

Abstract
Purpose
Despite significant progress in the molecular understanding of medulloblastoma, stratification of risk in patients remains a challenge. Focus has shifted from clinical parameters to molecular markers, such as expression of specific genes and selected genomic abnormalities, to improve accuracy of treatment outcome prediction. Here, we show how integration of high-level clinical and genomic features or risk factors, including disease subtype, can yield more comprehensive, accurate, and biologically interpretable prediction models for relapse versus no-relapse classification. We also introduce a novel Bayesian nomogram indicating the amount of evidence that each feature contributes on a patient-by-patient basis.
Patients and Methods
A Bayesian cumulative log-odds model of outcome was developed from a training cohort of 96 children treated for medulloblastoma, starting with the evidence provided by clinical features of metastasis and histology (model A) and incrementally adding the evidence from gene-expression–derived features representing disease subtype–independent (model B) and disease subtype–dependent (model C) pathways, and finally high-level copy-number genomic abnormalities (model D). The models were validated on an independent test cohort (n = 78).
Results
On an independent multi-institutional test data set, models A to D attain an area under receiver operating characteristic (au-ROC) curve of 0.73 (95% CI, 0.60 to 0.84), 0.75 (95% CI, 0.64 to 0.86), 0.80 (95% CI, 0.70 to 0.90), and 0.78 (95% CI, 0.68 to 0.88), respectively, for predicting relapse versus no relapse.
Conclusion
The proposed models C and D outperform the current clinical classification schema (au-ROC, 0.68), our previously published eight-gene outcome signature (au-ROC, 0.71), and several new schemas recently proposed in the literature for medulloblastoma risk stratification.
INTRODUCTION
Medulloblastomas are primitive embryonal tumors of the CNS arising in the cerebellum and disseminating throughout the CNS. Over the past 15 years, significant progress has been made in understanding the biologic mechanisms driving these tumors.1 These advances provide a growing framework for new risk stratification schemas and targeted therapies. Efforts to determine risk in the context of current treatment seek to improve overall survival and decrease long-term deficits associated with multimodal treatment regimens based on conventional chemotherapy, surgical resection, and craniospinal irradiation.2–5
The current clinical medulloblastoma classification schema, based on age and metastasis status at diagnosis, extent of initial resection, and histology, has limited predictive power. However, 5-year survival rates for standard-risk patients can be up to 85%, with 60% to 80% for high-risk groups. Despite this relative success, survival almost universally comes at the expense of long-term neurologic and neurocognitive deficits resulting from the aggressiveness of the treatments. Importantly, the current clinical schema fails to identify a significant group of patients (approximately 20%) who are categorized as standard-risk but do not respond to treatment. Because of these limitations, the search for better risk stratification schemas based on molecular markers and genomic abnormalities have become the focus of interest in the last decade.6,7
Histology has been associated with clinical outcome8–10; however, because tumors display highly variable degrees of heterogeneity, subtyping via traditional histopathology is especially difficult. Several molecular markers and genomic abnormalities have been shown to correlate with poor clinical outcome, including c-Myc amplification,11 17p loss/i(17)q,12 concomitant expression of LDHB/CCNB1,13 gain of 6q/17q,14 and overexpression of CDK615 and survivin.16 Similarly, β-catenin mutations and monosomy 617,18 and overexpression of TrkC19 have been associated with good clinical outcome. However, statistical association with clinical outcome does not necessarily yield accurate classification on a case-by-case basis. In addition, many of these genomic markers suffer from low penetrance and modest sensitivity/specificity. Thus, they are rather limited as prognostic markers alone and are not routinely used in the clinic to evaluate risk.
We previously introduced an eight-gene molecular signature6 that was effective at separating patients in standard-risk and high-risk groups (80% v 17% 5-year overall survival, respectively) and outperformed the current clinical schema but was less accurate for data sets beyond the original study (Data Supplement). Moreover, while we observed a number of molecular subtypes of medulloblastoma in that study, we had insufficient numbers of samples to assess whether outcome signatures varied by subtype.
In the ensuing years, there have been a number of advances in molecular signature analysis. In our own work, we established the increased strength of using sets of genes rather than individual genes to distinguish biologic phenotypes.20,21 We extended this method to evaluate the activity of a set of genes in a single sample and used it effectively in two recent studies.22,23 Other signature approaches were also introduced.24–26 More recently, methods integrating clinical data and genomic features13,14 have been applied to a variety of clinical prediction problems.27–31
Here, we describe a novel method for predicting response to therapy (relapse/no relapse) for medulloblastoma and establish its efficacy on independent test data. We combine clinical data with molecular subtypes, pathway activation signatures, and copy number data. Importantly, our method goes beyond establishing association with these risk factors and instead gives a predictive probability of relapse within 30 months of treatment. The method is based on a Bayesian cumulative log-odds model that computes the total evidence for relapse in each patient from the status of clinical and genomic features. This approach is general and provides a paradigm for genomic-based clinical prediction applicable to other tumor types. We also introduce a novel Bayesian nomogram32 for assessing a patient's overall risk on the basis of the positive/negative contribution of individual risk factors (the model features).
With better classification performance than the current clinical schema in use by the Children's Oncology Group (COG) and other cooperatives, the approach presented here shows promise to improve risk stratification for standard therapeutic protocols. The deployment of our model will require expression array and single nucleotide polymorphism–array profiling of each tumor, or alternatively, the development of clinical assays for rapid evaluation of expression and copy-number alteration in a few hundred genes. The subtypes identified in this model could also be relevant to future therapeutic strategies that directly target the molecular mechanisms of tumorigenesis.
PATIENTS AND METHODS
Patients
Tumor samples were obtained through the COG Tumor Bank (protocol ACNS02B3) and from Children's Hospital Boston, University of Washington Medical Center, Texas Children's Hospital, and The Johns Hopkins University Medical Center under approval from the respective institutional review boards. The training set consisted of 96 samples for which relapse status at 30 months post-treatment was known. Matched normal blood samples were collected through the COG Tumor Bank (protocol ACNS02B3) and from Children's Hospital Boston under institutional review board approval. The test set included 78 samples: 47 samples from our original study6 not used for training, 16 samples from Kool et al,33 and 15 samples from the COG Tumor Bank. All training and test samples correspond to patients at least 3 years old treated with conventional chemotherapy, surgical resection, and craniospinal irradiation (Table 1; Data Supplement). For all samples, we generated gene-expression and copy-number data using Affymetrix HT-HG-U133A2 and Affymetrix 250k and 6.0 arrays (Affymetrix, Santa Clara, CA), respectively (see Appendix), and we assigned risk categories according to current clinical criteria.2,7
Table 1.
