductance upon polarity reversal is rapid and thought to result from electrophoretic withdrawal of the unstructured, positively charged N terminus of LFN via the mouth of the pore. These findings support the hypothesis of a disulfide bridge forming between the Cys residues of the mutated LFN and the heteroheptameric pore.
Figure 3.

Kinetics of interactions between PA63 [WT]6[F427C]1 and LFN A1C. Three parameters were measured over time: occlusion of the PA [WT]6[F427C]1 channel by LFN A1C (triangles), translocation of LFN A1C through the PA [WT]6[F427C]1 channel after the pH gradient was applied (squares), and relief of blockage of the PA [WT]6[F427C]1 channel by LFN A1C after reversal of the membrane potential (circles). At each time point, the conductance was measured over a period of 1 min and normalized to account for total PA channels. Each point represents an average of three independent experiments, with standard deviation shown as error bars.
As an orthogonal test of disulfide bond formation between the PA [WT]6[F427C]1 pore and LFN A1C, we conducted gel shift assays. Briefly, [WT]7 or [WT]6[F427C]1 PA63 heptamers were mixed with WT LFN, LFN A1C, LFN A1C-alkylated, or LFN R263C at pH 8.5 in the presence of DTT to limit nonspecific disulfide bond formation during the binding step. DTT was then removed, and the pH was dropped to pH 5.5 to promote conversion to the pore. The samples were allowed to incubate 1 h at RT before being treated with 2-bromoacetamide, precipitated with TCA, and analyzed by SDS-PAGE (Figure 4). Two distinct high molecular weight bands were observed with [WT]6[F427C]1 + LFN A1C (Figure 4, lane 1). We hypothesized that these bands corresponded to disulfide cross-linked PA63−LFN and disulfide cross-linked PA63 dimers, respectively. In support of this hypothesis, both high molecular weight bands were eliminated by the addition DTT (Figure 4, lane 2), and only the upper band (corresponding to cross-linked PA−PA) was present with [WT]6[F427C]1 + LFN WT (Figure 4, lane 3); this band was also eliminated by the addition of DTT (Figure 4, lane 4). As expected, and in agreement with our planar lipid bilayer data, the PA−LFN cross-link was only observed with [WT]6[F427C]1 and LFN A1C; no PA−LFN cross-link was seen with WT LFN, LFN A1C-alkylated, or LFN R263C (Figure 4, lanes 5, 6, and 7).
Figure 4.

Coomassie-stained gel shift analysis shows formation of a disulfide cross-link between PA [WT]6[F427C]1 and LFN A1C. Lanes: 1, PA [WT]6[F427C]1 + LFN A1C; 2, PA [WT]6[F427C]1 + LFN A1C + DTT; 3, PA [WT]6[F427C]1 + LFN WT; 4, PA [WT]6[F427C]1 + LFN WT + DTT; 5, PA WT + LFN A1C; 6, PA [WT]6[F427C]1 + LFN A1C-alkylated; 7, PA [WT]6[F427C]1 + LFN R263C. DTT was added, as shown, to 15 mM in all samples that included DTT.
To confirm the identities of the bands, we transferred the proteins to nitrocellulose and performed Western blots using an antibody specific to LFN (Figure 5). As predicted, the proposed PA−LFN band was present in only in the lane that contained both PA [WT]6[F427C]1 and LFN A1C without DTT (Figure 5A). Additionally, a band consistent with a disulfide forming between two molecules of LFN A1C (LFN−LFN) was only observed in lanes that contained LFN A1C (Figure 5A,C), and that band was reduced dramatically in the presence of 15 mM DTT. Together, these data indicate that a disulfide cross-link forms selectively between PA [WT]6[F427C]1 pore and LFN A1C.
Figure 5.

Western blot probed with an anti-LFN antibody to reveal LFN-containing bands resulting from a gel shift analysis. Lanes: A, PA [WT]6[F427C]1 + LFN A1C; B, PA [WT]6[F427C]1 + LFN WT; C, PA WT + LFN A1C; D, PA WT + WT LFN. DTT was added, as shown, to 15 mM in all samples that included DTT.