Features or Risk Factors Used by the Bayesian Cumulative Log-Odds Model to Predict Relapse
| Feature | Values | Patients (%) | Posterior Log-Odds Ratio Ev (r | x; 95% CI) | Average Absolute Evidence (AvEv) | Description | Source | Gene Set Reference | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Relapse (prior) | −0.28 | 0.28 | Prior | ||||||||||
| Clinical | |||||||||||||
| Histology | |||||||||||||
| Classic | 68 | 0.0033 ± 0.06 | 0.094 | Tumor histology at diagnosis | |||||||||
| Desmo | 14 | −0.34 ± 0.20 | |||||||||||
| LCA | 17 | 0.27 ± 0.02 | |||||||||||
| Metastasis | Yes M1-4 | 17 | 0.58 ± 0.07 | 0.236 | Metastasis status at diagnosis | ||||||||
| No M0 | 75 | −0.16 ± 0.18 | |||||||||||
| Subtype-independent expression signature | |||||||||||||
| c-Myc activation | |||||||||||||
| High | 44 | 0.53 ± 0.22 | 0.485 | Genes upregulated (43) and downregulated (65) by c-Myc | MSigDB v2.5/C2 | YU_CMYC_UP/DN | |||||||
| Low | 56 | −0.45 ± 0.25 | Yu et al34 | ||||||||||
| Disease subtype (c1-c6) | |||||||||||||
| c1 | 15 | 0.69 ± 0.03 | 0.352 | Disease subtype as determined by gene expression | |||||||||
| c2 | 18 | −0.25 ± 0.13 | See Appendix | ||||||||||
| c3 | 29 | 0.23 ± 0.23 | |||||||||||
| c4 | 19 | −0.33 ± 0.16 | |||||||||||
| c5 | 9.4 | 0.08 ± 0.22 | |||||||||||
| c6 | 10 | −0.68 ± 0.52 | |||||||||||
| DNA copy number gains or losses | |||||||||||||
| amp(8q24.21) | Amp | 3.1 | 0.44 ± 0.40 | 0.113 | Amplification of chr8q24.21. Locus of c-Myc14 | ||||||||
| (c-MYC) | Norm | 36 | −0.085 ± 0.83 | Amplification of chr2p24.3. Locus of N-Myc14 | |||||||||
| amp(2p24.3) | Amp | 15 | 0.50 ± 0.36 | 0.421 | Deletion of chr6q. Monosomy 614 | ||||||||
| (N-MYC) | Norm | 25 | −0.37 ± 1.53 | ||||||||||
| del(6q) | Del | 6.2 | −0.12 ± 0.88 | 0.047 | Deletion of chr16q | ||||||||
| (monosomy 6) | Norm | 33 | 0.033 ± 0.68 | ||||||||||
| del(16q) | Del | 15 | 0.95 ± 0.24 | 0.855 | Deletion of chr16q23.3 | ||||||||
| Norm | 25 | −0.80 ± 3.8 | |||||||||||
| del(16q23.3) | Del | 16 | 0.83 ± 0.26 | 0.786 | Amplification of chr7q21.3 | ||||||||
| Norm | 24 | −0.76 ± 3.84 | |||||||||||
| amp(7q21.3) | Amp | 15 | 0.73 ± 0.22 | 0.63 | Amplification of chrq26.32. Locus of p110a/PI3K | ||||||||
| Norm | 25 | −0.57 ± 4.03 | |||||||||||
| amp(3q26.32) | Amp | 8.3 | 1.1 ± 0.25 | 0.581 | |||||||||
| Norm | 0.31 | −0.45 ± 2.05 | |||||||||||
| c1 | c2 | c3 | c4 | c5 | c6 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Subtype-dependent expression signatures | ||||||||||||
| mTOR induced | High | 42 | 0.44 ± 0.11 | 0.81 ± 0.22 | 0.20 ± 0.18 | 0.47 ± 0.22 | −0.11 ± 0.03 | 0.69 ± 0.22 | 0.382 | Genes upregulated (200) and downregulated (200) by mTOR | OPAM v3 | mTOR_UP.v1 |
| Low | 58 | 0.59 ± 0.22 | −0.61 ± 0.59 | −0.25 ± 0.03 | −0.22 ± 0.23 | 0.12 ± 0.23 | −0.22 ± 0.38 | Genes (25) directly regulated and induced by anti-CD44 | Majumder et al35 | |||
| Anti-CD44 regulated | High | 57 | −0.37 ± 0.22 | −0.05 ± 0.16 | 0.00 ± 0.22 | −0.22 ± 0.23 | −0.98 ± 1.12 | −0.12 ± 0.29 | 0.22 | Genes upregulated (20) and downregulated (7) by c-MYC | MSigDB v2.5/C2 | HOGERKORP_CD44_UP; Högerkorp et al36 |
| Low | 43 | 0.14 ± 0.09 | 0.12 ± 0.06 | 0.00 ± 0.22 | 0.47 ± 0.22 | 0.81 ± 0.05 | 0.29 ± 0.10 | Histidine metabolism genes (25) | MSigDB v2.5/C2 | COLLER_MYC | ||
| c-MYC activated (v2) | High | 43 | 0.10 ± 0.08 | −0.11 ± 0.14 | 1.20 ± 0.10 | −0.22 ± 0.23 | −0.39 ± 0.18 | 0.69 ± 0.19 | 0.441 | MSigDB v2.5/C2 | Coller et al37 | |
| Low | 57 | −0.14 ± 0.23 | 0.05 ± 0.06 | −0.63 ± 0.27 | 0.29 ± 0.03 | 0.34 ± 0.22 | −0.69 ± 1.41 | Genes (30) downregulated by Gli1 | OPAM v3 | HISTIDINE_METABOLISM | ||
| Histidine metabolism | High | 56 | 1.3 ± 1.00 | −0.15 ± 0.13 | 0.00 ± 0.22 | 0.47 ± 0.22 | 0.12 ± 0.18 | 0.29 ± 0.10 | 0.341 | KEGG pathway38 | ||
| Low | 44 | 0.64 ± 0.11 | −0.12 ± 0.06 | 0.00 ± 0.22 | −0.92 ± 1.88 | −0.11 ± 0.03 | −0.41 ± 0.58 | Ribavirin/RSV-induced upregulated (22) and downregulated (43) response | MSigDB v2.5/C2 | GLI1_UP.v1_DN | ||
| Gli1 downregulated | High | 57 | 0.08 ± 0.04 | 0.54 ± 0.18 | 0.00 ± 0.22 | 0.11 ± 0.08 | −0.44 ± 0.21 | −0.22 ± 0.38 | 0.275 | Yoon et al 39 | ||
| Low | 43 | 0.14 ± 0.08 | −0.98 ± 2.22 | 0.00 ± 0.22 | −0.22 ± 0.23 | 0.81 ± 0.05 | 0.69 ± 0.19 | RIBAVIRIN_RSV_UP(DN) | ||||
| Ribavirin/RSV- induced response | High | 56 | 0.23 ± 0.23 | 0.22 ± 0.02 | 0.00 ± 0.22 | −0.54 ± 0.56 | −0.17 ± 0.06 | 0.29 ± 0.11 | 0.25 | Zhang et al 40 | ||
| Low | 44 | 0.55 ± 0.12 | −0.18 ± 0.18 | 0.00 ± 0.22 | 0.65 ± 0.18 | 0.12 ± 0.18 | −0.41 ± 0.58 |
Abbreviations: Desmo, desmoplastic; LCA, large-cell/anaplastic; MSigDB, Molecular Signatures Database; Amp, amplified; Norm, normal; Del, deletion; RSV, respiratory syncytial virus.
Determining Molecular Subtype
To predict the relapse status of a new sample (in the test set) its disease subtype (c) must first be determined. Six molecular subtypes {c1, … ,c6} of the samples in the training set were defined by clustering in a separate study (see Appendix) on a larger collection of about 200 medulloblastoma samples.41 These disease subtypes are linked to molecular mechanisms of tumorigenesis (see Appendix) and are consistent with the findings of several independent studies.17,33 We then used these labels to train a disease-subtype prediction model for new samples.
To build the subtype prediction model, we used a collection of 2,599 gene sets drawn from our Molecular Signatures Database (MSigDB)21 and manually curated gene sets derived from data in the Gene Expression Omnibus (GEO; see Appendix). We estimated the activation score (degree of upregulation) of these sets in each training set sample by using a single-sample version of Gene Set Enrichment Analysis (ssGSEA).21,22 We dichotomized the activation scores as low or high (see Appendix). Using this gene set view of the data, for each disease subtype, we chose six expression signatures that best discriminate its previously assigned subtype label within the training set according to the area under receiver operating characteristic (au-ROC) curve (Data Supplement).
Bayesian Predictive Model
Our model predicts relapse status, r = {yes, no}, by accumulating the relevant log-odds evidence implied by the molecular subtype and the values of clinical and genomic features for a specific patient sample. We start from the prior, which can be thought of as the probability of relapse/no relapse based on the proportion of patients in the training set who relapse/do not relapse. Formally, we use the log-odds prior, Ev(r). We then define four nested submodels (A to D; Fig 1 and Table 1). Each model incrementally incorporates additional evidence associated with relapse from a different type of genomic feature: model A adds clinical attributes, ai, including histology (classic, desmoplastic, and large-cell/anaplastic) and metastasis status (M0 v M1-4). Model B adds disease subtype–independent gene-expression signatures, ei, that is, those associated with relapse across all tumor samples without dividing the cohort into molecular subtypes. Model C adds disease subtype–dependent gene-expression signatures, sei, that are specific to the sample's molecular subtype {c1, … ,c6}. Model D adds disease subtype–independent DNA copy-number gains and losses (genomic abnormalities), gi. This incremental approach allows us to compare what we gain in accuracy with each addition of new evidence.
Fig 1.

Bayesian cumulative log-odds model (probabilistic network) that integrates clinical and high-level genomic information to predict the probability of relapse. The submodel of model C corresponding to subtype determination is shown in a different color because it is applied separately and in advance for any unlabeled sample. RSV, respiratory syncytial virus.
Model Feature Selection
We chose model features in two ways: by their association with clinical outcome in past studies and by their strong correlation with the relapse/no relapse phenotype in the training data.
For gene-expression signatures, ei and sei, we used the 2,599 gene sets and the gene set view of the data as described above. We chose one disease subtype–independent expression signature representing c-Myc activation34 because c-Myc overexpression has been shown to be associated with poor outcome in medulloblastomas.42 In addition, we confirmed that the signature was in the top 20 pathway discriminators of relapse in the training set according to the au-ROC curve (Data Supplement). For the disease subtype–dependent expression signatures, we selected one of the top 20 discriminators of relapse inside each disease subtype within the training set according to the au-ROC curve (Data Supplement). In all these selections, we favored signatures with a clear biologic interpretation and/or those obtained from published activation and repression experiments.
For genomic abnormalities, we selected DNA copy number gains or losses by using single nucleotide polymorphism array data that we generated in our separate study41 (see Appendix) and from which we identified statistically significant focal amplifications and deletions using Genomic Identification of Significant Targets in Cancer (GISTIC).43 We then selected loci in two ways: (1) high association with relapse in the training data set [del(16q), del(16q23.3), amp(7q21.3), and amp(3q26.32); see Appendix] and (2) association with outcome in past studies [amp(8q24.21/c-Myc), amp(2p24.3/N-Myc) and del(6q/monosomy 6)]. Table 1 gives the log-odds association of each amplification or deletion with relapse. We used disease subtype–independent features because of the small number of samples with copy number data in the training set (38 of 96).
Training the Model
To set the parameters of the models, we evaluated the log-odds ratio, or conditional evidence, Ev(r | x), for relapse, r, conditional on each feature x. We compute the contingency tables of feature versus relapse status in the training set where the value of r is known for each sample. This yields the probability of relapse of the training samples conditional on the value of that feature, P(r | x), from which Ev(r | x) can be computed (see Appendix).
Model Evaluation: Predicting Relapse
To predict relapse for a patient sample, we combine all of the evidence via a Bayesian cumulative log-odds model given by equation (1) with the value of each Ev(r | x) determined by the value of each feature for the given sample.
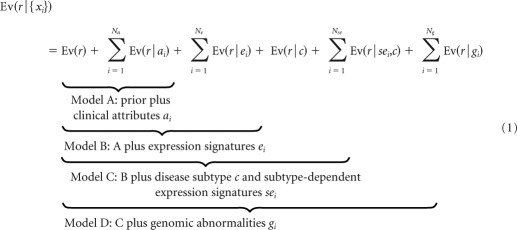 |
To facilitate the interpretation of each prediction, we represent the individual log-odds evidence from each feature value graphically as arms in a Bayesian nomogram.32 Note that the nomogram is not the standard type used in regression models and that to predict the relapse status of a new (test) sample, its disease subtype c must first be determined separately as described above.
RESULTS
To validate models A to D, we assessed how well each model recapitulated the relapse end points of the 96 training samples. Next we used the independent set of 78 test patient samples to evaluate how well each model generalized to new data and how well it predicted patient relapse. To make a predictive call, we estimated the probability of relapse using the feature values of each patient sample in equation (1).
Model Fit on Training Data
Training performance improves as the models incorporate more feature types (Table 2, Fig 2A; Data Supplement). For example, the au-ROC curve values for models A to D are 0.58, 0.66, 0.87, and 0.84, respectively. The differences between models C and D are small, presumably because of the limited number of samples with known genomic abnormality status (see Discussion).
Table 2.
Summary of Performance for the Current Clinical Schema (and model C inside the standard and high-risk groups) and Our Bayesian Cumulative Log-Odds Models A to D
| Model | Description | Training Set |
Test Set |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| au-ROC Curve | P | Error Rate | KM P | au-ROC Curve | 95% CI | P | Error Rate | KM P | ||
| S | Clinical schema | 0.62 | .075 | 0.368 | .0012 | 0.68 | 0.55 to 0.79 | .0044 | 0.288 | .045 |
| A | Clinical features (histology and metastasis) | 0.58 | .101 | 0.406 | .0767 | 0.73 | 0.61 to 0.85 | .000601 | 0.321 | .186 |
| B | A + disease subtype–independent pathway: c-Myc | 0.66 | .00321 | 0.365 | .0254 | 0.75 | 0.64 to 0.85 | .000177 | 0.346 | .000441 |
| C | B + disease subtype–dependent pathways | 0.87 | 5.2 × 10−10 | 0.219 | 7.62 × 10−9 | 0.80 | 0.7 to 0.89 | 1.07 × 10−6 | 0.256 | 1.96 × 10−8 |
| D | C + DNA copy number gains or losses | 0.84 | 4.94 × 10−9 | 0.281 | 5.61 × 10−6 | 0.78 | 0.67 to 0.88 | 5.56 × 10−6 | 0.256 | 2.14 × 10−8 |
| C | Inside standard-risk group | 0.88 | 2.8 × 10−7 | 0.207 | .0047 | 0.72 | 0.56 to 0.87 | .0066 | 0.255 | 7 × 10−4 |
| C | Inside high-risk group | 0.95 | 2.9 × 10−6 | 0.138 | .00038 | 0.83 | 0.56 to 0.86 | .0085 | 0.273 | .0076 |
Abbreviations: au-ROC, area under the receiver operating characteristic curve; KM, Kaplan-Meier.
Fig 2.

(A) Receiver operating characteristic (ROC) plots (empirical, dashed line; binormal, solid line) and area under the ROC (au-ROC) curve performance of the current clinical schema and models A to D in the training set. (B) ROC plot and au-ROC curve performance of the current clinical schema and models A to D in the independent test set. (C) ROC plots and au-ROC curve performance for model C in the independent test set for the standard-risk and (D) high-risk patient groups as defined by the current clinical schema. Note that only 73 of the test samples had corresponding clinical annotation that allowed categorization as standard risk or high risk. The model will still make a predictive call based on the genomic data. The 95% CIs in Figures 2B and 2C are estimates based on bootstrap sampling and are affected by small sample size. N, no; Y, yes.
Performance on Test Data Sets
On the independent test set of 78 samples, the performance of models A to C increases overall (Table 2, Fig 2B) as more information is made available, with a small decrease in model D (see Discussion), demonstrating the benefits of cumulative integration of several sources of information including disease subtypes. For example, the au-ROC curve values for models A to D in the test set are 0.73, 0.75, 0.80, and 0.78, respectively. The corresponding Kaplan-Meier log-rank P values show a similar trend (Data Supplement) as do the relative utility curves44 (Appendix and Data Supplement). Models C and D outperform the current clinical schema (au-ROC curve, 0.68; Fig 2B and Data Supplement), model A on the basis of clinical features alone (au-ROC curve, 0.73; Fig 2B and Data Supplement), and our previously published eight-gene outcome signature (au-ROC curve, 0.71; Data Supplement). Importantly, the largest increase in performance occurs in model C, which incorporates markers of outcome within molecular subtypes.
Improved Risk Stratification Using Molecular Markers
Figure 2C shows the breakdown of standard-risk and high-risk test set patients for model C (best-performing model). Fifteen patients categorized as standard-risk by the current clinical schema actually relapsed. Of those, model C correctly predicts that six (true-positive rate, 0.40; 97.5% CI, 0.14 to 0.71; false-positive rate, 0.11; 97.5% CI, 0.025 to 0.28) will relapse. Despite the large CIs, the difference between the clinical schema and model C remained significant using both DeLong et al45 (P = .04) and Integrated Discriminant Improvement (IDI) criteria46 (see Appendix). These preliminary results show potential practical value for identifying standard-risk patients who may benefit from treatment more suitable for high-risk patients.
DISCUSSION
Our model is one of the first medulloblastoma risk models that maintains good performance on an independent multi-institutional test set, suggesting its generalizability for use in future medulloblastoma clinical trials. Models C and D appear to outperform other state-of-the-art models reported in the literature, such as those of Pfister et al14 and de Haas et al.13 The similarity of our training and test results indicates that our strategy of selecting high-level features and our model architecture control for overtraining. The significant improvement of model C over the current clinical schema and model A (based on clinical features) and our previously published eight-gene outcome signature can be attributed to the use of highly informative expression signatures, specifically those conditional on disease subtype. Models that do not take into account disease subtype (eg, model B) are inherently less accurate. The use of disease subtype–specific features effectively addresses this problem.
To establish the method's effectiveness on independently acquired data and other acquisition platforms, we applied a simple normalization procedure based on a 0-to-1/min-max rescaling of the expression signatures' activation scores (see Appendix). For eventual clinical deployment, in which a single patient sample must be evaluated, a central laboratory and single platform would eliminate this step, subject to final validation of the model.
Three genomic abnormalities were associated with medulloblastoma outcome in past studies: c-Myc and N-Myc amplification and monosomy 6 correlate with relapse status but have low penetrance in our training set (8%, 36%, and 16%, respectively). This low penetrance combined with the asymmetry of their corresponding nomogram arms in Figure 3(indicating low predictive value when absent) limits their value as overall predictors of outcome. In contrast, the overexpression of the c-Myc expression signature (third row of Fig 4A) is a significant predictor of relapse with higher penetrance (43%) and has higher predictive value when absent, making it a better predictor of relapse. Our results suggest that in medulloblastomas, multiple mechanisms of Myc activation might be at play and would show the advantage of considering catch-all functional readouts of oncogene activation rather than relying solely on the status of known genomic abnormalities. The relevance of c-Myc activation as a marker of poor outcome in our study is consistent with its previously implicated role as a significant risk factor12,13,42 and a general regulator of poor-prognosis metastatic state.23
Fig 3.