In summary, our results in artificial membranes and by gel-shift analysis support the hypothesis that there is a direct interaction between the N-terminus of LFN and the Phe clamp of the PA pore during initiation of translocation and thereby validate the concept that the N-terminus of LFN plays an important role in initiating translocation through the PA pore. These results are complemented by two recent reports: (i) site-directed spin labeling studies demonstrating proximity of the N terminus of bound LF to the Phe clamp of the PA pore,(13) and (ii) the crystal structure of LFN bound to the octameric prepore showing, as mentioned above, that helix α1 of the bound cargo domain is bound within the mouth of the pore. The latter report localizes the unstructured N terminus to the vicinity of the Phe clamp.(9)
Acknowledgments
We thank Dr. Robin Ross and the Biomolecule Production Core of NERCE for helping with the production of proteins used in this study. We thank A. Fischer, B. Pentelute, and O. Sharma for valuable comments and discussions and Caroline Amiot for assistance in performing some of the experiments.
Glossary
Abbreviations
- Vm
membrane potential
- DPhPC
1,2-diphytanoyl-sn-glycerol-3-phophocholine
- DTT
dithiothreitol
- EDTA
ethylenediaminetetraacetic acid
- EF
edema factor
- LF
lethal factor
- LFN
N-terminal domain (residues 1−263) of LF
- SUMO
small ubiquitin modifier
- MES
2-(N-morpholino)ethanesulfonic acid
- n
number of experiments
- PA
protective antigen
- RT
room temperature
- t1/2
half-time of completion of translocation
- TCEP
tris(2-carboxyethyl)phosphine
- WT
wildtype
- Tris
tris(hydroxymethyl)aminomethane
- TCA
trichloroacetic acid
- SDS
sodium dodecyl sulfate
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis.
Author Present Address
Silver Creek Pharmaceuticals, San Francisco, CA 94158.
Author Present Address
† Department of Biology, Saint Louis University, St. Louis, MO 63103.
This research was funded by NIH grants (1F32 AI077280 to B.E.J., 5R01 AI022021 to R.J.C.) and NERCE A1057159. R.J.C. holds equity in PharmAthene, Inc.
Funding Statement
National Institutes of Health, United States
References
- Kintzer A. F.; Thoren K. L.; Sterling H. J.; Dong K. C.; Feld G. K.; Tang I. I.; Zhang T. T.; Williams E. R.; Berger J. M.; Krantz B. A. (2009) The protective antigen component of anthrax toxin forms functional octameric complexes. J. Mol. Biol. 392, 614–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogridge J.; Cunningham K.; Collier R. J. (2002) Stoichiometry of anthrax toxin complexes. Biochemistry 41, 1079–1082. [DOI] [PubMed] [Google Scholar]
- Young J. A.; Collier R. J. (2007) Anthrax Toxin: Receptor-Binding, Internalization, Pore Formation, and Translocation. Annu. Rev. Biochem. 76, 243–265. [DOI] [PubMed] [Google Scholar]
- Lacy D. B.; Mourez M.; Fouassier A.; Collier R. J. (2002) Mapping the anthrax protective antigen binding site on the lethal and edema factors. J. Biol. Chem. 277, 3006–3010. [DOI] [PubMed] [Google Scholar]
- Pannifer A. D.; Wong T. Y.; Schwarzenbacher R.; Renatus M.; Petosa C.; Bienkowska J.; Lacy D. B.; Collier R. J.; Park S.; Leppla S. H.; Hanna P.; Liddington R. C. (2001) Crystal structure of the anthrax lethal factor. Nature 414, 229–233. [DOI] [PubMed] [Google Scholar]
- Pentelute B. L.; Barker A. P.; Janowiak B. E.; Kent S. B.; Collier R. J. (2010) A semisynthesis platform for investigating structure-function relationships in the N-terminal domain of the anthrax Lethal Factor. ACS Chem. Biol. 5, 359–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoren K. L.; Worden E. J.; Yassif J. M.; Krantz B. A.. Lethal factor unfolding is the most force-dependent step of anthrax toxin translocation. Proc. Natl. Acad. Sci. U.S.A 2009, not supplied. [DOI] [PMC free article] [PubMed]