Bayesian nomogram showing the amount of evidence that each feature (risk factor) provides in the context of a specific patient's feature values. The arms of the nomogram represent the values of the posterior log odds ratio evidence, Ev(r | x), for each feature's values. The actual values taken by each feature are shown in blue or red according to the sign of Ev(r | x): positive magnitude to the right side is evidence for relapse (red) and negative magnitude to the left is evidence for no relapse (blue). The final sum of Ev(r | x) provides the final probability of relapse, which is 0.89 (95% CI, 0.65 to 0.97). OR, odds ratio; Lods, log-odds; Prob, probability; RSV, respiratory syncytial virus; H, high; L, low; DESMO, desmoplastic; LCA, large-cell/anaplastic (lymphoma); N, no; Y, yes.
Fig 4.

Heat map of 38 samples showing the Genomic Identification of Significant Targets in Cancer (GISTIC) amp(8q24.21/c-Myc) status, the GISTIC amp(2p24.3/N-Myc) status, the expression of the disease-independent c-Myc pathway, the del(6q/monosomy 6) GISTIC status, and the expression of the beta-catenin pathway.
We found that the addition of genomic amplifications and deletions (model D) does not improve accuracy, possibly because we had such data for only about 40% of training and test samples or that the information these loci carry is already subsumed in the expression signatures. For example, the expression of the β-catenin pathway, one of our disease subtype signatures, follows closely the status of del(6q). Unfortunately, we have too few samples with copy-number data to address this issue conclusively. Moreover, there were insufficient copy-number data within each subtype to evaluate whether subtype-dependent amplification or deletion has higher predictive value.
One of the four amplifications and deletions we identified as associated with outcome, amp(3q26.32/p110a/PI3K locus) has been associated with poor outcome in endometrial cancer.47 A detailed biologic interpretation of the disease subtype–dependent pathways will require a follow-up study. In particular, the mTOR pathway, highly upregulated in the disease subtype c1, is a central integrator of signals and AKT phosphorylation and has been demonstrated to be involved in medulloblastoma.48
We introduced a Bayesian nomogram similar to those based on regression models49,50 but containing additional elements to represent disease subtype–dependent pathways. For example, the patient shown in Figure 3 has the standard-risk profile, and the current clinical schema incorrectly predicts no relapse. In contrast, models C and D correctly predict relapse because of the cumulative log-odds evidence derived from the genomic features, including c-Myc activation. This nomogram shows only the relevant disease subtype (c1) arms; the full nomogram including all the disease subtype–dependent arms is shown in the Data Supplement.
In summary, we developed a model that predicts relapse in medulloblastoma, retaining high accuracy when applied to an independent multi-institutional validation test cohort. A key feature of the model is the combination of clinical parameters with molecular markers representing gene-expression signatures of mechanisms and pathways that are specific to disease subtypes. The model relies on variable relapse-associated levels of expression signatures within disease subtypes. Considered from the perspective of individual tumor samples, the model allows for outcome predictions along with a measure of the contribution of each individual risk factor. This goes well beyond methods that simply establish association of marker expression levels with disease outcome and gives a clear sense of the possible value of the method in a clinical setting. For example, the model correctly reclassified six of 15 patients who were standard-risk by clinical criteria as high-risk. These six patients represent > 10% of the standard-risk patients in our validation cohort who, up-front, should have been offered more aggressive therapy and a better chance of progression-free survival than what stratification based on clinical risk criteria offered.
The model described here represents a first step in obtaining a more accurate, quantitative risk stratification for medulloblastoma patients by taking advantage of multiple sources of information: clinical, molecular, and genetic. While not yet ready for clinical use, with further refinement it might lead to a real-time Clinical Laboratory Improvement Amendments (CLIA) –certified test that clinicians can use to help guide their treatment decisions (see Appendix). Precedent has already been established in breast cancer risk stratification by the Oncotype DX51 and MammaPrint52 tests currently in prospective clinical trials. Breast cancer has heterogeneity similar to that of medulloblastomas and thus serves as a good metric. These tests have shown that a gene expression–based assay can be performed with rapid turnaround and high sensitivity/specificity. Thus efforts to refine, transfer, and apply our proposed model to medulloblastomas should be a priority.
Supplementary Material
Acknowledgment
We thank James Olson, Ching Lau, Charlie Eberhart, Stefano Monti, Adam Margolin, Jonathan Jesneck, and Todd Golub for useful discussions; Jeff Michalski, the study chair of ACNS0331; and Jon Bistline for figure and bibliographic preparation.
Appendix
Sample Preparation
Training set.
All nucleic acid preparation was performed at Children's Hospital Boston with the exception of samples obtained from Texas Children's Hospital where samples were similarly processed. Briefly, primary tumor samples were partitioned for DNA and RNA extraction. RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's protocol. DNA was prepared using the Puregene DNA Extraction Kit (Gentra Systems, Minneapolis, MN) according to the manufacturer's protocol. Gene expression data were generated by hybridizing labeled RNAs to Affymetrix U133A2 high-throughput arrays (Affymetrix, Santa Clara, CA). Samples meeting a minimum of 37% p-calls were used in this analysis. Data were preprocessed by using the robust multichip average (Irizarry RA et al: Biostatistics 4:249-264, 2003) to generate the.gct files used in subsequent analyses. DNA copy number data were generated by using Affymetrix 250K StyI arrays (Affymetrix) or Affymetrix 6.0 arrays (Affymetrix) to obtain signal intensities and genotype calls. Briefly, genomic DNA was digested, adaptor-ligated, and amplified by polymerase chain reaction to achieve fragments ranging from 200 to 1,100 base pairs. These fragments were pooled, concentrated, and further fragmented with DNaseI (Affymetrix) followed by labeling, denaturing, and hybridization to arrays in batches of 96 samples. Arrays were then scanned using the GeneChip Scanner 3000 7G (Affymetrix). Signal intensities were normalized against a reference set of normal genomic DNA data generated through the Human HapMap project. The number of samples in the 96-sample training set with DNA copy number data is 38 (40%).
Test set.
In the test set, 15 samples (Children's Oncology Group Tumor Bank) were processed as described above. Of those 15, a total of six have DNA copy number data. Another set of 47 samples was taken from the data set from Pomeroy et al6 (Affymetrix Hu6800; Affymetrix). None of those samples had DNA copy number data. The remaining 16 samples, all with DNA copy number data, came from Kool et al33 (Affymetrix U133; Affymetrix).
Disease Subtypes
To define the disease subtype (c1-c6), we used the results of a separate study of a larger collection of about 200 medulloblastoma tumors described in Cho et al.41 The following is a summary of the two-step clustering methodology. First, we reduce the dimensionality of the expression data from thousands of genes to a few metagenes by applying nonnegative matrix factorization (Brunet JP, et al: Proc Natl Acad Sci U S A 101:4164-4169, 2004). We compute multiple factorization decompositions of an expression matrix A that contains our samples. For each factorization decomposition rank k between 2 and 20, we compute 100 decompositions: A ≈ W × H. We assess the stability of the factorization decompositions for each rank k and select the best factorization decomposition (in terms of estimation error) from each of the most stable three ranks for further analysis.
The second step involves the clustering of the samples in the much smaller metagene space represented by the matrix H by a technique similar to consensus clustering (Monti S, et al: Machine Learning 52:91-118, 2003). For each number of clusters n from 2 to 20, we cluster a random subsample of the data containing 85% of the samples by using the partitioning around medoids algorithm (Kaufman L, et al: Finding Groups in Data: An Introduction to Cluster Analysis. New York, NY, Wiley-Interscience, 1990). We repeat this process 500 times and measure the stability of the clustering solutions for each number of clusters by computing a consensus matrix and its cophenetic coefficient (as described in Brunet JP, et al: Proc Natl Acad Sci U S A 101:4164-4169, 2004).
Using the method described above, we clustered a data set containing the expression data for 198 medulloblastoma primary tumors (which included our training set), 11 normal cerebellum samples, 46 cell lines, and other tumors.33 Factorization decompositions using four, seven, and 11 metagenes were the most stable, but because of the diversity of the samples processed, we performed the clustering step on the representation of the data in a seven metagenes space and not a four metagenes space. The most stable solutions were achieved for partitioning of the samples into six, 11, and 15 clusters. All of the normal cerebellum samples were assigned to the same cluster (in the most stable clustering solution into 11 clusters). Medulloblastoma samples that were assigned to the cluster of the normal samples were removed from further analysis.