- Zhang S.; Finkelstein A.; Collier R. J. (2004) Evidence that translocation of anthrax toxin’s lethal factor is initiated by entry of its N terminus into the protective antigen channel. Proc. Natl. Acad. Sci. U.S.A. 101, 16756–16761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feld G. K.; Thoren K. L.; Kintzer A. F.; Sterling H. J.; Tang I. I.; Greenberg S. G.; Williams E. R.; Krantz B. A. (2010) Structural basis for the unfolding of anthrax lethal factor by protective antigen oligomers. Nat. Struct. Mol. Biol. 17, 1383–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krantz B. A.; Finkelstein A.; Collier R. J. (2006) Protein translocation through the anthrax toxin transmembrane pore is driven by a proton gradient. J. Mol. Biol. 355, 968–979. [DOI] [PubMed] [Google Scholar]
- Krantz B. A.; Melnyk R. A.; Zhang S.; Juris S. J.; Lacy D. B.; Wu Z.; Finkelstein A.; Collier R. J. (2005) A phenylalanine clamp catalyzes protein translocation through the anthrax toxin pore. Science 309, 777–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J.; Lang A. E.; Aktories K.; Collier R. J.. Phenylalanine-427 of anthrax protective antigen functions in both pore formation and protein translocation. Proc. Natl. Acad. Sci. U.S.A 2008, not supplied. [DOI] [PMC free article] [PubMed]
- Jennings-Antipov L. D.; Song L.; Collier R. J. (2011) Interactions of anthrax lethal factor with protective antigen defined by site-directed spin labeling. Proc. Natl. Acad. Sci. U.S.A. 108, 1868–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard A., Payton M. (1995) Fermentation and Growth of Escherichia coli for Optimal Protein Production, in Current Protocols in Protein Science, pp 5.3.1−5.3.18, John Wiley and Sons, Inc., New York. [DOI] [PubMed] [Google Scholar]
- Miller C. J.; Elliott J. L.; Collier R. J. (1999) Anthrax protective antigen: prepore-to-pore conversion. Biochemistry 38, 10432–10441. [DOI] [PubMed] [Google Scholar]
- Wigelsworth D. J.; Krantz B. A.; Christensen K. A.; Lacy D. B.; Juris S. J.; Collier R. J. (2004) Binding stoichiometry and kinetics of the interaction of a human anthrax toxin receptor, CMG2, with protective antigen. J. Biol. Chem. 279, 23349–23356. [DOI] [PubMed] [Google Scholar]
- Janowiak B. E.; Finkelstein A.; Collier R. J. (2009) An approach to characterizing single-subunit mutations in multimeric prepores and pores of anthrax protective antigen. Protein Sci. 18, 348–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janowiak B. E.; Fischer A.; Collier R. J. (2010) Effects of introducing a single charged residue into the phenylalanine clamp of multimeric anthrax protective antigen. J. Biol. Chem. 285, 8130–8137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller P.; Rudin D. O. (1963) Induced excitability in reconstituted cell membrane structure. J. Theor. Biol. 4, 268–280. [DOI] [PubMed] [Google Scholar]
- Juris S. J.; Melnyk R. A.; Bolcome R. E. 3rd; Chan J.; Collier R. J. (2007) Cross-linked forms of the isolated N-terminal domain of the lethal factor are potent inhibitors of anthrax toxin. Infect. Immun. 75, 5052–5058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacy D. B.; Wigelsworth D. J.; Melnyk R. A.; Harrison S. C.; Collier R. J. (2004) Structure of heptameric protective antigen bound to an anthrax toxin receptor: a role for receptor in pH-dependent pore formation. Proc. Natl. Acad. Sci. U.S.A. 101, 13147–13151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama H.; Janowiak B. E.; Brzozowski M.; Juryck J.; Falke S.; Gogol E. P.; Collier R. J.; Fisher M. T. (2008) GroEL as a molecular scaffold for structural analysis of the anthrax toxin pore. Nat. Struct. Mol. Biol. 15, 754–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyre D. E.; Le Trong I.; Merritt E. A.; Eccleston J. F.; Green N. M.; Stenkamp R. E.; Stayton P. S. (2006) Cooperative hydrogen bond interactions in the streptavidin-biotin system. Protein Sci. 15, 459–467. [DOI] [PMC free article] [PubMed] [Google Scholar]