Next, we performed a similar analysis on the remaining medulloblastoma samples (n = 194) and rhabdoid samples (n = 9). The most stable factorization decompositions were achieved using 12, eight, and five metagenes. Following the factorization decomposition using 12 metagenes, the most stable clustering solution was achieved when grouping into eight clusters.
We noticed that one of the clusters contained heterogeneous samples and was highly enriched with samples collected at Texas Children's Hospital, suggesting a batch effect. We identified one metagene as capturing a signature of the Texas Children's Hospital samples and so removed its value from all of our analyzed samples, yielding a metagene space of only 11 dimensions. We clustered the samples in this space into seven clusters. All rhabdoid samples were assigned to the same cluster. Medulloblastoma samples that were assigned to the rhabdoid cluster were removed from further analysis (n = 5). The six clusters of medulloblastoma left are those we use to define disease subtypes c1-c6.
A detailed analysis of these subtypes produced this interpretation41: c1: photoreceptor activation and c-Myc amplification; c2: first subgroup of classic tumors; c3: sonic hedgehog (SHH) pathway activation; c4: second group of classic tumors; c5: photoreceptor activation without c-Myc amplification; and c6: monosomy 6 and β-catenin activation.
Expression signatures database.
We created a database of molecular signatures by combining the Broad Institute's Molecular Signatures Database C2 collection, release 2 (MSigDB: http://www.broadinstitute.org/msigdb), containing 1,892 gene sets, with our own Oncogene Pathway Activation Database (version 3), which is a manually curated collection of 273 gene sets defined from gene expression data sets from Gene Expression Omnibus (GEO) and the biomedical literature, representing oncogene activation or tumor suppressor dysregulation. These two collections combined produced a total of 2,165 gene sets. The signatures from MSigDB are taken as defined in the MSigDB with no change. The signatures from our Oncogene Pathway Activation Database are defined by selecting the top 100-150-200 or 300 upregulated (UP) and downregulated (DN) genes according to the difference of means across the relevant phenotypes. The exact number was defined on a case-by-case basis according to the strength of the marker genes in terms of their predictive accuracy on the different subsets as measured by the area under the receiver operating characteristic (au-ROC) curve. The single-sample procedure (see below) computes enrichment scores for each of the 2,165 signatures but also produces additional combined signatures for those that have both UP and DN versions to produce a grand total of 2,599 signatures.
Single-Sample Gene Set Enrichment Analysis Procedure to Transform Data From Gene to Gene Set Space
We used the same procedure as described in Barbie et al.22 First we preprocessed the training and test data sets by mapping from probe IDs to gene symbols by using the probe collapse by max procedure as is done in Gene Set Enrichment Analysis (GSEA; Subramanian et al21 and www.broadinstitute.org/gsea). Then for each of the gene sets in the database (seee above), we define an enrichment score that represents the degree of absolute enrichment of a given gene set in each of the samples in the training or test data sets. The gene expression values for a given sample are first rank-normalized and then an enrichment score is produced by evaluating an enrichment statistic that is a function of the differences in the empirical cumulative distribution functions (ECDFs) of the genes in the gene set and the remaining genes. This procedure is similar to the one used in GSEA,21 but instead of using a gene list ranked by differential expression, the list is ranked by absolute expression (in one sample). The enrichment score is obtained not by a weighted Kolmogorov-Smirnov statistic as in GSEA but by an integration of the difference between the ECDFs. For a given gene set G of size NG and single sample S of the training or test data sets of N genes, the genes are replaced by their ranks according their absolute expression from high to low: L = {r1, r2, r3,…, rN}. An enrichment score ES(G, S) is obtained by a weighted sum (integration) of the difference between the ECDFs of the genes in the gene set and the ECDFs of the remaining genes:
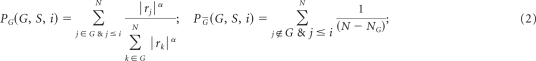 |
This calculation is repeated for each signature and each sample in the data set. Notice that this quantity is signed and that the exponent α = 3/4 adds a weight to the rank. This quantity is slightly more robust and more sensitive to differences in the tails than the Kolmogorov-Smirnov statistic. It is particularly well suited to represent the activation score of gene sets that are based on a relatively small subset of the genes attaining high expression values. For gene sets with both UP and DN versions, besides their independent UP and DN enrichment scores, a combined score is computed by ES(GUP, S) – ES(GDN, S).
Feature selection of expression signatures.
Once the single-sample versions of GSEA (ssGSEA) scores were computed for all samples in the training data set, we computed the au-ROC curve for each feature with respect to the relapse phenotype in the entire data set and in each disease subtype. We sorted the 2,599 features and chose one in the top 20 to provide the disease subtype–independent and disease subtype–dependent expression signatures (Table 1 and Data Supplement). The top 20 features have similar high values for the au-ROC curve and are roughly equivalent in their predictive value. Rather than choosing the top feature, we chose one that had a clear biologic annotation and/or represented a direct experiment in which a pathway is being induced or deregulated by a single gain or loss of function. Our motivation for this was to make the model accurate in terms of the predictive power and transparent and interpretable in terms of the meaning of the features.
Dichotomization of expression-signature features.
For each expression-signature feature, we slid a decision boundary from the lowest to the highest ssGSEA score value across the training set, rescaled to [0,1], and computed the discrimination error at both sides of the boundary. The dichotomization threshold was set at the point where the classification errors induced by the dichotomization were equal on both sides of the boundary. By using this threshold, each expression-signature value was redefined as H (high) or L (low) according to which side of the threshold it fell on. To dichotomize the variables in the test set in a similar way, each expression signature was also rescaled to the [0,1] range.
Scaling of test set expression signatures
We applied a simple normalization procedure based on a 0-to-1/min-max rescaling because of the cross-platform nature of our test set, which included gene-expression profiles derived from multiple data sets obtained by using different platforms (ie, Affymetrix U133A2 and HU6800). This was necessary to establish that the method was general enough to work on independently acquired data and other acquisition platforms. The cross-platform nature of this work requires the use of some normalization method so that the enrichment scores and their dichotomization thresholds across the training set and the different data sets that compose the test set have a similar dynamic range.
The efficacy of the scaling was established by the results on the test set, which did not differ substantially from more sophisticated normalization approaches. A limitation of this approach is that it cannot be used on a single-patient sample presented to the model in isolation. If our approach is deployed via a central laboratory on a single gene-expression profiling platform, then scaling should not be necessary. In this scenario, the same data acquisition platform would be used to construct a refined stratification model and to test patient samples. The raw enrichment scores could then be used without any rescaling, subject to validation.
There are numerous approaches for mapping the empirical density distributions of enrichment scores across data sets. We opted for a simple 0-to-1/min-max rescaling procedure applied to each data set (Jain AK, et al: Encyclopedia of Biometrics. New York, NY, Springer, 2009). It has the advantage of working quite well, as our testing ROCs indicate. We note that extreme values can have the disadvantage of having slower convergence and larger variation when estimated from finite samples, but this did not prove to be a problem in practice. At an earlier stage of developing our method, we experimented with other normalization procedures, such as standardizing the enrichment scores or matching the mean of the positive (negative) Kolmogorov-Smirnov–like empirical density distributions of enrichment scores. There was no significant difference in the overall results using this more sophisticated method instead of 0-to-1 scaling.
Cumulative log-odds model.
As described in the article, the model integrated all of the evidence via a Bayesian cumulative log-odds model. This model was a simple application of Bayes rule plus assumptions about the independence of features, given disease subtype. It was an example of an extended naive Bayes classifier or a directed acyclic Bayesian network that used dichotomized variables (Bishop CM: Pattern Recognition and Machine Learning. Heidelberg, Germany, Springer, 2006; Pearl J: Probabilistic Reasoning in Intelligent Systems: Networks of Plausible Inference. San Francisco, CA, Morgan Kaufmann, 1988).
We evaluate the conditional evidence, Ev(r | x), for relapse, r, conditional on each feature x using the posterior log-odds ratio,
The conditional probabilities P(r | x) were computed from contingency tables of feature versus relapse. Because some contingency table entries contain zeroes, a simple smoothing procedure consisting of adding two counts to each contingency matrix entry is implemented as part of the model. For example, for the feature “histology” based on the counts in the training set we have the values provided in Appendix Table A1. The values in the last column are reported in column 4 of Table 1 and also provide the length of the arms in the corresponding entry of the nomogram as shown in Appendix Figure A1.
As can be seen in the nomogram, the desmoplastic histology, known in the past to have an association with a more favorable clinical outcome, decreases the risk of relapse by 0.34 log-odds units. The large-cell/anaplastic (LCA) histology, as expected because of its association with poor outcome, increases the risk by 0.27 log-odds units. The classic histology produces only a small amount of log-odds evidence for relapse (0.033) and is therefore not very informative. Thus, a test sample with LCA histology will contribute 0.27 toward the log-odds evidence for relapse. This process is repeated for all features in Table 1 to arrive at the final log-odds evidence for relapse for the sample.
The cumulative log-odds model is also equivalent to a likelihood ratio (Lachenbruch P et al: Biometrics 35:69-85, 1979) computed as a product of posterior odds ratios:
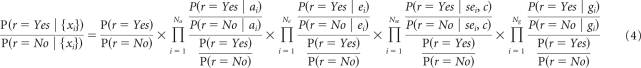 |
Nomogram.
Our nomogram is based on the Bayesian nomogram introduced in Možina et al,11 including the use of CIs. We introduced a novel feature in our use of conditional arms (lines) to display an extra level of depth for the disease subtype–dependent expression signatures (Data Supplement).
Predicting disease subtype.
The disease-subtype feature is known for the training set but has to be inferred in the test set. This is done by using the 36 subtype expression signatures mi (Data Supplement), six for each subtype (as described in the article), that provide the probabilities of membership of a test sample to each disease subtype. For example, the conditional evidence of a test sample belonging to class c1, given the values of the six subtype expression signatures that discriminate c1, is:
and similarly for each subtype.
Methodology flow chart.
The entire computational methodology is summarized in the Data Supplement.
Software.
The cumulative log-odds model is implemented by using the gRaphical Models in R packages: gRain and gRbase (Højsgaard S: Graphical independence networks with the gRain package for R. J Stat Software [in press]; Dethlefsen C et al: J Stat Soft 14, 2005). We compute the conditional probability tables by using the compileCPT function and the log-odds model building a graphical model network by using the grain function. The probability of relapse is computed by marginalization (querygrain) of the root node (relapse) after adding all the feature evidence by calling the setFinding function. Additional functionality is provided by the verification R package (ROC plots), GenePattern (Reich M, et al: Nat Genet 38:500-501, 2006) modules, and custom code for the nomograms. All code is available on request to the authors.
Feature selection of DNA copy number gains or losses.
As described in Patients and Methods, we select the expression signatures from the ones that best discriminate the relapse versus no-relapse status according to the au-ROC curve. To select the Genomic Identification of Significant Targets in Cancer (GISTIC) genomic abnormalities, we used their average absolute evidence (AvEv) with respect to the relapse status defined by:
Here, the sum is over all the k distinct values Xi of the feature. This positive quantity quantifies the amount of evidence the given feature contributes on average to the model. This is the metric we used to rank the top copy number gains or losses described in Patients and Methods. We exclude whole chromosome features and concentrate on the features that represent more focal copy number gains or losses and choose the top four features with the highest AvEv to be part of the model (model D).
Details on the Improved Risk Stratification Using Molecular Markers
In the article, we point out our reassignment of test set patients clinically categorized as standard risk to the high-risk group. Figure 2C shows that we have a 40% true-positive rate (six of 15) and an 11% false-positive rate (four of 36) for prediction of relapse (high-risk) restricting our attention to these standard-risk patients. The exact values are true-positive rate of 0.40 (97.5% CI, 0.14 to 0.71) and false-positive rate of 0.11 (97.5% CI, 0.025 to 0.28) where we computed CIs following the approach of Simel et al (Simel DL, et al: J Clin Epidemiol 44:763-770, 1991).
The wide CIs for the model's au-ROC curve tend to underestimate model differences. They are very conservative because of small sample size and the bootstrapping method used to generate them. A formal and systematic way to estimate the significance of the improvement going from the clinical schema to model C is to use either a test of correlated au-ROC curves45 or the Integrated Discriminant Improvement (IDI) introduced by Pencina et al.46 We describe the results of both approaches in the next two paragraphs.
Test of correlated au-ROC curve.
Here, we use a test that specifically takes into account the fact that the clinical schema and model C au-ROC curve plots are correlated because they correspond to the same patients (test set). We follow the approach of DeLong et al.45 We computed this using the R package pROC. The differences between the clinical schema and model C are significant (P = .04).
IDI.
The IDI46 can be defined as a difference in model discrimination slopes (ie, the difference of mean predicted probabilities of relapses and nonrelapses across models). The IDI values are calculated by using the means of predicted probabilities for relapses and no relapses for the two models (equations 12 and 13 in Pencina et al46):
Therefore, to evaluate the IDI, we need to use model-generated predicted probabilities. Because the clinical schema lacks such probabilities, we approximate them by using model A. Using the IDI approach, we can analyze the improvement going from any of our models to any other model. The detailed IDI results are provided in Appendix Table A2.
The relevant improvement of model C over model A, relevant to the data shown in Figure 2C, is significant for the standard-risk group and also over the entire validation test set. The IDI approach is more sensitive because it can detect improvements in classification performance between models that do not appear significantly different in terms of their au-ROC curves. This motivated its introduction by Pencina et al.46 Both the au-ROC curve and the IDI can be interpreted as corrected average sensitivities but using different weights or corrections (see Fig 3 and Appendix equations A3-A10 in Pencina et al46). The au-ROC curve is an average sensitivity weighted by the derivative of the specificity. The IDI is an average sensitivity corrected by the decrease in (1 – specificity). The values of sensitivity corresponding to small cutoffs are weighted more heavily by the au-ROC curves. This means that some improvement in sensitivity, at higher cutoffs, would not contribute as much. In contrast, the IDI weights more evenly the gains in sensitivity across all cutoff values.
Relative utility curves.
These curves show the relative utility (ie, the fraction of the utility of perfect prediction) that is achieved at the optimal cut point for a risk prediction model versus risk threshold.45 We compute the relative utility curves by using the false-positive rates (FPRs) and true-positive rates (TPRs) that are computed to generate the binormal ROC plots, and we used those values to evaluate formula (15) in Baker et al44:
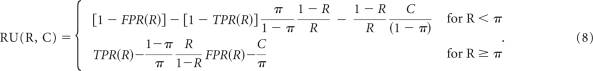 |
Therefore the RU values are computed from each row in the binormal ROC table, generated by the verification R package, which has entries for the ROC threshold, R, and the FPR and TPR obtained from the fitted binormal model. As prevalence, π, we used the prior probability of relapse, and we set the C parameter (cost of test) to zero. The relative utility curve is therefore generated by plotting RU(R, C = 0), according to the formula above, versus R (ROC threshold) using all the points (rows) in the binormal ROC table. These curves emphasize the significant difference going from model B to model C and the equivalence of model C and model D. They also show a significant improvement of models B to D versus the current schema.
Clinical deployment.
As mentioned in the main text, the proof of concept using real-time gene expression–based risk prediction has been established in breast cancer with the Oncotype DX (Genomic Health, Redwood City, CA) and MammaPrint (Agendia, Amsterdam, the Netherlands) platforms. Samples are sent directly to a commercial reference laboratory run by these companies. The turnaround time quoted by Genomic Health for the Oncotype DX platform is 10 to 14 days from the date the specimen is received. Agendia quotes 10 days for their MammaPrint technology. Each company provides specific containers, protocols, and shipping instructions for use in collection. While the Oncotype DX platform can be run on formalin-fixed paraffin-embedded samples, the MammaPrint uses only samples collected within 1 hour of resection.
For pediatric cancer, processes for handling and shipping samples to core laboratories where they are tested for clinical decision making have been developed for several cancers. Within the Children's Oncology Group, for example, protocols are in place for childhood leukemia and neuroblastoma. When fully implemented, we envision our approach deployed in a similar setup in which a national reference laboratory or core is established to which samples are shipped using protocols, shipping containers, and logistics that have already been developed for other childhood cancers. The techniques for measuring gene expression can yield data within 2 to 3 days. With the clinical and gene expression data in hand, our probabilistic model can be run in minutes. It is, therefore, certainly reasonable to conclude that these methods can be used with turnaround time to a result in 10 to 14 days.
Table A1.
Conditional Probabilities and Evidence for Histology Feature
| Feature Value Xi | Counts Relapse |
Counts + 2 (smoothed) Relapse |
Probability P (r x = Xi) |
Odds |
Posterior Odds Ratio |
Evidence (posterior log-odds ratio) Ev(r = Yes x = Xi) | |||
|---|---|---|---|---|---|---|---|---|---|
| No | Yes | No | Yes | No | Yes | ||||
| LCA | 8 | 8 | 10 | 10 | 0.5000 | 0.5000 | 1.0 | 1/0.7668 = 1.3041 | 0.27 |
| Classic | 37 | 28 | 39 | 30 | 0.5652 | 0.4340 | 0.7693 | 0.7693/0.7668 = 1.0033 | 0.0033 |
| Desmoplastic | 9 | 4 | 11 | 6 | 0.6471 | 0.3529 | 0.5454 | 0.5454/0.7668 = 0.7113 | −0.34 |
| Prior | 54 | 40 | 60 | 46 | 0.5660 | 0.4340 | 0.7668 | — | — |
Abbreviation: LCA, large-cell/anaplastic.
Table A2.
Integrated Discriminant Improvement Results
| Improvement:From Model X to Model Y | Entire Test Cohort |
Standard-Risk Group |
||
|---|---|---|---|---|
| IDI | P | IDI | P | |
| A to B | 0.061 | .0050* | 0.016 | .30 |
| A to C | 0.19 | 5 × 10−5* | 0.14 | .014* |
| A to D | 0.20 | 1.6 × 10×4* | 0.16 | .013* |
| B to C | 0.13 | .0008* | 0.13 | .0091* |
| C to D | 0.0061 | .40 | 0.019 | .28 |
Abbreviation: IDI, integrated discriminant improvement.
Significant.
Fig A1.

Nomogram entry for histology feature. DESMO, desmoplastic; LCA, large-cell/anaplastic (lymphoma).
Footnotes
Supported by Grants No. R01-CA109467, R01-CA105607, R01-CA121941, R01-GM074024, P50-CA112962, and P30-HD018655.
Authors' disclosures of potential conflicts of interest and author contributions are found at the end of this article.
Clinical trial information can be found for the following: NCT00919750.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The author(s) indicated no potential conflicts of interest.
AUTHOR CONTRIBUTIONS
Conception and design: Pablo Tamayo, Yoon-Jae Cho, Scott L. Pomeroy, Jill P. Mesirov
Financial support: Scott L. Pomeroy, Jill P. Mesirov
Provision of study materials or patients: Yoon-Jae Cho, Heidi Greulich, Lauren Ambrogio, Netteke Schouten-van Meeteren, Tianni Zhou, Allen Buxton, Marcel Kool, Matthew Meyerson, Scott L. Pomeroy
Collection and assembly of data: Yoon-Jae Cho, Heidi Greulich, Lauren Ambrogio, Netteke Schouten-van Meeteren, Tianni Zhou, Allen Buxton, Marcel Kool, Matthew Meyerson, Scott L. Pomeroy
Data analysis and interpretation: Pablo Tamayo, Yoon-Jae Cho, Aviad Tsherniak, Scott L. Pomeroy, Jill P. Mesirov
Manuscript writing: Pablo Tamayo, Yoon-Jae Cho, Aviad Tsherniak, Heidi Greulich, Lauren Ambrogio, Netteke Schouten-van Meeteren, Tianni Zhou, Allen Buxton, Marcel Kool, Matthew Meyerson, Scott L. Pomeroy, Jill P. Mesirov
Final approval of manuscript: Pablo Tamayo, Yoon-Jae Cho, Aviad Tsherniak, Heidi Greulich, Lauren Ambrogio, Netteke Schouten-van Meeteren, Tianni Zhou, Allen Buxton, Marcel Kool, Matthew Meyerson, Scott L. Pomeroy, Jill P. Mesirov
REFERENCES
- 1.Cho YJ, Pomeroy SL. Targeted therapy in medulloblastoma. In: Houghton PJ, Arceci R, editors. Molecularly Target Therapy for Childhood Cancer. New York, NY: Springer Verlag; 2009. p. 350. [Google Scholar]
- 2.Packer RJ, Goldwein J, Nicholson HS, et al. Treatment of children with medulloblastomas with reduced-dose craniospinal radiation therapy and adjuvant chemotherapy: A Children's Cancer Group Study. J Clin Oncol. 1999;17:2127–2136. doi: 10.1200/JCO.1999.17.7.2127. [DOI] [PubMed] [Google Scholar]
- 3.Albright AL, Wisoff JH, Zeltzer PM, et al. Effects of medulloblastoma resections on outcome in children: A report from the Children's Cancer Group. Neurosurgery. 1996;38:265–271. doi: 10.1097/00006123-199602000-00007. [DOI] [PubMed] [Google Scholar]
- 4.Thomas PR, Deutsch M, Kepner JL, et al. Low-stage medulloblastoma: Final analysis of trial comparing standard-dose with reduced-dose neuraxis irradiation. J Clin Oncol. 2000;18:3004–3011. doi: 10.1200/JCO.2000.18.16.3004. [DOI] [PubMed] [Google Scholar]
- 5.Bailey CC, Gnekow A, Wellek S, et al. Prospective randomised trial of chemotherapy given before radiotherapy in childhood medulloblastoma: International Society of Paediatric Oncology (SIOP) and the (German) Society of Paediatric Oncology (GPO)—SIOP II. Med Pediatr Oncol. 1995;25:166–178. doi: 10.1002/mpo.2950250303. [DOI] [PubMed] [Google Scholar]
- 6.Pomeroy SL, Tamayo P, Gaasenbeek M, et al. Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature. 2002;415:436–442. doi: 10.1038/415436a. [DOI] [PubMed] [Google Scholar]
- 7.Crawford JR, MacDonald TJ, Packer RJ. Medulloblastoma in childhood: New biological advances. Lancet Neurol. 2007;6:1073–1085. doi: 10.1016/S1474-4422(07)70289-2. [DOI] [PubMed] [Google Scholar]
- 8.Giangaspero R, Perry R, Kelly P, et al. Tumours of the central nervous system. In: Kleihues P, Cavenee W, editors. World Health Organization Classification of Tumours: Pathology and Genetics—Tumours of the Nervous System. Lyon, France: IARC Press; 2000. [Google Scholar]
- 9.Eberhart CG, Kepner JL, Goldthwaite PT, et al. Histopathologic grading of medulloblastomas: A Pediatric Oncology Group study. Cancer. 2002;94:552–560. doi: 10.1002/cncr.10189. [DOI] [PubMed] [Google Scholar]
- 10.Gilbertson R, Wickramasinghe C, Hernan R, et al. Clinical and molecular stratification of disease risk in medulloblastoma. Br J Cancer. 2001;85:705–712. doi: 10.1054/bjoc.2001.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rutkowski S, von Bueren A, von Hoff K, et al. Prognostic relevance of clinical and biological risk factors in childhood medulloblastoma: Results of patients treated in the prospective multicenter trial HIT'91. Clin Cancer Res. 2007;13:2651–2657. doi: 10.1158/1078-0432.CCR-06-1779. [DOI] [PubMed] [Google Scholar]
- 12.Lamont JM, McManamy CS, Pearson AD, et al. Combined histopathological and molecular cytogenetic stratification of medulloblastoma patients. Clin Cancer Res. 2004;10:5482–5493. doi: 10.1158/1078-0432.CCR-03-0721. [DOI] [PubMed] [Google Scholar]
- 13.de Haas T, Hasselt N, Troost D, et al. Molecular risk stratification of medulloblastoma patients based on immunohistochemical analysis of MYC, LDHB, and CCNB1 expression. Clin Cancer Res. 2008;14:4154–4160. doi: 10.1158/1078-0432.CCR-07-4159. [DOI] [PubMed] [Google Scholar]
- 14.Pfister S, Remke M, Benner A, et al. Outcome prediction in pediatric medulloblastoma based on DNA copy-number aberrations of chromosomes 6q and 17q and the MYC and MYCN loci. J Clin Oncol. 2009;27:1627–1636. doi: 10.1200/JCO.2008.17.9432. [DOI] [PubMed] [Google Scholar]
- 15.Mendrzyk F, Radlwimmer B, Joos S, et al. Genomic and protein expression profiling identifies CDK6 as novel independent prognostic marker in medulloblastoma. J Clin Oncol. 2005;23:8853–8862. doi: 10.1200/JCO.2005.02.8589. [DOI] [PubMed] [Google Scholar]
- 16.Haberler C, Slavc I, Czech T, et al. Histopathological prognostic factors in medulloblastoma: High expression of survivin is related to unfavourable outcome. Eur J Cancer. 2006;42:2996–3003. doi: 10.1016/j.ejca.2006.05.038. [DOI] [PubMed] [Google Scholar]
- 17.Thompson MC, Fuller C, Hogg TL, et al. Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. J Clin Oncol. 2006;24:1924–1931. doi: 10.1200/JCO.2005.04.4974. [DOI] [PubMed] [Google Scholar]
- 18.Fattet S, Haberler C, Legoix P, et al. Beta-catenin status in paediatric medulloblastomas: Correlation of immunohistochemical expression with mutational status, genetic profiles, and clinical characteristics. J Pathol. 2009;218:86–94. doi: 10.1002/path.2514. [DOI] [PubMed] [Google Scholar]
- 19.Segal RA, Goumnerova LC, Kwon YK, et al. Expression of the neurotrophin receptor TrkC is linked to a favorable outcome in medulloblastoma. Proc Natl Acad Sci U S A. 1994;91:12867–12871. doi: 10.1073/pnas.91.26.12867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mootha VK, Lindgren CM, Eriksson KF, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 21.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barbie DA, Tamayo P, Boehm JS, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462:108–112. doi: 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wolfer A, Wittner BS, Irimia D, et al. MYC regulation of a “poor-prognosis” metastatic cancer cell state. Proc Natl Acad Sci U S A. 2010;107:3698–3703. doi: 10.1073/pnas.0914203107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bild AH, Yao G, Chang JT, et al. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature. 2006;439:353–357. doi: 10.1038/nature04296. [DOI] [PubMed] [Google Scholar]
- 25.Edelman E, Porrello A, Guinney J, et al. Analysis of sample set enrichment scores: Assaying the enrichment of sets of genes for individual samples in genome-wide expression profiles. Bioinformatics. 2006;22:e108–e116. doi: 10.1093/bioinformatics/btl231. [DOI] [PubMed] [Google Scholar]
- 26.Lee E, Chuang HY, Kim JW, et al. Inferring pathway activity toward precise disease classification. PLoS Comput Biol. 2008;4:e1000217. doi: 10.1371/journal.pcbi.1000217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burnside ES, Rubin DL, Fine JP, et al. Bayesian network to predict breast cancer risk of mammographic microcalcifications and reduce number of benign biopsy results: Initial experience. Radiology. 2006;240:666–673. doi: 10.1148/radiol.2403051096. [DOI] [PubMed] [Google Scholar]
- 28.Gevaert O, De Smet F, Timmerman D, et al. Predicting the prognosis of breast cancer by integrating clinical and microarray data with Bayesian networks. Bioinformatics. 2006;22:e184–e190. doi: 10.1093/bioinformatics/btl230. [DOI] [PubMed] [Google Scholar]
- 29.Nagl S, Williams M, Williamson J. Objective Bayesian nets for systems modelling and prognosis in breast cancer. In: Holmes DE, Jain LC, editors. Innovations in Bayesian Networks: Theories and Applications. Berlin, Germany: Springer; 2008. pp. 131–167. [Google Scholar]
- 30.Sebastiani P, Nolan VG, Baldwin CT, et al. A network model to predict the risk of death in sickle cell disease. Blood. 2007;110:2727–2735. doi: 10.1182/blood-2007-04-084921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Swartz RJ, West LA, Boiko I, et al. Classification using the cumulative log-odds in the quantitative pathologic diagnosis of adenocarcinoma of the cervix. Gynecol Oncol. 2005;99(suppl 1):S24–S31. doi: 10.1016/j.ygyno.2005.07.038. [DOI] [PubMed] [Google Scholar]
- 32.Možina M, Deműar J, Kattan M, et al. Nomograms for visualization of naive Bayesian classifier. Presented at the 8th European Conference on Principles and Practice of Knowledge Discovery in Databases; September 20-24, 2004; Pisa, Italy. [Google Scholar]
- 33.Kool M, Koster J, Bunt J, et al. Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PLoS One. 2008;3:e3088. doi: 10.1371/journal.pone.0003088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu D, Cozma D, Park A, et al. Functional validation of genes implicated in lymphomagenesis: An in vivo selection assay using a Myc-induced B-cell tumor. Ann N Y Acad Sci. 2005;1059:145–159. doi: 10.1196/annals.1339.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Majumder PK, Febbo PG, Bikoff R, et al. mTOR inhibition reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nat Med. 2004;10:594–601. doi: 10.1038/nm1052. [DOI] [PubMed] [Google Scholar]
- 36.Högerkorp CM, Bilke S, Breslin T, et al. CD44-stimulated human B cells express transcripts specifically involved in immunomodulation and inflammation as analyzed by DNA microarrays. Blood. 2003;101:2307–2313. doi: 10.1182/blood-2002-06-1837. [DOI] [PubMed] [Google Scholar]
- 37.Coller HA, Grandori C, Tamayo P, et al. Expression analysis with oligonucleotide microarrays reveals that MYC regulates genes involved in growth, cell cycle, signaling, and adhesion. Proc Natl Acad Sci U S A. 2000;97:3260–3265. doi: 10.1073/pnas.97.7.3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kegg: Histidine metabolism: Reference pathway. http://www.genome.jp/kegg/pathway/map/map00340.html.
- 39.Yoon JW, Kita Y, Frank DJ, et al. Gene expression profiling leads to identification of GLI1-binding elements in target genes and a role for multiple downstream pathways in GLI1-induced cell transformation. J Biol Chem. 2002;277:5548–5555. doi: 10.1074/jbc.M105708200. [DOI] [PubMed] [Google Scholar]
- 40.Zhang Y, Jamaluddin M, Wang S, et al. Ribavirin treatment up-regulates antiviral gene expression via the interferon-stimulated response element in respiratory syncytial virus-infected epithelial cells. J Virol. 2003;77:5933–5947. doi: 10.1128/JVI.77.10.5933-5947.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cho J-Y, Tsherniak A, Tamayo P, et al. Integrative genomic analysis of medulloblastoma identifies a molecular subgroup that drives poor clinical outcome. J Clin Oncol. 2011;29:1424–1430. doi: 10.1200/JCO.2010.28.5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Herms J, Neidt I, Lüscher B, et al. C-MYC expression in medulloblastoma and its prognostic value. Int J Cancer. 2000;89:395–402. [PubMed] [Google Scholar]
- 43.Beroukhim R, Getz G, Nghiemphu L, et al. Assessing the significance of chromosomal aberrations in cancer: Methodology and application to glioma. Proc Natl Acad Sci U S A. 2007;104:20007–20012. doi: 10.1073/pnas.0710052104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baker SG, Cook NR, Vickers A, et al. Using relative utility curves to evaluate risk prediction. J R Stat Soc Ser A Stat Soc. 2009;172:729–748. doi: 10.1111/j.1467-985X.2009.00592.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.DeLong ER, DeLong DM, Clarke-Pearson DL. Comparing the areas under two or more correlated receiver operating characteristic curves: A nonparametric approach. Biometrics. 1988;44:837–845. [PubMed] [Google Scholar]
- 46.Pencina MJ, D'Agostino RB, Sr, D'Agostino RB, Jr, et al. Evaluating the added predictive ability of a new marker: From area under the ROC curve to reclassification and beyond. Stat Med. 2008;27:157–172. doi: 10.1002/sim.2929. discussion 207-212. [DOI] [PubMed] [Google Scholar]
- 47.Salvesen HB, Carter SL, Mannelqvist M, et al. Integrated genomic profiling of endometrial carcinoma associates aggressive tumors with indicators of PI3 kinase activation. Proc Natl Acad Sci U S A. 2009;106:4834–4839. doi: 10.1073/pnas.0806514106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dickson BC, Mulligan AM, Zhang H, et al. High-level JAG1 mRNA and protein predict poor outcome in breast cancer. Mod Pathol. 2007;20:685–693. doi: 10.1038/modpathol.3800785. [DOI] [PubMed] [Google Scholar]
- 49.Harrell FE. Logistic Regression, and Survival Analysis. New York, NY: Springer-Verlag; 2001. Regression Modeling Strategies: With Applications to Linear Models. [Google Scholar]
- 50.Iasonos A, Schrag D, Raj GV, et al. How to build and interpret a nomogram for cancer prognosis. J Clin Oncol. 2008;26:1364–1370. doi: 10.1200/JCO.2007.12.9791. [DOI] [PubMed] [Google Scholar]
- 51.Cronin M, Sangli C, Liu ML, et al. Analytical validation of the Oncotype DX genomic diagnostic test for recurrence prognosis and therapeutic response prediction in node-negative, estrogen receptor-positive breast cancer. Clin Chem. 2007;53:1084–1091. doi: 10.1373/clinchem.2006.076497. [DOI] [PubMed] [Google Scholar]
- 52.Slodkowska EA, Ross JS. MammaPrint 70-gene signature: Another milestone in personalized medical care for breast cancer patients. Expert Rev Mol Diagn. 2009;9:417–422. doi: 10.1586/erm.09.32. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.